
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bipyridine complexes of E3+ (E = P, As, Sb, Bi): strong Lewis acids, sources of E(OTf)3 and synthons for EI and EV cations†
Saurabh S.
Chitnis
a,
Alasdair P. M.
Robertson
a,
Neil
Burford
*a,
Brian O.
Patrick
b,
Robert
McDonald
c and
Michael J.
Ferguson
c
aDepartment of Chemistry, University of Victoria, Victoria, British Columbia V8W 3V6, Canada. E-mail: nburford@uvic.ca; Fax: +1 250 721 7147; Tel: +1 250 721 7150
bDepartment of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada
cDepartment of Chemistry, University of Alberta, Edmonton, Alberta T6G 2T2, Canada
First published on 3rd August 2015
Abstract
Triflate salts of trications [(bipy)2E]3+ ([6E][OTf]3) and [(tbbipy)2E]3+ ([6′E][OTf]3) (bipy = 2,2′-bipyridine, tbbipy = 4,4′-di-tbutyl-2,2′-bipyridine; E = P, As, Sb, Bi) have been synthesized and comprehensively characterized. The unique molecular and electronic structures of this new class of complexes involving pnictogen Lewis acids has been assessed in the solid, solution and gas phases to reveal systematic variations in metric parameters, ligand lability and charge concentration. While the Lewis acidity of E3+ has the trend E = Bi < Sb < As < P as determined by gas-phase calculations and 1H NMR spectroscopy, the Lewis acidity of [6E]3+ has the trend E = P < As < Sb < Bi according to gas-phase calculations. Derivatives of [6′E][OTf]3 (E = P, As) are latent sources of E(OTf)3 as demonstrated by their reactions with dmap, which give the corresponding derivatives of [(dmap)3E][OTf]3. The highly oxidizing nature of P(OTf)3 and As(OTf)3 is evidenced in reactions of [6′E][OTf]3 (E = P, As) with phosphines, which give EI-containing monocations [(R3P)2E]1+ and oxidatively coupled dications [R3PPR3]2+, illustrating new P–P and P–As bond forming strategies. Cations [6′E]3+ (E = P, As) are C–H bond activating agents that dehydrogenate 1,4-cyclohexadiene, with higher activity observed for E = P. Combinations of [6′E]3+ and tBu3P activate H2 and D2 under mild conditions, evidencing frustrated Lewis pair activity. Oxidation of [6′P][OTf]3 with SO2Cl2 gives [(tbbipy)2PCl2][OTf]3, containing a PV-trication, but there is no evidence of the analogous reaction with [6′As][OTf]3. The observations highlight new directions in the chemistry of highly charged cations and reveal a rich reactivity for p-block triflates E(OTf)3, which can be accessed through derivatives of [6E][OTf]3 and [6′E][OTf]3.
Introduction
Numerous monocationic and dicationic p-block element centered complexes are known,1 but structurally authenticated salts containing trications are rare, because the charge concentration often results in oxidation of the ligands. For example, the trisphosphine-antimony trication 1 (Chart 1) undergoes reductive elimination of a diphosphonium dication below room temperature.2,3 In this context, the pyridine ligands in 2,4 the tris-pyrazole based ligands in 3a,b,5 the carbene based ligands in 4a–c,6 and the crown ether ligands in 5E7 illustrate types of oxidatively resistant donors that enable studies of such reactive coordination centers.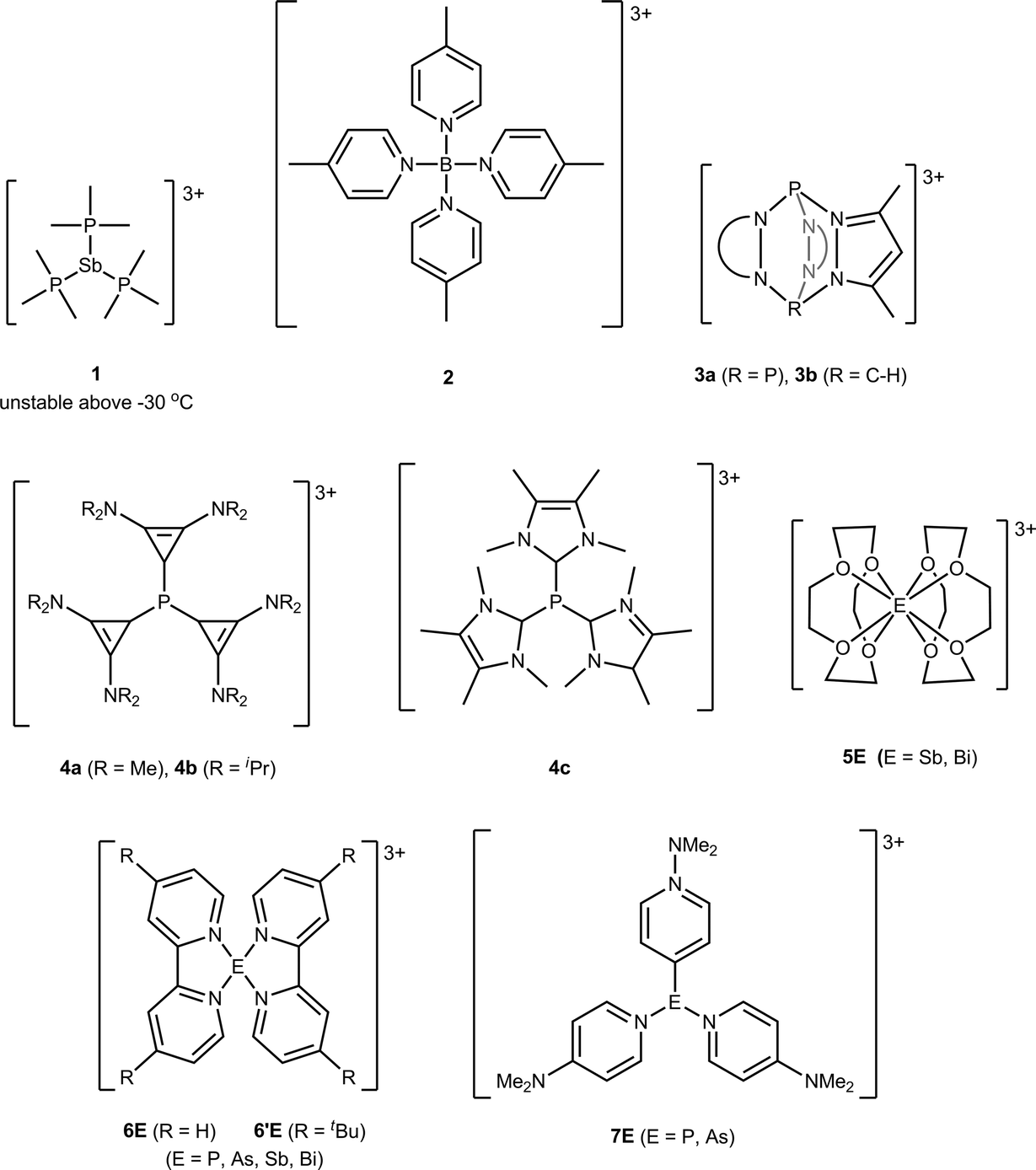 | ||
| Chart 1 p-Block element centered tricationic complexes. | ||
As a prototypical ligand for transition metal acceptors in a variety of oxidation states, 2,2′-bipyridine (bipy) offers relatively high basicity and oxidative resistance, which we have now exploited to enable a comprehensive study of a series of compounds of generic formulae [(bipy)2E][OTf]3, [6E][OTf]3, and [(tbbipy)2E][OTf]3, [6′E][OTf]3 (E = P, As, Sb, Bi; tbbipy = 4,4′-di-tbutyl-2,2′-bipyridine).8 The compounds are characterized as salts containing trications that represent bipy or tbbipy complexes of E3+. A diverse reactivity is evident for these complexes, including ligand exchange, which provides access to the dmap complexes [7E][OTf]3 (E = P, As). Element triflates, E(OTf)n, are widely employed as Lewis acids,9 oxidizing agents10–12 and latent sources of En+.13–15 Interesting examples of small-molecule activation and catalysis effected by p-block element triflate salts are also well documented,16–18 including those involving Sb(OTf)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and Bi(OTf)3,20 which can be isolated on preparative scales,21,22 enabling their ubiquitous use. Lighter congeners featuring the more electronegative P and As centers have not yet been reported, precluding assessment of their reactivity. In this context, we demonstrate that derivatives of [6E][OTf]3 and [6′E][OTf]3 represent examples of E(OTf)3 transfer reagents, C–H and H–H bond activating reagents, and synthons for EI- and EV-centered cations.
19 and Bi(OTf)3,20 which can be isolated on preparative scales,21,22 enabling their ubiquitous use. Lighter congeners featuring the more electronegative P and As centers have not yet been reported, precluding assessment of their reactivity. In this context, we demonstrate that derivatives of [6E][OTf]3 and [6′E][OTf]3 represent examples of E(OTf)3 transfer reagents, C–H and H–H bond activating reagents, and synthons for EI- and EV-centered cations.
Results and discussion
Complexes of E(OTf)3 with bipy or tbbipy were prepared according to Scheme 1 and isolated as crystalline solids. While all derivatives decompose to give the protonated ligand on exposure to ambient atmosphere, they can be stored indefinitely under inert atmosphere at room temperature. Derivatives of [6E][OTf]3 are less soluble in MeCN and CH2Cl2 than derivatives of [6′E][OTf]3, due to the presence of four tbutyl groups in the latter. Interestingly, while the phosphorus derivatives are yellow due to a HOMO–LUMO transition centered around 300 nm,23 all other derivatives are colourless as solids or in MeCN solutions. | ||
| Scheme 1 Synthesis of [6E][OTf]3 and [6′E][OTf]3. | ||
The solid-state structures of [6P][OTf]3·2MeCN, [6′P][OTf]3·MeCN, [6′As][OTf]3·2.83MeCN, [6Sb][OTf]3·MeCN, [6′Sb][OTf]3·MeCN, and [6Bi][OTf]3·MeCN have been determined to reveal spirocyclic environments for E with four E–N bonds (10-E-4 as per the Arduengo nomenclature24) and varying degrees of E–O triflate contacts, as shown in Fig. 1. Selected metric parameters for derivatives of [6E][OTf]3 and [6′E][OTf]3 are collated in Table 1, where computationally determined (gas phase) values are listed for [6As]3+ and [6′Bi]3+, for which experimental solid state data are not available. In all cases, the structures indicate the stereochemical presence of a lone pair at the acceptor pnictogen centre. The intermolecular MeCN⋯P interaction for [6′P][OTf]3·MeCN (coordinated solvent) and interion O⋯E interactions (triflate anions) in [6P][OTf]3, [6′P][OTf]3·MeCN, [6′As][OTf]3·2.83MeCN, and [6′Sb][OTf]3·MeCN are closer in magnitude to ∑r,vdW than to ∑r,cov for the elements involved. In contrast, short Bi⋯O interactions are observed in [6Bi][OTf]3, representing elongated Bi–O covalent bonds rather than triflate anions interacting with a bismuth cation. We therefore classify all derivatives as ionic except [6Bi][OTf]3 which is best described in the solid state as a bis-bipy adduct of Bi(OTf)3.
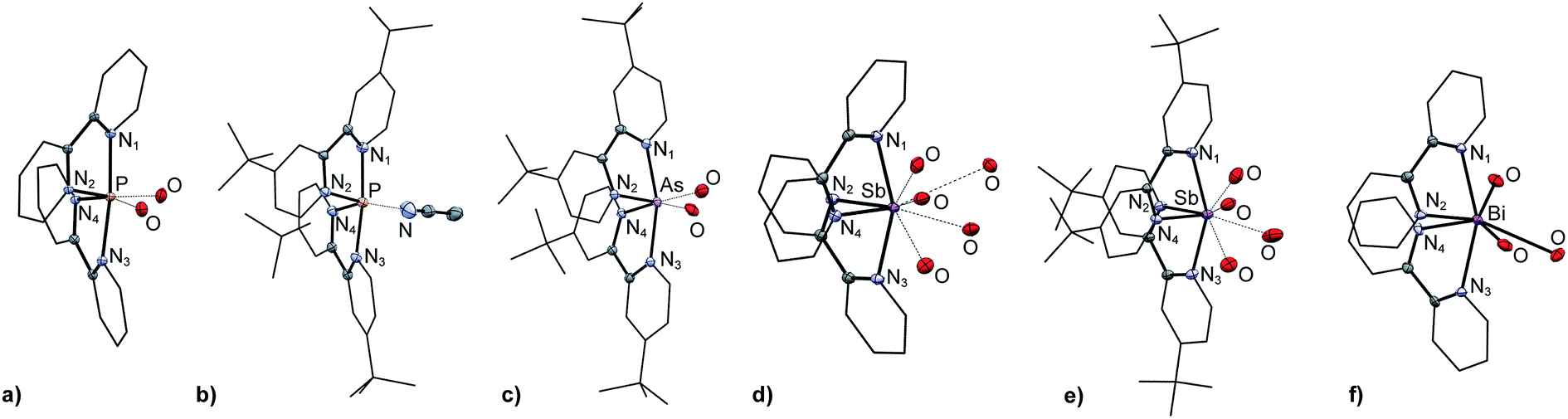 | ||
| Fig. 1 Solid-state molecular structures of the cations in (a) [6P][OTf]3·2MeCN, (b) [6′P][OTf]3·MeCN, (c) [6′As][OTf]3·2.83MeCN, (d) [6Sb][OTf]3·MeCN, (e) [6′Sb][OTf]3·MeCN, and (f) [6Bi][OTf]3. Hydrogen atoms, non-interacting portions of the triflate anions and solvent molecules have been omitted for clarity. | ||
| [6P][OTf]3 ·2MeCN | [6′P][OTf]3 ·MeCN | [6As]3+ | [6′As][OTf]3 ·2.83MeCN | [6Sb][OTf]3 ·MeCN | [6′Sb][OTf]3 ·MeCN | [6Bi][OTf]3 ·MeCN | [6′Bi]3+ | |
|---|---|---|---|---|---|---|---|---|
| E–N1 | 1.939(2) | 1.971(2) | 2.0993 | 2.124(2) | 2.284(2) | 2.269(2) | 2.454(6) | 2.3461 |
| E–N2 | 1.811(2) | 1.812(2) | 1.9714 | 1.997(2) | 2.233(2) | 2.198(2) | 2.364(6) | 2.2519 |
| E–N3 | 1.974(2) | 1.959(2) | 2.0993 | 2.125(2) | 2.332(2) | 2.310(2) | 2.430(6) | 2.3461 |
| E–N4 | 1.816(2) | 1.811(2) | 1.9714 | 1.999(2) | 2.243(2) | 2.218(2) | 2.372(6) | 2.2519 |
| ∑r,cov(E, N) | 1.82 | 1.82 | 1.92 | 1.92 | 2.11 | 2.11 | 2.22 | 2.22 |
| ∑r,vdW(E, N) | 3.35 | 3.35 | 3.40 | 3.40 | 3.61 | 3.61 | 3.62 | 3.62 |
| E–OOTf | 3.006(2) | — | 2.705(2) | 2.598(2) | 2.586(2) | 2.607(6) | — | |
| 3.109(2) | 2.742(2) | 2.650(2) | 3.113(2) | 2.532(6) | ||||
| 3.077(1) | 2.767(2) | 2.893(6) | ||||||
| 3.247(1) | 3.343(2) | |||||||
| 3.367(1) | ||||||||
| ∑r,cov(E, O) | 1.74 | 1.74 | 1.84 | 1.84 | 2.03 | 2.03 | 2.14 | 2.14 |
| ∑r,vdW(E, O) | 3.32 | 3.32 | 3.37 | 3.37 | 3.58 | 3.58 | 3.59 | 3.59 |
| N1–E–N2 | 82.20(7) | 81.61(8) | 78.86 | 77.67(8) | 72.09(4) | 72.43(5) | 68.1(2) | 71.53 |
| N3–E–N4 | 82.07(7) | 82.25(8) | 78.86 | 77.76(8) | 71.53(4) | 68.2(2) | 71.53 | |
| N1–E–N3 | 173.09(8) | 173.10(8) | 168.47 | 162.75(8) | 156.02(4) | 153.45(5) | 154.4(2) | 152.92 |
| N1–E–N4 | 92.91(7) | 92.63(8) | 93.47 | 89.96(8) | 87.87(4) | 89.17(5) | 94.1(2) | 90.16 |
| N2–E–N3 | 93.36(7) | 94.44(8) | 93.47 | 90.45(8) | 91.42(4) | 86.70(5) | 88.8(2) | 90.16 |
| N2–E–N4 | 99.57(8) | 97.56(8) | 97.39 | 91.54(8) | 78.66(4) | 81.98(5) | 74.7(2) | 95.78 |
The bond angles for a given E in [6E][OTf]3 and [6′E][OTf]3 are expected to be very similar because the divergent planes defined by N1–E–N2 and N3–E–N4 result in the tBu groups facing away from each other, so that steric repulsion between them is minimal. For example, the quaternary carbon centers in the tBu groups that are para to N2 and N4 in [6′P][OTf]3 are separated by nearly 10 Å, and the bond angles within the disphenoidal frames of [6P]3+ and [6′P]3+ are essentially identical, as they are for [6Sb]3+ and [6′Sb]3+. The inductive effect of a tBu group para to the nitrogen atoms is expected to make the tbbipy ligands more basic compared to bipy and lead to stronger E–N interactions. Consistently, the 31P NMR spectrum of a CD3CN mixture containing tbbipy and [6P][OTf]3 in a 2:1 stoichiometry showed a broad peak corresponding to [6P′][OTf]3. The E–N distances in [6P][OTf]3·2MeCN and [6′P][OTf]3·MeCN are similar, although the presence of a coordinated MeCN donor in the latter may reduce the electrophilicity of the phosphorus center and offset the expected E–N shortening. Better suited for direct comparison are [6Sb][OTf]3·MeCN and [6′Sb][OTf]3·MeCN, where the MeCN molecule in the lattice does not interact with the Sb centers. Evidencing the inductive effect of the para-tBu group, E–N distances in [6′Sb][OTf]3·MeCN are on average 0.1 Å shorter than the bipy derivative, which also shows two more Sb–O interion contacts than does the tbbipy derivative. Moreover, the Sb–O contacts in [6′Sb][OTf]3·MeCN are on average 0.05 Å longer than those in [6Sb][OTf]3·MeCN. These observations support a slightly greater Lewis basicity for tbbipy, which is discernible in the E–N distances, in the absence of additional donors (e.g. coordinated solvent) at E.
The trans configured E–N bonds in all derivatives are ca. 0.1 Å longer than the E–N bonds in equatorial positions due to the mutual trans influence of the E–N1 and E–N3 interactions (Fig. 2). For the equatorial positions, the trend in E–N bond lengths, [6P]3+ ≈ [6′P]3+ < [6′As]3+ < [6Sb]3+ ≈ [6′Sb]3+ < [6Bi]3+, reflects the sum of the respective covalent radii (∑r,cov). The trend in the axial (N1–E–N3) and equatorial interligand angles (N2–E–N4), [6P]3+ ≈ [6′P]3+ > [6′As]3+ > [6Sb]3+ ≈ [6′Sb]3+ > [6Bi]3+, is consistent and is attributed to the extent of triflate anion association, which is greater for an atom with a larger atomic radius. The chelation angles (N1/3–E–N2/4) exhibit the trend [6P]3+ ≈ [6′P]3+ > [6′As]3+ > [6Sb]3+ ≈ [6′Sb]3+ > [6Bi]3+ as the N–E–N interaction subtends smaller angles for longer E–N bonds.
 | ||
| Fig. 2 Definition of key angles in the disphenoidal geometry of [6E]3+. | ||
Infrared spectra for derivatives of [6′E][OTf]3 enable quantification of the interion coordination in the solid state. The symmetric SO3 stretch, νs(SO3), in several triflate salts has been studied previously and appears as a characteristically sharp absorbance in the 1020–1050 cm−1 range.28 The portions of the infrared spectra of [6′E][OTf]3 shown in Fig. 3 illustrate a trend in ν(SO3) of E = P > As > Sb > Bi, which we attribute to the degree of charge transfer from the anion to the pnictogen centre, which influences the S–O bond order. The broader bands for the heavier homologues are attributed to the loss of C3v symmetry due to cation–anion interaction. The spectra for ligand-free Sb(OTf)3 and Bi(OTf)3, for which extensive Sb–O and Bi–O interactions are predicted, exhibit similarly broad S–O stretching bands (see Fig. S2, ESI†) that are shifted to lower frequencies (958 cm−1 and 1000 cm−1, respectively) than the analogous value in [Bu4N][OTf] (1032 cm−1),28 which features a weakly coordinating cation, and the calculated value for an isolated triflate anion in the gas phase (1035 cm−1, PBE0/aug-cc-pVTZ).
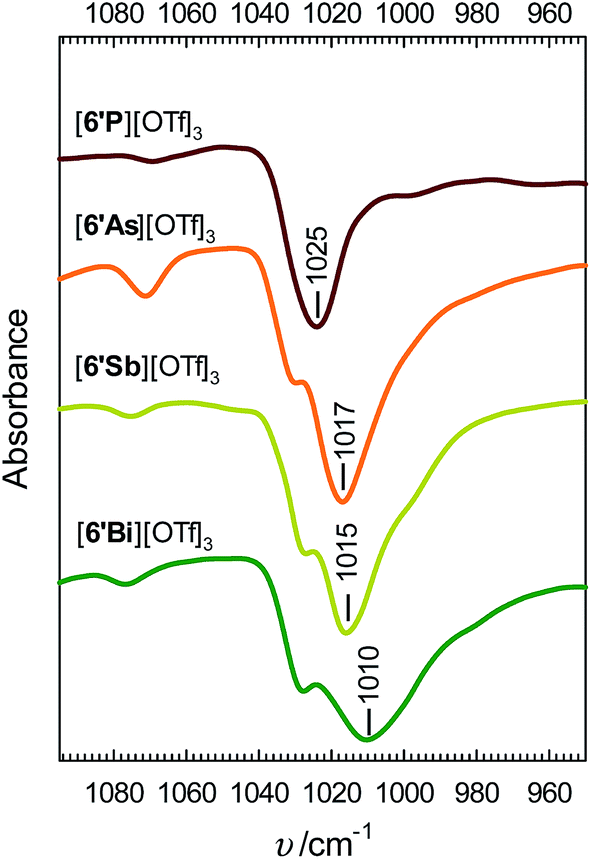 | ||
| Fig. 3 Infrared spectra (950–1100 cm−1) of [6′E][OTf]3 obtained on powdered salts using an ATR module. | ||
Gas-phase structures, bonding, and Lewis acidity of [6E]3+
Optimized structures for [6E]3+ in the gas phase adopt a disphenoidal C2 symmetry for all derivatives, consistent with the observed solid-state structures. Selected calculated bond lengths and angles are given in Table 2. As observed experimentally in the solid state, the computed structures reveal axial E–N distances that are longer by ca. 0.1 Å than the equatorial E–N distances and average E–N bond distances that are primarily determined by the respective covalent radii. For pnictogen centers with a larger covalent radius, the bite angle N1/3–E–N2/4 and the equatorial and the axial interligand angles are smaller. The equatorial interligand angle in the solid-state structures of Sb and Bi derivatives are significantly smaller than in the anion-free gas-phase structures of [6Sb]3+ and [6Bi]3+, suggesting that the steric pressure of the interion contacts present in the solid state influence this angle. By comparison the axial interligand angle N1–E–N3 in the experimental and calculated structures are essentially identical, implying minimal distortion due to interion contacts.| Cation | N1/3–E | N2/4–E | N1/3–E–N2/4 | N2–E–N4 | N1–E–N3 |
|---|---|---|---|---|---|
| [6P]3+ | 1.9511 | 1.8224 | 82.72 | 100.61 | 175.68 |
| [6As]3+ | 2.0993 | 1.9714 | 78.86 | 97.39 | 168.47 |
| [6Sb]3+ | 2.2545 | 2.1677 | 73.96 | 95.19 | 157.34 |
| [6Bi]3+ | 2.3607 | 2.2678 | 71.50 | 95.12 | 153.59 |
Natural Bond Orbital (NBO) partial charges and Wiberg Bond Indices (WBI) for the gas-phase cations are listed in Table 3, evidencing a high positive charge for the central pnictogen centre, which is greater for heavier elements, as expected on the basis of relative electronegativities. Consistently, the WBIs for the N–E interaction has the trend P > As > Sb > Bi, implying a more ionic E–N bond for the heavier pnictogens. For a given derivative, the WBI value for the axial E–N interactions is smaller than the equatorial interactions, indicating less effective bonding along the N1–E–N3 axis than in the N2–E–N4 plane. Noting that the equatorial interligand angles range from 95 to 100° in all cases, we surmise that of the three mutually perpendicular p-orbitals that serve as acceptor orbitals at E3+, two are engaged by N2 and N4 in the equatorial plane, while the third accommodates two strained trans interactions involving N1 and N3.
| E | Charge (E) | Charge (N1/3) | Charge (N2/4) | WBI (N1/3–E) | WBI (N2/4–E) |
|---|---|---|---|---|---|
| P | +1.40 | −0.53 | −0.52 | 0.49 | 0.69 |
| As | +1.58 | −0.54 | −0.54 | 0.43 | 0.61 |
| Sb | +1.78 | −0.55 | −0.56 | 0.39 | 0.58 |
| Bi | +1.86 | −0.54 | −0.55 | 0.36 | 0.50 |
To assess the relative Lewis acidities of E3+, we have calculated the enthalpies for the heterolytic removal of both bipy ligands from [6E]3+. Scheme 2a represents removal of the ligands and relaxation of their geometries to the C2h minimum for free bipy, and Scheme 2b represents removal of the ligands with retention of the geometry observed in [6E]3+. The difference between the two enthalpies represents the energy required for two non-interacting bipy molecules (C2h) to adopt the (bipy)2 geometry in each complex (Scheme 2c). The ΔHrxn values in Table 4 show that the enthalpic requirement for ligand dissociation from E3+ has the trend E = Bi < Sb < As < P, irrespective of whether or not steric factors are considered. Values for ligand strain show a parallel trend, but the range (98–181 kJ mol−1) is small compared to the range for the overall ligand dissociation process (2302–3575 kJ mol−1). We therefore conclude that steric effects have a minor influence on the calculated enthalpies of ligand dissociation in Scheme 2a, which are dominated by electronic effects.
 | ||
| Scheme 2 (a) Dissociation of two bipy ligands in [6E]3+, (b) dissociation of two bipy ligands in [6E]3+ with retention of the (bipy)2 geometry of [6E]3+, (c) organization of two bipy ligands to the (bipy)2 geometry found in [6E]3+, and (d) dissociation of OPMe3 from [(bipy)2E(OPMe3)]3+. | ||
We rationalize the calculated trend in dissociation enthalpies on the basis of atomic size, with a smaller atom having a higher charge concentration and the best orbital match in the N(sp3)→E(np) HOMO–LUMO interaction (cf. N(sp3)→P(3p) vs. As(4p) vs. Sb(5p) vs. Bi(6p)). The electrostatic and orbital interactions are both expected to weaken as atomic radii and the number of nodes in the acceptor p-orbitals increase. The trend in ligand strain is presumably related to the N1–E–N3 angle, which shows the most dramatic variation amongst all parameters in the calculated structures of [6E]3+, and decreases over a 22° range from phosphorus (175.68°) to bismuth (153.59°). We propose that the strained ligand geometry in [6P]3+ is enforced by orbital interactions involving three mutually perpendicular 3p acceptor orbitals at the P3+ centre. By comparison, in [6Bi]3+, where E–N bonding is calculated to be more ionic (Table 3), the preference for an N1–E–N3 angle of 180° is lowest.
While reaction enthalpies for Scheme 2a and b represent the Lewis acidity of monoatomic trications E3+, ΔHrxn for Scheme 2d assesses the Lewis acidities of complexes [6E]3+ by measuring the energy required for removal of a prototypical ligand, OPMe3, from hypothetical complexes [(bipy)2E(OPMe3)]3+. The enthalpies for this process indicate that the Lewis acidity of complexes [6E]3+ has the trend E = P < As < Sb < Bi, which is the opposite trend to that of monoatomic E3+, and is rationalised on steric grounds acknowledging the trend in atomic radii and consequential coordination sphere. Consistently, the range of enthalpy values calculated for Scheme 2d (178–255 kJ mol−1) is much smaller than that observed for Scheme 2a (2302–3575 kJ mol−1) and is comparable to the ligand strain enthalpies calculated for Scheme 2c (98–181 kJ mol−1). In addition, comparison of the optimized structures for [6E]3+ and [(bipy)2E(OPMe3)]3+ shows that the greatest geometric deformation upon complexation with OPMe3 is compression of the interligand angle N2–E–N4 (see Fig. 2 for definition). The magnitude of this geometric adjustment, which leads to steric clash between the bipy ligands, is greatest for E = P (15°) and least for E = Bi (7°), consistent with the calculated trend for Scheme 2d.
NMR characterization of [6E][OTf]3 and [6′E][OTf]3
CD3CN solutions of [6E][OTf]3 and [6′E][OTf]3 exhibit 19F NMR chemical shift values for all species in the range −78.9 to −79.5 ppm (cf. −79.4 for [PPh4][OTf]), indicative of dissociated triflate ions. In addition, solutions of all derivatives polymerize THF within hours of mixing, implicating a high Lewis acidity29,30 in coordinating solvents. No significant change was observed in the 1H or 31P NMR shifts of salts [6′E][OTf]3 over a broad concentration range, implying the absence of a bimolecular association process as might be expected from an equilibrium between the anion-bound and anion-free cations (see representative data for [6′Bi][OTf]3 in Fig. S3, ESI†). We conclude that CD3CN solutions of [6E][OTf]3 and [6′E][OTf]3 contain solvated trications and triflate anions with minimal interion interaction.The aromatic resonances in the 1H NMR spectra of derivatives of [6E][OTf]3 in CD3CN are shown in Fig. 4. As predicted for a C2 symmetric bis-bipy complex, eight aromatic resonances are detected for [6P][OTf]3 at 25 °C. For [6As][OTf]3, four broad peaks are observed, which broaden further upon cooling to 0 °C and resolve into additional peaks upon cooling to −35 °C. Only four aromatic resonances are detected for [6Sb][OTf]3 and [6Bi][OTf]3, at 25 °C and at −35 °C. While the solid-state structure, featuring eight unique hydrogen environments for the cations in [6E][OTf]3 (Fig. 1), is apparently retained in solution for E = P, the mobility of the bipy ligands at 25 °C is sufficiently high for E = As, Sb, and Bi that only four hydrogen environments are detected. At −35 °C, ligand mobility is partially restricted for [6As][OTf]3 leading to additional signals but complete resolution of eight hydrogen environments, as in [6P][OTf]3, was not detected. The observations indicate a mobility for the bipy ligands around E with the trend E = P < As < Sb ≈ Bi and parallels the trend in ionicity for the E–N bond (Table 3). We propose that the more covalent N–P and N–As bonds are conformationally rigid due to the directional requirements of efficient orbital overlap to make a covalent bond (three mutually perpendicular p-orbitals), whereas the more ionic N–Sb and N–Bi interactions have a smaller barrier to motion due to the absence of a directional component for electrostatic interactions (point charges).
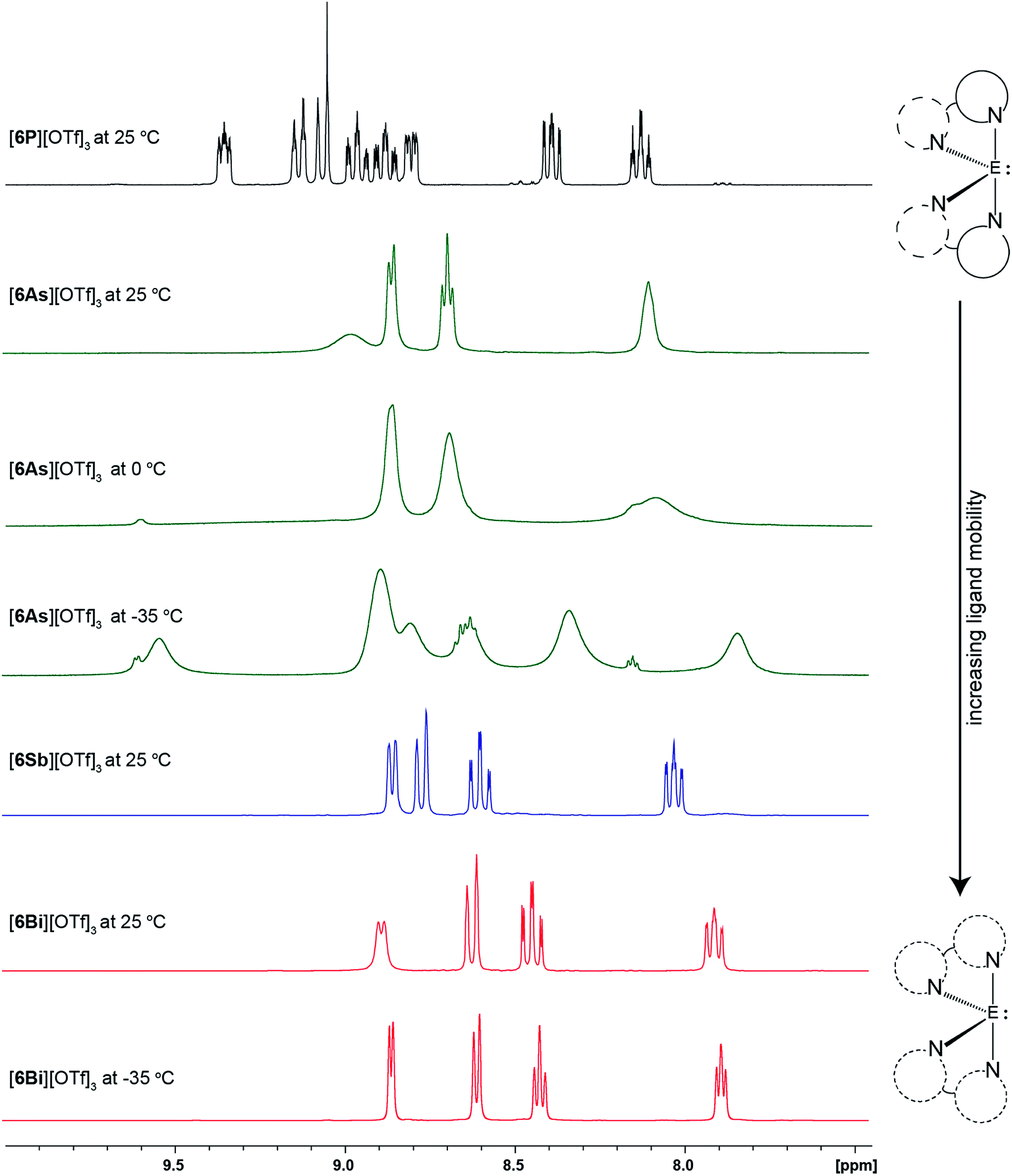 | ||
| Fig. 4 1H NMR resonances in the aromatic region for CD3CN solutions of [6P][OTf]3 (black), [6As][OTf]3 (green), [6Sb][OTf]3 (blue), and [6Bi][OTf]3 (red). | ||
The difference between the 31P NMR chemical shift of free Et3PO and that of its adduct with a Lewis acid has been correlated with the strength of the Lewis acid (Gutmann–Beckett method).31,32 No systematic trend was observed in the chemical shifts observed (see Fig. S4, ESI†) in 31P NMR assays of solutions containing equimolar amounts of Et3PO and [6′E][OTf]3.33 Moreover, in the case of [6′P][OTf]3, a complex spectrum showing a mixture of products was obtained, none of which could be assigned to the phosphine oxide adduct. Deoxygenation of Et3PO by electrophilic phosphorus cations has been reported recently and may be operative.34 Moreover, as a wide range in covalent radii (1.11–1.51 Å)25 is spanned going from P to Bi, the steric influence on 31P chemical shifts may be greater than those due to differing Lewis acidities, confounding a straightforward assessment due to steric factors, as highlighted recently for borane Lewis acids.35
Reactivity of [6′P][OTf]3 and [6′As][OTf]3
The structures of [6P][OTf]3 and [6′P][OTf]3 represent rare examples of hypervalent phosphorus(III) acceptor centres, and are comparable to those involving N-heterocyclic carbene (NHC),36 phosphine,37,38 catecholate,39 and phenylpyrazole40 ligands. Moreover, electron precise (8 valence electron) phosphorus based frameworks 3 and 4 (Chart 1) are the only phosphorus(III) centred trications that have been structurally characterized.41 By comparison, the 10 valence electron count imposed by the two chelate ligands at phosphorus in [6P]3+ and [6′P]3+ render these trications as novel examples of electron-rich phosphorus Lewis acids. Examples of arsenic(III)-centred mono- and dications featuring phosphine42 or bipy43 ligands have been reported as well as two-coordinate arsenium monocations.44–46 However, [6′As][OTf]3 is the first structurally authenticated example of an arsenic-centred trication.The reactivity of Sb(OTf)3 and Bi(OTf)3 has been studied previously, leading to their widespread use as Lewis acid catalysts,19,20 but the absence of synthetic routes to P(OTf)3 and As(OTf)3 has precluded investigations of these potential synthetic reagents. Phosphorus polycations have been used as a precursors to cationic bicyclophosphines and cyclic phosphorus oxides,47 and derivatives of 4 (Chart 1) have been shown to bind transition metal centers via the lone pair at the phosphorus(III) center to give highly effective precatalysts for C–C bond forming reactions.48 Intrigued by the unique intersection of molecular and electronic structures represented by the trications in [6′P][OTf]3 and [6′As][OTf]3, and envisioning these salts as in situ equivalents of E(OTf)3 (E = P, As), we have conducted an initial survey of their reactivity.
[6′E][OTf]3 as E(OTf)3 transfer reagents
Reactions of [6′E][OTf]3 (E = P, As) with three equivalents of 4-dimethylaminopyridine (dmap) quantitatively (by 31P and 1H NMR) yield [(dmap)3E][OTf]3, [7E][OTf]3, (E = P, As) and free tbbipy (Scheme 3a). Neither dmap complexes could be isolated from the reaction mixtures but their identities were definitively established by independent syntheses (Scheme 3b) and structural elucidation (Fig. 5). While the [7P]3+ ion has previously been detected spectroscopically in mixtures of PCl3 and dmap,49,50 the 31P NMR chemical shift attributed to the trichloride salt was reported to vary widely (δ = 79–114 ppm) depending upon concentration, suggesting a dynamic process.51 By comparison, [7P][OTf]3 exhibits a 31P NMR chemical shift (δ = 101.7 ppm) for the redissolved crystals that does not vary over a broad concentration range.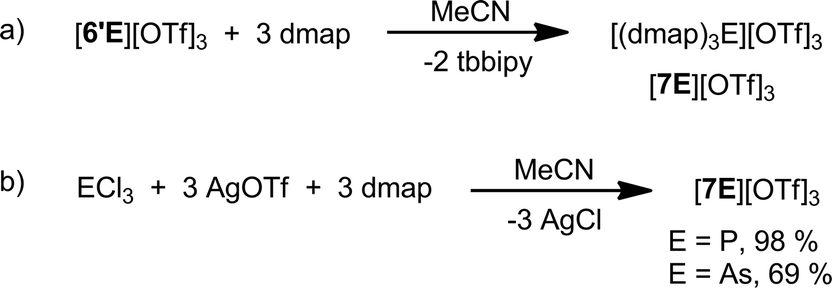 | ||
| Scheme 3 (a) Formation of [7E][OTf]3 from [6′E][OTf]3. (b) Independent synthesis of [7E][OTf]3 (E = P, As). | ||
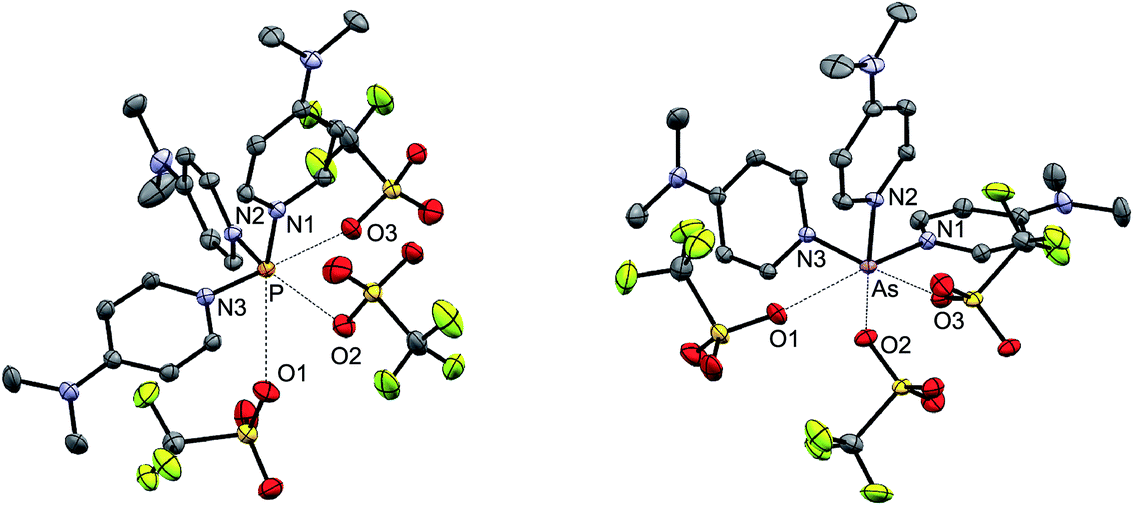 | ||
| Fig. 5 Solid state structures of [7P][OTf]3·1.5MeCN (left, one of two crystallographically distinct molecules shown) and [7As][OTf]3·2MeCN (right). Hydrogen atoms and solvent molecules have been omitted for clarity. | ||
The solid-state structures of [7E][OTf]3 reveal three dmap ligands bound to the pnictogen centers giving a trigonal pyramidal geometry at the pnictogen centre for the [7E]3+ ions.52 Three weak contacts with the triflate anions are evident, giving a six-coordinate geometry that is distorted by the presence of a stereochemically active lone pair in each case. The three weak E⋯O contacts are trans configured with respect to the three E–N bonds. The E–N bond lengths (Table 5) reflect the relative atomic radii of the phosphorus and arsenic atoms and are 0.1–0.2 Å shorter than the corresponding values in [6P][OTf]3, [6′P][OTf]3, and [6′As][OTf]3 due to the greater basicity and lesser steric demands of dmap compared to bipy or tbbipy. The N–E–N bond angles in [7E][OTf]3 are in the 90–100° range consistent with values observed for the cis-configured N–E–N angles in derivatives of [6E][OTf]3 and [6′E][OTf]3 (Table 5).
| [7P][OTf]3·1.5MeCN | [7As][OTf]3·2MeCN | |
|---|---|---|
| E–N1 | 1.7635(17) | 1.9157(17) |
| E–N2 | 1.7578(16) | 1.9447(16) |
| E–N3 | 1.7588(17) | 1.9174(16) |
| E–OOTf | 3.0462(18) | 2.8428(15) |
| 3.2615(17) | 2.654(2) | |
| 3.0395(19) | 2.969(2) | |
| N1–E–N2 | 98.42(8) | 92.51(7) |
| N2–E–N3 | 97.40(8) | 92.64(7) |
| N1–E–N3 | 99.24(8) | 96.45(7) |
[6′E]3+ as synthons for EI cations
In contrast to ligand exchange with dmap, reaction of [6′P][OTf]3 with PMe3 yields products due to redox chemistry (Scheme 4a). The previously reported PI containing reduction product, [(Me3P)2P]1+ (δ = 15.0 and −156.3 ppm, 1JPP = 438 Hz)53 and the PIV containing oxidation product, [Me3PPMe3]2+ (δ = 28.4 ppm),54 have been definitively identified by 31P NMR spectroscopy (Fig. S5, ESI†). The analogous reaction with PPh3 yielded [(Ph3P)2P]1+ (δ = 30, −174, 1JPP = 502 Hz)55 as the major product, but a complex mixture of oxidation products was obtained, suggesting that [Ph3PPPh3]2+, which is isoelectronic with the metastable hexaphenylethane molecule,56 may also be unstable relative to its constitutional isomers (Scheme 4b). Similarly, a 31P NMR assay of the 1:3 reaction between [6′As][OTf]3 and PMe3 showed a singlet due to [Me3PPMe3]2+ together with a resonance at 22.4 ppm, tentatively assigned to the AsI cation, [(Me3P)2As]1+, which could not be isolated from the reaction mixture (Scheme 4c). In a parallel experiment, a 31P NMR assay of the 1:2 reaction between [6′As][OTf]3 and 1,2-bis(diphenylphosphino)ethane (dppe) showed a singlet at 60.5 ppm due to the previously reported AsI cation [(dppe)As]1+,57 and unidentified oxidation products (Scheme 4d). We conclude that trications [6′E]3+ are strong oxidizing agents owing to their formidable molecular charge and effect oxidative P–P coupling while being reduced to PI or AsI containing monocations. This redox outcome contrasts the ligand displacement observed in the presence of the more oxidatively resistant ligand dmap, and is analogous to reactivity patterns established for FSb(OTf)2 and Sb(OTf)3.21
 | ||
| Scheme 4 Reactions of [6′E][OTf]3 (E = P, As) with phosphines. Species in blue were definitively identified by their previously reported 1H or 31P NMR resonances. | ||
C–H and H–H bond activation by [6′E]3+
The equimolar reaction of [6′P][OTf]3 with 1,4-cyclohexadiene in CD3CN showed complete consumption of starting materials after 16 hours at 80 °C (Scheme 5a). The 1H NMR of the reaction mixture showed formation of benzene (δ = 7.38 ppm) and diprotonated tbbipy as the major products (>80%, Fig. S6, ESI†). The 31P NMR spectrum exhibits a mixture of unidentified products, none of which exhibit P–H couplings. The spectroscopic data are consistent with C–H bond activation involving dehydrogenation of 1,4-cyclohexadiene and sequestering of protons in [tbbipy-H2]2+. The analogous reaction with [6′As][OTf]3 showed only 10% conversion of 1,4-cyclohexadiene to benzene over 16 h at 80 °C, with concomitant formation of [tbbipy-H2]2+ and an insoluble black precipitate (Fig. S7, ESI†). C–H bond activation has recently been reported for mixtures of diphosphonium dications and tBu3P.58 Consistently, the 1:2 combinations of [6′E][OTf]3 (E = P, As) and tBu3P in CD3CN effect complete dehydrogenation of 1,4-cyclohexadiene to yield benzene and [tBu3P-H]1+ within 16 hours at 80 °C (Scheme 5b). In line with the expectation that these reactions proceed via formation of a frustrated Lewis pair59 between [6′E]3+ and tBu3P, 31P NMR spectra of equimolar reaction mixtures containing tBu3P and either [6′P][OTf]3 or [6′As][OTf]3 show no evidence of coordination between the strong Lewis acids and the bulky base pairs (see Fig. S8, ESI†). Frustrated Lewis pair activity is also evidenced by 1:2 mixtures of [6′P][OTf]3 and tBu3P in CD3CN with H2 or D2 (1 atm pressure) in a sealed NMR tube at 80 °C over 16 hours (Scheme 5c), which show complete conversion of tBu3P to [tBu3P-H]1+ or [tBu3P-D]1+ by 31P NMR spectroscopy (Fig. S9, ESI†).
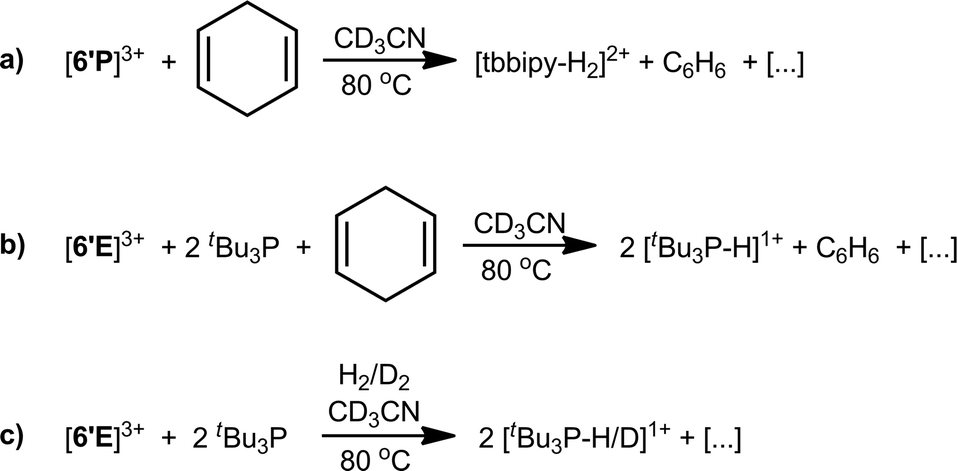 | ||
| Scheme 5 C–H and H–H bond activation by [6′E][OTf]3. | ||
[6′E]3+ as synthons for EV cations
The 31P NMR spectrum of an equimolar mixture of [6′P][OTf]3 and SO2Cl2 shows a single 31P NMR resonance at δ = −146.9 ppm, assigned to the PV containing [(tbbipy)2PCl2]3+. The upfield resonance is consistent with a five- or six-coordinate geometry and is similar to shifts reported for [(dmap)2PCl4]1+ (δ = −196 ppm)60 and [(bipy)PCl4]1+ (δ = −191 ppm).61 Moreover, the singlet at −146.9 ppm is also observed in the 31P NMR spectrum of a 2:1:3 mixture of tbbipy, PCl5 and TMSOTf.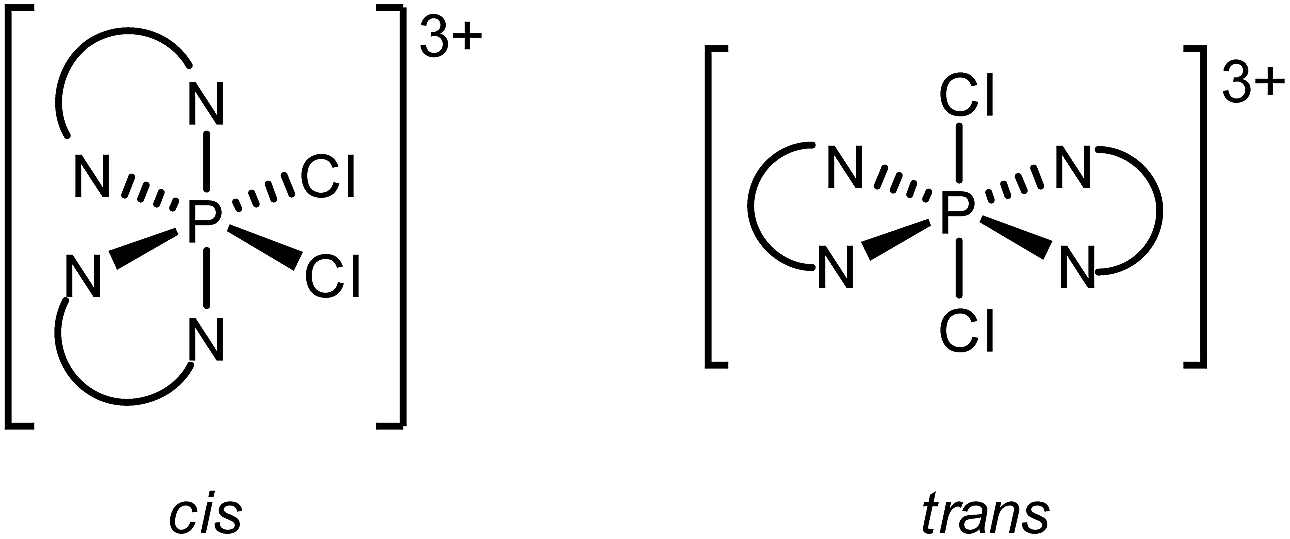
Two configurational outcomes are envisioned for the octahedral structure of [(tbbipy)2PCl2]3+, with a cis or trans arrangement of the chlorine atoms. The 1H NMR spectrum of the cation shows six resonances in the aromatic region (Fig. 6) and two resonances for the tBu groups, consistent with C2 symmetry, precluding a trans configuration of chlorine centres. Gas-phase calculations using bipy ligands revealed that both isomers are true energy minima (no negative vibrational frequencies), but a 64 kJ mol−1 preference for the cis isomer was calculated, arising from significant steric clash between the ortho hydrogen atoms of the ligands when a trans configuration is imposed (Fig. S10, ESI†). No cis/trans isomerism was detected experimentally upon heating a sample to 80 °C for an hour, consistent with the rigidity of the disphenoidal frame inferred for [6P]3+ from 1H NMR spectroscopy (Fig. 4).
 | ||
| Fig. 6 Portion of the 1H NMR spectrum (CD3CN, 298 K) of the crude reaction mixture containing equimolar amounts of [6′P][OTf]3 and SO2Cl2. | ||
Addition of excess Cl2 gas to MeCN solution of [6′P][OTf]3 yields a product with identical spectral features as those assigned to [(tbbipy)2PCl2]3+, as well as a number of unidentified byproducts. Interestingly, equimolar mixtures of [6′As][OTf]3 and SO2Cl2 showed no evidence of reaction even after heating to 80 °C for 2 hours. 1H NMR assays of these reaction mixtures showed only signals due to unreacted [6′As][OTf]3.
Conclusions
In summary, we have isolated and comprehensively characterized the bipyridine complexes [6E][OTf]3 and [6′E][OTf]3 for E = P, As, Sb, Bi, representing rare examples of salts containing trications and unique homologous series. The solid-state structures show systematic variations as a function of the atomic size of E. Larger element centers facilitate interion interactions for [6E][OTf]3 and [6′E][OTf]3 in the order E = P < As < Sb < Bi as determined by X-ray crystallography and infrared spectroscopy. Gas-phase calculations (PBE0/def2-TZVP) reveal a trend from polar covalent to ionic E–N bonds for [6E]3+ going from E = P to E = Bi, consistent with data from 1H NMR spectroscopy. The Lewis acidity of monoatomic trications E3+ exhibits the trend E = Bi < Sb < As < P based on calculation of charge densities and ligand dissociation energies in the gas phase. However the calculated Lewis acidity of complexes [6E]3+ towards a prototypical ligand, OPMe3, exhibit the opposite trend, E = P < As < Sb < Bi due to steric factors.Derivatives of [6′E][OTf]3 with E = P and As represent rare examples of non-metal triflates and E(OTf)3 transfer reagents, as illustrated by reactions with dmap, which proceed via ligand displacement to yield [(dmap)3E][OTf]3 and free tbbipy. Reactions of [6′E][OTf]3 with PR3 give access to EI-containing cations concomitant with oxidative P–P coupling. Cations [6′E]3+ (E = P, As) are single-component C–H bond activating agents as shown by dehydrogenation of 1,4-cyclohexadiene, which occurs more rapidly for E = P than for E = As. Both cations also dehydrogenate 1,4-cyclohexadiene in the presence of tBu3P, indicative of frustrated Lewis pair chemistry. Combinations of [6′E][OTf]3 (E = P, As) with tBu3P activate H2 or D2 under mild conditions to give [tBu3P-H/D]1+. While the reaction of [6′P][OTf]3 with SO2Cl2 furnished the PV-containing [(tbbipy)2PCl2][OTf]3, the analogous oxidation of [6′As][OTf]3 was not observed. These observations highlight a rich reaction chemistry for P(OTf)3 and As(OTf)3 (Scheme 6) that is rendered accessible in salts [6E][OTf]3 and [6′E][OTf]3.
 | ||
| Scheme 6 Reactivity of [6′E][OTf]3 (E = P, As). | ||
Acknowledgements
We thank the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Vanier Canada Graduate Scholarships Program for funding. We thank the referees for their valuable suggestions regarding additional reactivity studies of [6′E][OTf]3.References
- J. L. Dutton and P. J. Ragogna, Coord. Chem. Rev., 2011, 255, 1414–1425 CrossRef CAS.
- S. S. Chitnis, Y. Carpenter, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., Int. Ed., 2013, 52, 4863–4866 CrossRef CAS PubMed.
- S. S. Chitnis, A. P. M. Robertson, N. Burford, J. J. Weigand and R. Fischer, Chem. Sci., 2015, 6, 2559–2574 RSC.
- I. Vargas-Baca, M. Findlater, A. Powell, K. V. Vasudevan and A. H. Cowley, Dalton Trans., 2008, 6421–6426 RSC.
- (a) J. J. Weigand, K. Feldmann, A. K. C. Echterhoff, A. W. Ehlers and K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 6178–6181 CrossRef CAS PubMed; (b) L. Gu, G. Gopakumar, P. Gualco, W. Thiel and M. Alcarazo, Chem.–Eur. J., 2014, 20, 8575–8578 CrossRef CAS PubMed.
- (a) J. Petuskova, M. Patil, S. Holle, C. W. Lehmann, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2011, 133, 20758–20760 CrossRef CAS PubMed; (b) F. D. Henne, A. T. Dickschat, F. Hennesdorf, K.-O. Feldmann and J. J. Weigand, Inorg. Chem., 2015, 54, 6849–6861 CrossRef CAS PubMed.
- R. Garbe, B. Vollmer, B. Neumüller, J. Pehler and K. Dehnicke, Z. Anorg. Allg. Chem., 1993, 619, 271–276 CrossRef CAS.
- Preliminary communication: S. S. Chitnis, N. Burford and M. J. Ferguson, Angew. Chem., Int. Ed., 2013, 52, 2042–2045 CrossRef CAS PubMed.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W. L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS PubMed.
- I. Tsuneo, Y. Koide and S. Hiyama, Chem. Lett., 1990, 19, 1445–1446 CrossRef.
- R. Corbo, T. P. Pell, B. D. Stringer, C. F. Hogan, D. J. D. Wilson, P. J. Barnard and J. L. Dutton, J. Am. Chem. Soc., 2014, 136, 12415–12421 CrossRef CAS PubMed.
- M. Donath, M. Bodensteiner and J. J. Weigand, Chem.–Eur. J., 2014, 20, 17306–17310 CrossRef CAS PubMed.
- C. D. Martin, C. M. le and P. J. Ragogna, J. Am. Chem. Soc., 2009, 131, 15126–15127 CrossRef CAS PubMed.
- J. Beckmann, J. Bolsinger, A. Duthie, P. Finke, E. Lork, C. Lüdtke, O. Mallow and S. Mebs, Inorg. Chem., 2012, 51, 12395–12406 CrossRef CAS PubMed.
- P. A. Rupar, V. N. Staroverov and K. M. Baines, Science, 2008, 322, 1360–1363 CrossRef CAS PubMed.
- C. L. B. Macdonald, A. M. Corrente, C. G. Andrews, A. Taylor and B. D. Ellis, Chem. Commun., 2004, 250–251 RSC.
- N. Li, R. Qiu, X. Zhang, Y. Chen, S. Yin and X. Xiu, Tetrahedron, 2015, 71, 4275–4281 CrossRef CAS.
- J. B. Hendrickson and M. S. Hussoin, J. Org. Chem., 1987, 52, 4137–4139 CrossRef.
- S. Kobayashi and I. Komoto, Tetrahedron, 2000, 6463–6465 CrossRef CAS.
- H. Gaspard-Iloughmane and C. le Roux, Eur. J. Org. Chem., 2004, 2517–2532 CrossRef CAS.
- S. S. Chitnis, A. P. M. Robertson, N. Burford, J. J. Weigand and R. Fischer, Chem. Sci., 2015, 6, 2559–2574 RSC.
- M. Peyronneau, C. Arrondo, L. Vendier, N. Roques and C. le Roux, J. Mol. Catal. A: Chem., 2004, 211, 89–91 CrossRef CAS.
- λ max = 300 nm for [6′P]3+. For model cation [6P]3+, time-dependent DFT (PBE0/def2-TZVP, acetonitrile field) calculations yield a λmax of 366 nm corresponding to a HOMO–LUMO transition. See Fig. S1, ESI† for the experimental absorbance spectrum and views of the HOMO and LUMO in [6P]3+.
- A. J. Arduengo III and C. A. Stewart, Chem. Rev., 1994, 94, 1215–1237 CrossRef.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- D. H. Johnston and D. F. Shriver, Inorg. Chem., 1993, 32, 1045–1047 CrossRef CAS.
- G. A. Olah, O. Farooq, C. X. Li, M. A. M. F. Farnia and J. J. Aklonis, J. Appl. Polym. Sci., 1992, 45, 1355–1360 CrossRef CAS.
- M. E. Woordhouse, F. D. Lewis and T. J. Marks, J. Am. Chem. Soc., 1982, 104, 5586–5594 CrossRef.
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- Derivatives of [6′E][OTf]3 were used since the poor solubility of [6P][OTf]3 makes 31P NMR measurements challenging.
- M. H. Holthausen, R. R. Hiranandani and D. W. Stephan, Chem. Sci., 2015, 6, 2016–2021 RSC.
- A. E. Ashley, T. J. Herrington, G. C. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed.
- K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 14204–14208 CrossRef CAS PubMed.
- G. Muller, H. Matheus and M. Winkler, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 1155–1162 CAS.
- P. Wawrzyniak, A. L. Fuller, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2009, 48, 2500–2506 CrossRef CAS PubMed.
- I. Granoth and J. C. Martin, J. Am. Chem. Soc., 1979, 101, 4623–4626 CrossRef CAS.
- A. N. Kornev, V. V. Sushev, Y. S. Panova, N. V. Zolotareva, E. V. Baranov, G. J. Fukin and G. A. Abakumov, Eur. J. Inorg. Chem., 2015, 2057–2066 CrossRef CAS.
- K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 6566–6568 CrossRef CAS PubMed.
- E. Conrad, N. Burford, U. Werner-Zwanziger, R. McDonald and M. J. Ferguson, Chem. Commun., 2010, 46, 2465–2467 RSC.
- A. L. Brazeau, A. S. Nikouline and P. J. Ragogna, Chem. Commun., 2011, 47, 4817–4819 RSC.
- S. G. Baxter, A. H. Cowley and S. K. Mehrotra, J. Am. Chem. Soc., 1981, 103, 5572–5573 CrossRef CAS.
- N. Burford, T. M. Parks, B. W. Royan, B. Borecka, T. S. Cameron, J. F. Richardson, E. J. Gabe and R. Hynes, J. Am. Chem. Soc., 1992, 114, 8147–8153 CrossRef CAS.
- C. Hering, J. Rothe, A. Schulz and A. Villinger, Inorg. Chem., 2013, 52, 7781–7790 CrossRef CAS PubMed.
- K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 7545–7549 CrossRef CAS PubMed.
- J. Carreras, M. Patil, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2012, 134, 16753–16758 CrossRef CAS PubMed.
- R. Weiss and S. Engel, Synthesis, 1991, 12, 1077–1079 CrossRef.
- R. Weiss and S. Engel, Angew. Chem., Int. Ed., 1993, 31, 216–217 CrossRef.
- L. V. Bezgubenko, S. E. Pipko and A. D. Sinista, Russ. J. Gen. Chem., 2009, 79, 911–918 CrossRef CAS.
- We note that the crystal structure of [7P][OTf]3 was previously presented by M. Donath, K. Schwedtmann, A. K. C. Echterhoff, R. Panzer, S. Schulz, F. Hennesdorf, and J. J. Weigand, at the International Conference on Sustainable Phosphorus Chemistry (ICSPC), Florence, December 04, 2014.
- S. S. Chitnis, E. MacDonald, N. Burford, U. Werner-Zwanziger and R. McDonald, Chem. Commun., 2012, 48, 7359–7361 RSC.
- J. J. Weigand, S. D. Riegel, N. Burford and A. Decken, J. Am. Chem. Soc., 2007, 129, 7969–7976 CrossRef CAS PubMed.
- A. Schmidpeter, S. Lochschmidt and W. S. Sheldrick, Angew. Chem., Int. Ed., 1985, 24, 226–227 CrossRef.
- J. M. McBride, Tetrahedron, 1974, 2009–2022 CrossRef CAS.
- B. D. Ellis and C. L. B. MacDonald, Inorg. Chem., 2004, 43, 5981–5986 CrossRef CAS PubMed.
- M. H. Holthausen, J. M. Bayne, I. Mallov, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 7298–7301 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- S. E. Pipko, L. Bezgubenko, A. D. Sinista, E. B. Rusanov, E. G. Kapustin, M. I. Povolotskii and V. V. Schvadchak, Heteroat. Chem., 2008, 19, 171–177 CrossRef CAS.
- K. B. Dillon, R. N. Reeve and T. Waddington, J. Chem. Soc., Dalton Trans., 1977, 2382–2388 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1410568–1410574. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02423d |
| This journal is © The Royal Society of Chemistry 2015 |