
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive species involved in the regioselective photooxidation of heptamethine cyanines†
Roger R.
Nani
a,
James A.
Kelley
a,
Joseph
Ivanic
*b and
Martin J.
Schnermann
*a
aChemical Biology Laboratory, Center for Cancer Research, National Cancer Institute at Frederick, Frederick, Maryland 21702, USA. E-mail: martin.schnermann@nih.gov
bAdvanced Biomedical Computing Center, DSITP, Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702, USA. E-mail: joseph.ivanic@nih.gov
First published on 18th August 2015
Abstract
Heptamethine cyanines are important near-IR fluorophores used in many fluorescence applications. Despite this utility, these molecules are susceptible to light-promoted reactions (photobleaching) involving photochemically generated reactive oxygen species (ROS). Here, we have sought to define key chemical aspects of this nearly inescapable process. Near-IR photolysis of a model heptamethine cyanine leads to the regioselective oxidative cleavage of the characteristic polyene. We report the first quantitative analysis of the major reaction pathway following either photolysis or exposure to candidate ROS. These studies clearly indicate that only singlet oxygen (1O2), and not other feasible ROS, recapitulates the direct photolysis pathway. Computational studies were employed to investigate the regioselectivity of the oxidative cleavage process, and the theoretical ratio is comparable to observed experimental values. These results provide a more complete picture of heptamethine cyanine photooxidation, and provide insight for the design of improved compounds for future applications.
Introduction
The light-induced degradation (photobleaching) of fluorescent small molecules is a pervasive issue that often interferes with modern fluorescence techniques. These reactions generally involve photooxidation pathways in which the photo-excited molecule interacts with nearly ubiquitous O2 to form reactive oxygen species (ROS).1 Reactions between the self-generated ROS and the fluorophore lead to the loss of the desired optical signal.2 While the general pathway is well accepted,3 the precise reactions, and particularly the identity of the products, involved in these oxidative events are often only poorly understood.4,5 A detailed understanding of these ubiquitous processes would provide a framework from which to develop improved fluorescent scaffolds and to create new fluorophore-dependent applications.6Cyanines are important fluorophores that find extensive use across a range of modern biological procedures. Particularly useful are the heptamethine cyanines, which contain a 7-carbon linker connecting two indolenine heterocycles. The absorption and emission maxima of these molecules are generally around 800 nm, occupying the center of the near-IR window where tissue penetration is maximized and autofluorescence is minimized.7 One example, indocyanine green, is an FDA approved diagnostic agent used in numerous clinical contexts and new variants continue to be developed for advanced optical applications.8–11
The photodegradation of cyanines is a frequent issue that can impede fluorescence readout. Not only deleterious, this chemistry has been used deliberately for productive applications. We recently demonstrated that the photooxidation of C4′-dialkylamine heptamethine cyanines could be used as the central element of a biologically compatible near-IR uncaging approach.12 Other investigators have employed the ROS-mediated oxidation of cyanines for sensing methods.13,14 Prior studies examining cyanine photodegradation have shown that photolysis leads to oxidative cleavage of the polyene linker to provide carbonyl products (eqn (1)).15–19 These studies, while providing a key initial description of this process, have either not addressed or provided conflicting information regarding the observed products and the nature of the oxidant(s). Other uncertain aspects of cyanine reactivity include the effect of key ROS and the basis of the high regioselectivity observed in the oxidative cleavage reaction. Clear answers to these fundamental questions would enable efforts that seek to expand the utility of heptamethine cyanines.
 | (1) |
To address these questions, we have employed a combination of experimental and theoretical approaches. Our experimental efforts have centered on mass spectral studies and the first quantitative accounting of cyanine oxidation products, using both direct irradiation and exogenous reactive oxygen species. Photolysis at the cyanine absorption maximum induces C–C cleavage at two of the four feasible reaction sites in the cyanine polyene. We show that only singlet oxygen (1O2), generated either chemically or through photosensitization, and not other common ROS, provides the photolysis products in similar yield. We have also carried out the first quantum mechanical computational analysis of the major oxidative cleavage reaction pathway. Our computational studies have provided a clear justification for both the origin of the regioselectivity (i.e. the relative stability of possible intermediate dioxetanes) and the relative ratio of the observed products for the major 1O2-dependent pathway. In total, we have defined key aspects of this photodegradation process, which is encountered in nearly any application of these broadly employed molecules.
Results and discussion
Photooxidation: experimental results
We sought to address the regioselectivity of the photooxidative C–C cleavage, which, in the case of symmetrical heptamethine cyanines, could lead to four regioisomeric product mixtures. Prior reports on several different heptamethine cyanine substrates provided conflicting data with three studies suggesting that oxidative cleavage occurs at a single terminal position in the cyanine polyene,15,20,21 another cleavage occurs at only a central position in the cyanine polyene,22 and two recent studies, including our own, observing cleavage at two sites.12,23 Certainly some of this apparent ambiguity is derived from the slightly different substrates and conditions used between the different studies. Moreover, these studies often employed only mass spectral data to assign the photooxidative products, and in no instances were reaction yields determined.We have chosen 1, which represents the simplest heptamethine indocyanine, as a model system for our efforts.24 To accurately measure yields of reactions run at low micromolar concentrations, we employed triplicate HPLC analysis calibrated using standard curves generated from independently prepared standards of previously known 2 and 6 (see ESI†). Exposing a solution of 1 (50% MeCN/H2O, 20 μM) to modest fluence (20 mW cm−2) of 740 nm light for 2 hours produces 2 and 6 in 57% and 12% yields, respectively (Fig. 1A). We have also carried out HRMS studies and detected 3 and 7, validating that oxidative cleavage occurred between C2/C1′ and C2′/C3′, as well as [1 + O2] ions. Careful analysis of the HRMS spectra found no evidence for the compounds derived from cleavage between C1′/C2′ (4 and 5), and C3′/C4′ (8 and 9), or hydrolysis products thereof, supporting the notion that this reaction proceeds with high regioselectivity. We have also shown that 2 and 6 accumulate concurrently with the loss of the signal for 1 and that the ratio of the two intermediates (∼5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) is essentially constant throughout the reaction (Fig. S1†). Control experiments have determined that these photoproducts are stable for the duration of the photolysis experiment. While we account for a majority (∼70%) of the material through this method, there is a fraction of 1 that is not attributed to identifiable species. Despite attempts to purify and characterize other products using HPLC, we have not been able to account for the balance of the reaction mixture.
:1) is essentially constant throughout the reaction (Fig. S1†). Control experiments have determined that these photoproducts are stable for the duration of the photolysis experiment. While we account for a majority (∼70%) of the material through this method, there is a fraction of 1 that is not attributed to identifiable species. Despite attempts to purify and characterize other products using HPLC, we have not been able to account for the balance of the reaction mixture.
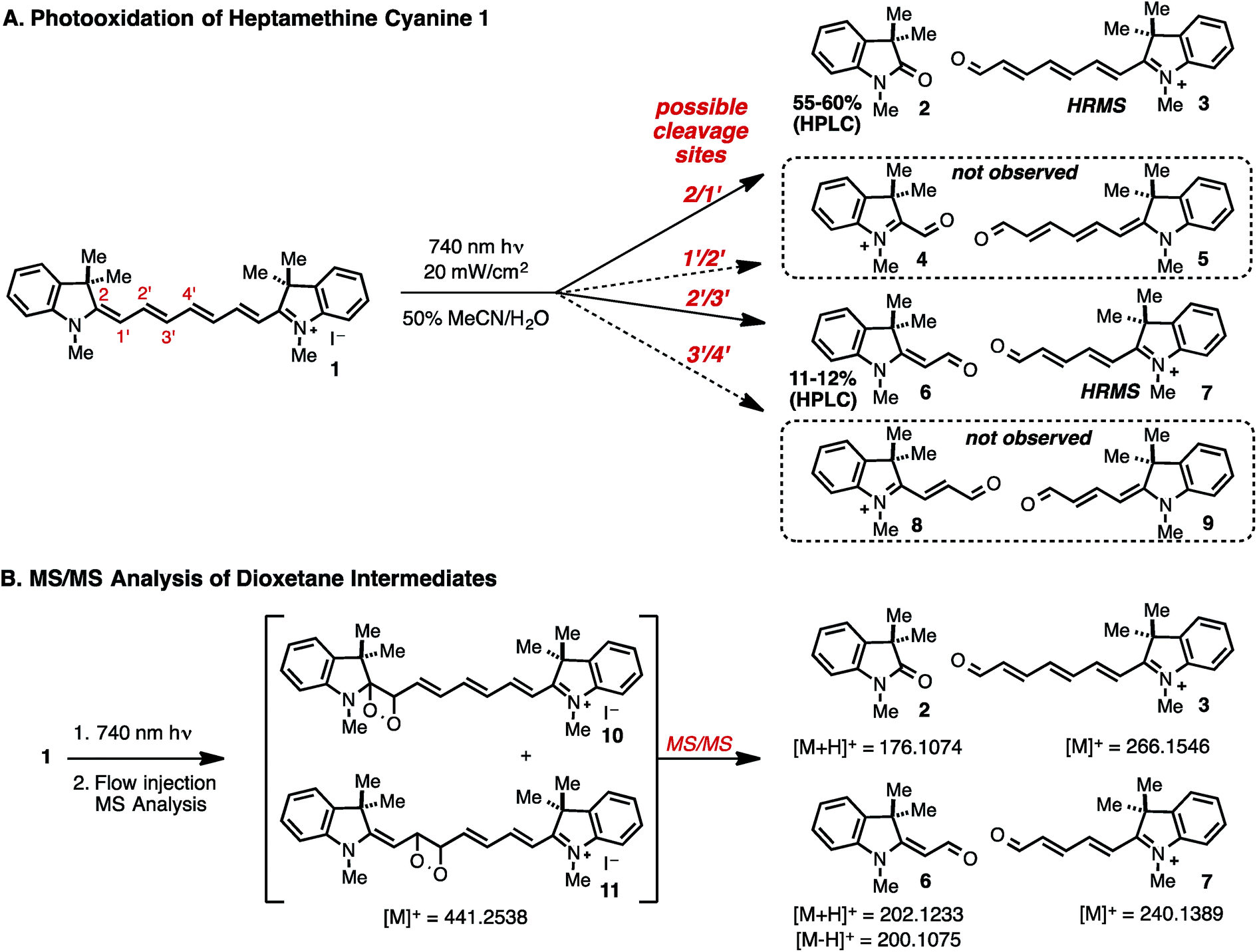 | ||
| Fig. 1 (A) Photoproducts observed by HRMS upon 740 nm light irradiation of 1 and (B) MS/MS [1+O2] adducts. | ||
We have carried out further studies on [1 + O2] ions to determine if these species represent dioxetane intermediates. As expected, the formation of these ions was observed only upon near-IR irradiation. MS/MS analysis following direct flow injection was performed to investigate accessible fragmentation pathways (Fig. 1B).25 Collision-induced dissociation (CID) of the [1 + O2] ions generated a product ion spectrum containing high-resolution masses that correspond to carbonyls 2 ([M + H]+), 3 ([M]+), 6 ([M − H]+, [M + H]+) and 7 ([M]+), among others. These results provide additional evidence to support the intermediacy of dioxetanes 10 and 11, as these represent the most reasonable structures of a [1 + O2] mass that could fragment directly to the observed carbonyl products.
To investigate the oxidant in this reaction, we have examined the reactivity of 1 with independently generated ROS. Our efforts commenced with 1O2, which was generated by two mechanistically distinct methods. The photosensitizer, tetraphenylporphyrin zinc (ZnTPP), is well established to cleanly form 1O2 in high quantum yield (ϕΔ = 0.7) upon irradiation with 420 nm light.26,27 This method afforded a similar product distribution of oxindole 2 (61–64%) and aldehyde 6 (13–15%) to that produced by direct cyanine excitation (entries 2 and 3, respectively, Table 1). Photolysis using only 420 nm light in the absence of a photosensitizer did induce photooxidation to form 2 and 6, albeit with dramatically slower kinetics (∼15 fold) to that observed in the presence of catalytic ZnTPP (Fig. S2†). This result is consistent with a small (∼2% relative to the λmax) absorption of 1 at 420 nm. To complement the photosensitizer-dependent photooxidation, we examined the impact of 1O2 generated via the thermal decomposition of endoperoxide 12.28 We found that exposure of 1 to 200 equivalents of 12 at 34 °C in 50% MeCN:H2O produced a high yield of oxindole 2 (71%) and aldehyde 6 (10%), collectively. Thus, 2 and 6 are formed in similar yields through direct near-IR photolysis and from independently generated 1O2, which was formed either thermally or photochemically. This observation provides significant support to the notion that 1O2-mediated dioxetane formation is the major pathway involved in cyanine photooxidation.29 Photosensitized formation of 1O2 is consistent with the small number of prior experimental measurements of heptamethine cyanine triplet state emission values (including one measurement on 1), which exceed the requisite 22.6 kcal mol−1 needed for 1O2 energy transfer.30,31
|
|
||||
|---|---|---|---|---|
| Entry | Conditionsa | Remaining 1b | 2 | 6 |
|
a Run at 22 °C in 1:1 MeCN:H2O unless noted otherwise.
b Determined by HPLC and reported in percent yield. Values are average of experimental triplicate. Error expressed as standard deviation in parentheses.
|
||||
| 1 | Dark | 99.5% (±1.6) | < 0.5% | <0.5% |
| 2 | 740 nm hν | 1.6% (±0.3) | 56.6% (±2.8) | 11.5% (±0.4) |
| 3 | Zn(TPP) 5 mol% 420 nm hν | <0.5% | 62.8% (±1.7) | 14.4% (±0.7) |
| 4 |

|
5.5% (±0.6) | 71.7% (±0.3) | 10.3% (±0.1) |
It has been suggested that cyanine photodegradation may involve superoxide (O2˙−), generated through electron transfer from the triplet state and, potentially, hydrogen peroxide (H2O2) or hydroxyl radical (OH˙), formed as downstream products of O2˙− .17,18,20,32–35 To investigate this possibility, the reactivity of 1 with these ROS was examined. In the case of OH˙ (entry 1, Table 2), 1 was consumed, but formed only trace quantities of 6 (1–3%) and barely detectable quantities of 2 (<0.5%). We have been unable to identify any of the numerous species that appear to be formed (see ESI† for representative HPLC traces), suggesting that complex radical-mediated pathways are involved. In contrast, O2˙− and H2O2 largely do not react with 1 under neutral aqueous conditions, even at concentrations that exceed those attainable by photolysis.36 Critically, control experiments showed that compounds 2 and 6 were unaffected by these or the other ROS generating conditions shown in Table 2. These control experiments ensure that the observed product ratio is indicative of reactions involving 1, and not of further oxidation of 2 and 6. In total, these results indicate that HO˙, H2O2, and O2˙− are not likely the major components of direct cyanine photooxidation, with all three providing product mixtures that are clearly distinct from that obtained upon direct near-IR photolysis. The critical observation is that, while other ROS can generate modest yields of oxindole 2, only 1O2, formed either chemically or photochemically, is capable of generating 6 in conversion comparable to that generated by direct photolysis.
|
|
||||
|---|---|---|---|---|
| Entry | Conditionsa | Remaining 1b | 2 | 6 |
|
a Run at 22 °C in 1:1 MeCN:H2O unless noted otherwise.
b Determined by HPLC and reported in percent yield. Values are average of experimental triplicate. Error expressed as standard deviation in parentheses.
|
||||
| 1 (HO˙) | FeCl2 200 μM H2O2 500 μM NaHPO4 pH = 6 | 3.4% (±1.2) | <2.7% (±0.3) | <0.5% |
| 2 (H2O2) | H2O2 100 μM PBS pH = 7.4 | 99.5% (±0.5) | <0.5% | <0.5% |
| 3 (H2O2) | H2O2 1 mM PBS pH = 7.4 | 99.0% (±0.5) | <0.5% | <0.5% |
| 4 (O2˙−) | KO2 100 μM | 99.2% (±1.1) | <0.5% | <0.5% |
| 5 (O2˙−) | KO2 1 mM | 84.1% (±0.2) | 10.6% (±0.1) | <0.6% (±0.1) |
| 6 (ClO−) | NaOCl 100 μM PBS pH = 7.4 | <0.5% | 19.5% (±0.7) | <0.5% |
| 7 (ONOO−) | NaONO2 150 μM PBS pH = 9 | <0.5% | 32.8% (±1.0) | <0.8% (±0.1) |
Recent studies have imaged oxidative flux by using oxidation of certain cyanines with biological ROS, specifically hypochlorite (NaOCl), peroxynitrite (ONOO−), OH˙, and O2˙−.13,14,37 The yields of products obtained from such ROS could provide insights useful in future efforts to develop these methods. Reaction of 1 with NaOCl generated meaningful quantities of oxindole 2, and no measureable 6 (entry 6, Table 2). While exposure of 1 to 150 μM NaONO2 under basic conditions produced over 30% yield of 2 (entry 7, Table 2), only trace amounts of 6 were observed.38 As with entry 1, we were unable to identify any other oxidation products formed in entries 6 and 7. Taken together with observations of Nagano and coworkers,13 these results provide additional evidence indicating that the reactivity of heptamethine cyanines is highly dependent on the ROS species.
Regioselectivity: quantum mechanical computational study
While the studies above clarified that the major photooxidation pathway of heptamethine cyanines involved singlet oxygen, the basis for the high regioselectivity of this process – with cleavage occurring at only two of four possible sites – was unclear. To address this, we pursued a computational approach. Although the distinctive optical properties of heptamethine cyanines have been examined computationally in several prior reports,39–43 our efforts are the first to examine the reactivity of cyanines.As a first step, we computed the total reaction energies (electronic) for the four possible cleavage pathways using the B3LYP and MP2 methods with the 6-31G(d) basis set (see Methods section for full details). We point out that we cannot explicitly compute MP2 and B3LYP 1Δg O2 energies since this electronic state is not a single-reference dominated wavefunction. As such, we have determined 1Δg O2 energies by adjustments (adding experimental electronic energy splitting) to readily computed 3Σg− O2 energies (see Methods). A consequence of this limitation is that we are unable to compute Gibbs free energy changes for these reactions. The π system of 1 is completely flat (C2v point group) at the B3LYP level of theory but very slightly twisted (C2 point group) when using the MP2 method. We have considered both cis and trans isomers (about the end C–C bond, where relevant) to identify the lowest energy product aldehydes. Fig. 2 clearly shows that all four reactions are significantly exothermic (ΔEe < −50 kcal mol−1 relative to reactants 1 + 1Δg O2), with cleavage at the C2/C1′ and C2′/C3′ sites leading to the most favorable products, having B3LYP-predicted energies of −89 kcal mol−1 and −66 kcal mol−1, respectively. We also point out that the MP2 and B3LYP results are in excellent agreement. While the two experimentally observed cleavage pathways are also the most favored in absolute terms, the observation that all pathways are highly exothermic suggested that this explanation is incomplete.
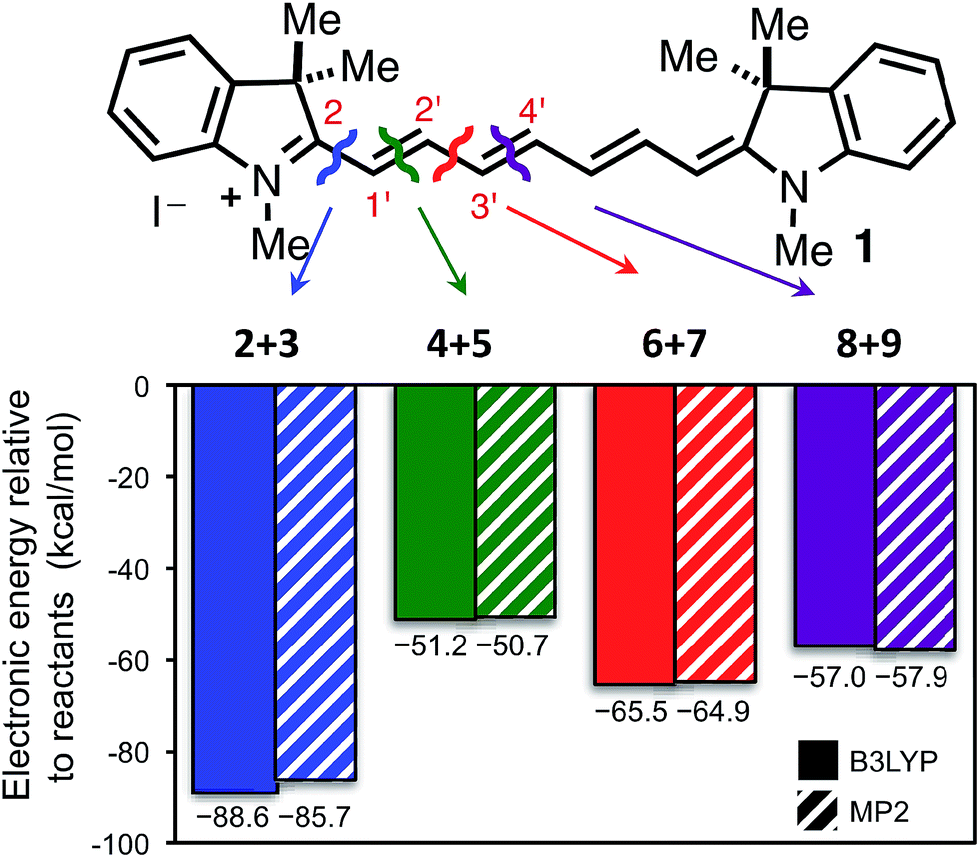 | ||
| Fig. 2 Computed reaction energies (electronic) for the four possible cleavage pathways of 1 + 1Δg O2 → R = O + [R′ = O]+. Blue, green, red, and indigo indicate cleavage at C2/C1′, C1′/C2′, C2′/C3′, and C3′/C4′ sites, respectively. | ||
The experimental results provided additional evidence to suggest that O2 addition and subsequent cleavage proceeds via dioxetane intermediates.44 Accordingly, we investigated whether the energetics of dioxetane formation is responsible for the observed regioselectivity. In order to locate the absolute lowest-energy structures for each of the four possible intermediates, we performed geometry optimizations starting from a very large number of initial structures that comprehensively spanned the potential energy surfaces. For each type of dioxetane intermediate, the only structural restrictions applied were to conserve (i) the initial trans conformation of the two carbon atoms that bind O2 and (ii) the trans backbones of the carbon chains (to rings) starting from the carbon atoms immediately attached to the C(OO)C dioxetane unit. Our reasoning being that O2 addition is expected to occur with high stereoselectivity. This prediction follows from seminal studies by Turro and coworkers on reactions of related ene-carbamates with 1O2.45 A full description, with informative illustrations, of the structural degrees of freedom that have been considered is provided in the ESI.† Geometry searches were performed independently at the MP2/6-31G(d) and B3LYP/6-31G(d) levels of theory and all discovered minima were confirmed by computations of vibrational frequencies. The numbers of minima located for each class of intermediate and method is summarized in Table 3. We find that the MP2 method predicts more minima for each intermediate class than B3LYP (no less than 21 for addition over C3′/C4′) and includes more dioxetane “twist” conformations and N-methyl proton orientations (typically staggered). The B3LYP/6-31G(d) method identified four minima for each intermediate except the second (addition over C1′/C2′), for which five were discovered and in each case only one N-methyl orientation was preferred (typically eclipsed).
| Method | 10: (C2/C1′) | 13: (C1′/C2′) | 11: (C2′/C3′) | 14: (C3′/C4′) |
|---|---|---|---|---|
| a 6-31G(d) basis set used for minima searches. b See Fig. 1A for carbon numbering scheme, whereby CX/CY indicates O–O addition locale. c Numbers in parentheses do not consider N-methyl orientation. d At the B3LYP/CCT level of theory one minimum optimizes to another leaving only four. | ||||
| MP2 | 12 (6) | 6 (6) | 18 (9) | 21 (12) |
| B3LYP | 4 (4) | 5 (5)d | 4 (4) | 4 (4) |
Fig. 3 illustrates the lowest energy (electronic) B3LYP/6-31G(d) structures for each of the four possible intermediates. The lowest energy MP2 structures (provided in ESI†) are commensurate except for 13 (addition over C1′/C2′), which differs by a 180° rotation about the C2–C1′ bond.
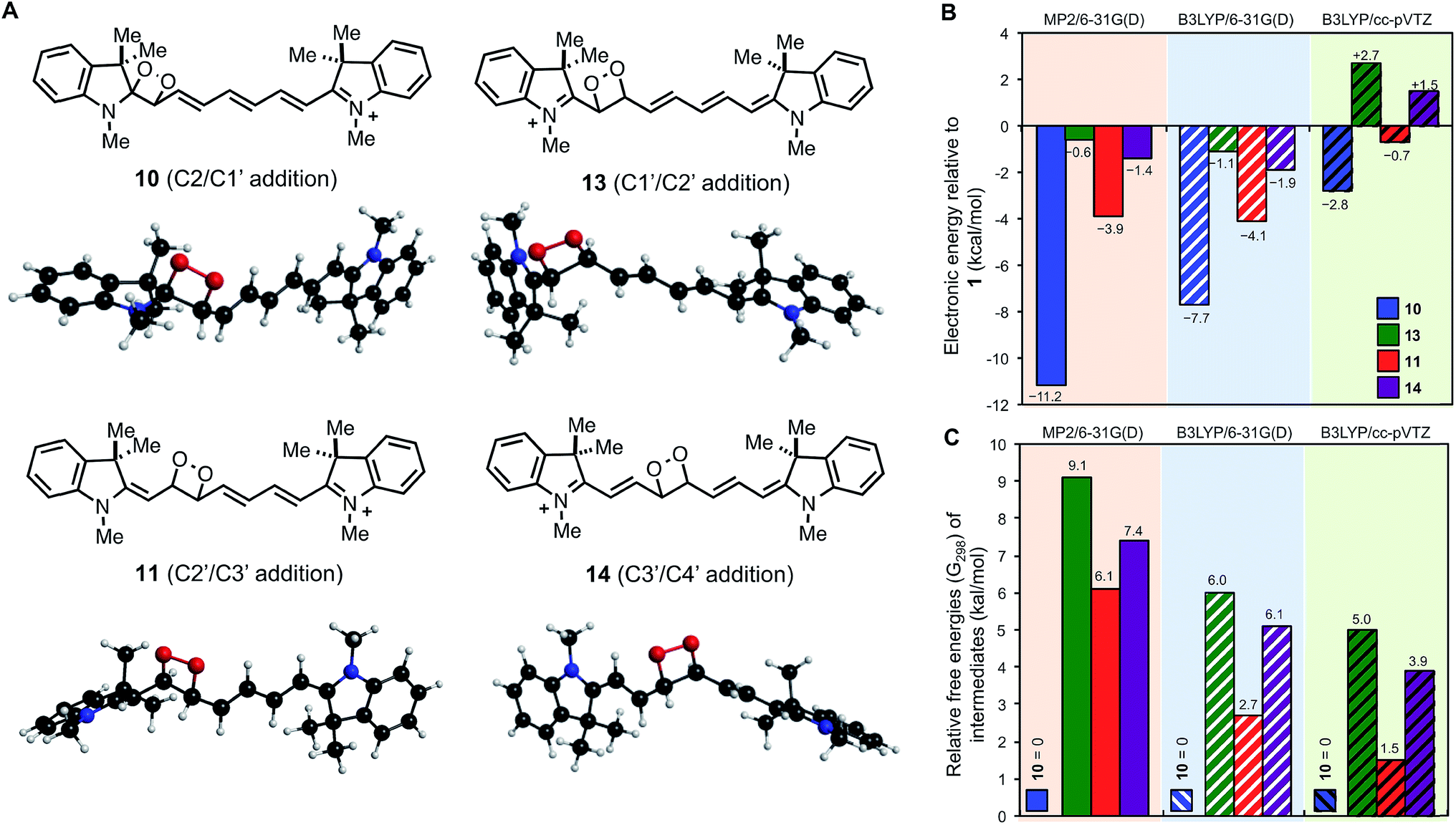 | ||
| Fig. 3 Lowest energy (electronic) B3LYP/6-31G(d) dioxetane intermediate structures (A) and computed stabilities relative to (B) reactants 1 + 1Δg O2 (electronic) and (C) each other (G298). | ||
All 17 dioxetane intermediate minima located using the B3LYP/6-31G(d) method were also optimized using the larger cc-pVTZ basis set where lowest energy structures for each of 10, 11, 13 and 14 were commensurate to those shown in Fig. 3A. However, one of the intermediate 13 species optimized to another, leaving only four. Fig. 3B shows the stabilities (electronic) of the dioxetane intermediates relative to the reactants 1 + 1Δg1O2. We find that the MP2/6-31G(d) and B3LYP/6-31G(d) results are comparable, predicting similar exothermic stabilities for each intermediate in the order 10 < 11 < 14 < 13, although MP2 predicts a more stable (by 3.5 kcal mol−1) intermediate 10. When advancing to the B3LYP/cc-pVTZ level of theory, the energetic order of the intermediates is preserved, however, now only intermediates 10 and 11 are computed to be exothermic by 2.8 kcal mol−1 and 0.7 kcal mol−1, respectively. Therefore, the computational results indicate that 1O2 addition and subsequent cleavage is energetically favored only via dioxetane intermediates 10 and 11. Not only does this prediction match which cleavage pathways are observed experimentally (C2/C1′ and C2′/C3′), it also correctly indicates that cleavage at (C2/C1′) should be the major reaction pathway.
We have also calculated Gibbs free energies (G298) at the MP2/6-31G(d) and B3LYP/6-31G(d) levels of theory for all discovered minima (B3LYP/6-31G(d) G298 corrections were used to estimate B3LYP/cc-pVTZ G298 energies). For the two methods MP2/6-31G(d) and B3LYP/cc-pVTZ it can be the case that the lowest G298 structure for an intermediate can vary from the corresponding lowest electronic energy structure and these differences are shown in Fig. S12 and S13.†Fig. 3C shows computed relative free energies (G298) of the dioxetane intermediates where 10 remains the most stable. We find that when going from the MP2/6-31G(d) to B3LYP/6-31G(d) to B3LYP/cc-pVTZ levels of theory that intermediates 11, 13, and 14 are successively stabilized. Our most reliable prediction, B3LYP/cc-pVTZ, indicates that 11 is most competitive with 10, lying only 1.5 kcal mol−1 higher. At room temperature (298 K), this energy difference amounts to a Boltzmann ratio of ∼13:1 that should transfer to the corresponding cleavage products 2 + 3 and 6 + 7. This theoretical prediction is remarkably similar to the experimentally determined ratio of ∼5:1, especially when taking into account that solvent effects have not been considered and energy margins are of the order of a kcal mol−1. In fact, the experimentally observed ratio translates to an energy difference (at 298 K) of 0.95 kcal mol−1 which is really not very different from 1.5 kcal mol−1 when one acknowledges that quantum chemical accuracy of 1 kcal mol−1 is typically reached only with benchmark methods that are currently impossible for this sized system.
Conclusions
The photooxidation of heptamethine cyanines accompanies nearly any use of these broadly employed molecules. These studies, which centered on quantitative accounting of the major products and detailed computational studies, have provided key insights into this process. Cyanine photolysis leads to the regioselective cleavage of the cyanine polyene at two of the four possible positions. Only exogenously generated 1O2, and not other common ROS, is capable of replicating this reaction pathway. We account for ∼70% of the molar balance of the starting cyanine, suggesting that at least this fraction goes through an energy transfer/1O2 pathway, while leaving open the possibility that the remainder reacts through other pathways (perhaps initiated by electron transfer from the triplet state).Our computational analysis suggests that regioselective cleavage at only two positions of the polyene, C2/C1′ and C2′/C3′, can be interpreted as being a consequence of the overall energetics of the dioxetane intermediates. At the highest level examined, our calculations indicate that O2 addition is exothermic only at the observed sites (C2/C1′ and C2′/C3′). By contrast, 1O2 additions at the remaining two sites, C1′/C2′ and C3′/C4′, are less favorable and may actually be endothermic. Work in progress is currently exploring the reaction coordinate for singlet oxygen addition to form the dioxetane intermediates, itself a complex process in spite of numerous prior investigations.46–48
The framework developed here should be of significant utility to efforts that seek to improve the photostability of cyanine fluorophores. Moreover, these studies provide a mechanistic foundation for our ongoing program harnessing the cyanine photooxidation reaction as the key step in a near-IR uncaging method. The systematic modification of the heptamethine cyanine scaffold is currently being pursued to access improved compounds for such applications.
Methods
Experimental
High-resolution LC/MS and MS/MS analyses were conducted on a Thermo-Fisher LTQ-Orbitrap-XL hybrid mass spectrometer system with an Ion MAX API electrospray ion source in positive ion mode. Flow injection analysis followed by collision-induced dissociation was utilized for MS/MS experiments. A quantitative HPLC assay to determine yields of 1, 2, and 6 was employed using an Agilent 1260 Infinity instrument. Samples were irradiated in HPLC vials using LED light sources obtained from Marubeni America Corporation or subjected to the reaction conditions in Tables 1 and 2.Quantum chemistry
The calculations in this study were executed using the GAMESS49 package. We have used the second-order Møller–Plesset perturbation theory (MP2)50–52 and density functional theory (DFT)53 methods where, for the latter, the B3LYP54,55 hybrid density functional (containing VWN5 functional) was utilized with a fine grid of 120 radial and 590 angular Lebedev points. Open-shell MP2-style calculations for the triplet oxygen species used the Z-averaged perturbation theory (ZAPT)56–58 flavor. The double-zeta 6-31G(d)59 and triple-zeta cc-pVTZ60 (spherical functions) basis sets were employed. Geometries were optimized to tight convergence (maximum Cartesian gradient < 1 × 10−5) using analytic gradients and Hessians were computed seminumerically (double differences) using analytic gradients for all stationary points located. We note that Fig. S3† shows data evaluating the B3LYP and MP2 (including a variety of open-shell types) methods against benchmark and experiment for the hypothetical model reaction: 3O2 + H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 → 2H2CO; all relevant references are provided therein. Based upon these results the ZAPT energy of the 3Σg− O2 molecule was used to determine reaction energies at the MP2 level of theory. Absolute energies of the 1Δg O2 molecule were determined by adding the experimental 3Σg− → 1Δg energy difference (22.639 kcal mol−1)61 to computed energies of the 3Σg− O2 system.
CH2 → 2H2CO; all relevant references are provided therein. Based upon these results the ZAPT energy of the 3Σg− O2 molecule was used to determine reaction energies at the MP2 level of theory. Absolute energies of the 1Δg O2 molecule were determined by adding the experimental 3Σg− → 1Δg energy difference (22.639 kcal mol−1)61 to computed energies of the 3Σg− O2 system.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, and the National Cancer Institute, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.References
- A. Diaspro, G. Chirico, C. Usai, P. Pamoino and J. Dobrucki, Photobleaching, in Handbook of Biological Confocal Microscopy, ed. J. B. Pawley, Springer, New York, 2006, pp. 690–702 Search PubMed.
- M. Levitus and S. Ranjit, Q. Rev. Biophys., 2011, 44, 123–151 CrossRef CAS PubMed.
- Q. S. Zheng, M. F. Juette, S. Jockusch, M. R. Wasserman, Z. Zhou, R. B. Altman and S. C. Blanchard, Chem. Soc. Rev., 2014, 43, 1044–1056 RSC.
- J. Widengren and R. Rigler, Bioimaging, 1996, 4, 149–157 CAS.
- I. Georgakoudi and T. H. Foster, Photochem. Photobiol., 1998, 67, 612–625 CAS.
- T. Ha and P. Tinnefeld, Annu. Rev. Phys. Chem., 2012, 63, 595–617 CrossRef CAS PubMed.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef CAS PubMed.
- J. T. Alander, I. Kaartinen, A. Laakso, T. Patila, T. Spillmann, V. V. Tuchin, M. Venermo and P. Valisuo, Int. J. Biomed. Imaging, 2012, 2012, 1–26 CrossRef PubMed.
- M. Henary and M. Mojzych, Stability and Reactivity of Polymethine Dyes, in Heterocyclic Polymethine Dyes: Synthesis, Properties and Applications, ed. L. Strekowski, Topics in Heterocyclic Chemistry 14, Springer-Verlag, Berlin, 2008, p. 221 Search PubMed.
- R. R. Nani, J. B. Shaum, A. P. Gorka and M. J. Schnermann, Org. Lett., 2015, 17, 302–305 CrossRef CAS PubMed.
- A. P. Gorka, R. R. Nani and M. J. Schnermann, Org. Biomol. Chem., 2015, 132, 7584–7598 Search PubMed.
- A. P. Gorka, R. R. Nani, J. Zhu, S. Mackem and M. J. Schnermann, J. Am. Chem. Soc., 2014, 136, 14153–14159 CrossRef CAS PubMed.
- D. Oushiki, H. Kojima, T. Terai, M. Arita, K. Hanaoka, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2010, 132, 2795–2801 CrossRef CAS PubMed.
- M. T. Sun, H. Yu, H. J. Zhu, F. Ma, S. Zhang, D. J. Huang and S. H. Wang, Anal. Chem., 2014, 86, 671–677 CrossRef CAS PubMed.
- S. Lepaja, H. Strub and D. J. Lougnot, Z. Naturforsch., A: Phys. Sci., 1983, 38, 56–60 Search PubMed.
- G. W. Byers, S. Gross and P. M. Henrichs, Photochem. Photobiol., 1976, 23, 37–43 CrossRef CAS PubMed.
- A. Toutchkine, D. V. Nguyen and K. M. Hahn, Org. Lett., 2007, 9, 2775–2777 CrossRef CAS PubMed.
- B. R. Renikuntla, H. C. Rose, J. Eldo, A. S. Waggoner and B. A. Armitage, Org. Lett., 2004, 6, 909–912 CrossRef CAS PubMed.
- N. J. Patel, E. Manivannan, P. Joshi, T. J. Ohulchanskyy, R. R. Nani, M. J. Schnermann and R. K. Pandey, Photochem. Photobiol., 2015 DOI:10.1111/php.12482.
- P. Chen, J. Li, Z. G. Qian, D. S. Zheng, T. Okasaki and M. Hayami, Dyes Pigm., 1998, 37, 213–222 CrossRef CAS.
- X. Y. Chen, X. J. Peng, A. J. Cui, B. S. Wang, L. Wang and R. Zhang, J. Photochem. Photobiol., A, 2006, 181, 79–85 CrossRef CAS PubMed.
- A. Samanta, M. Vendrell, R. Das and Y. T. Chang, Chem. Commun., 2010, 46, 7406–7408 RSC.
- E. Engel, R. Schraml, T. Maisch, K. Kobuch, B. Koenig, R. M. Szeimies, J. Hillenkamp, W. Baumler and R. Vasold, Invest. Ophthalmol. Visual Sci., 2008, 49, 1777–1783 Search PubMed.
- M. P. Samtsov, S. A. Tikhomirov, O. V. Buganov, K. N. Kaplevsky, D. G. Melnikov and L. S. Lyashenko, J. Appl. Spectrosc., 2009, 76, 783–790 CrossRef CAS.
- H. K. Ijuin, M. Yamada, M. Ohashi, N. Watanabe and M. Matsumoto, Eur. J. Mass Spectrom., 2008, 14, 17–25 CrossRef CAS PubMed.
- R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391–475 CrossRef CAS.
- F. Nifiatis, J. C. Athas, K. D. D. Gunaratne, Y. Gurung, K. M. Monette and P. J. Shivokevich, Open Spectrosc. J., 2011, 5, 1–12 CrossRef CAS.
- N. J. Turro, M. F. Chow and J. Rigaudy, J. Am. Chem. Soc., 1981, 103, 7218–7224 CrossRef CAS.
- E. L. Clennan and A. Pace, Tetrahedron, 2005, 61, 6665–6691 CrossRef CAS PubMed.
- V. A. Kuzmin, A. S. Tatikolov and Y. E. Borisevich, Chem. Phys. Lett., 1978, 53, 52–55 CrossRef CAS.
- J. P. Fouassier, D. J. Lougnot and J. Faure, Chem. Phys. Lett., 1975, 35, 189–194 CrossRef CAS.
- A. Toutchkine, V. Kraynov and K. Hahn, J. Am. Chem. Soc., 2003, 125, 4132–4145 CrossRef CAS PubMed.
- Q. Zheng, S. Jockusch, Z. Zhou and S. C. Blanchard, Photochem. Photobiol., 2014, 90, 448–454 CrossRef CAS PubMed.
- C. P. Chen, B. M. Zhou, D. H. Li and G. G. Xu, J. Photochem. Photobiol., A, 1995, 89, 25–29 CrossRef CAS.
- M. S. Baker and J. M. Gebicki, Arch. Biochem. Biophys., 1984, 234, 258–264 CrossRef CAS.
- M. S. Chan, D. Xu, L. Guo, D. Y. Tam, L. S. Liu, M. S. Wong and P. K. Lo, Org. Biomol. Chem., 2015, 13, 7307–7312 CAS.
- K. Y. Pu, A. J. Shuhendler and J. H. Rao, Angew. Chem., Int. Ed., 2013, 52, 10325–10329 CrossRef CAS PubMed.
- R. M. Uppu and W. A. Pryor, Anal. Biochem., 1996, 236, 242–249 CrossRef CAS PubMed.
- R. S. Lepkowicz, O. V. Przhonska, J. M. Hales, J. Fu, D. J. Hagan, E. W. Van Stryland, M. V. Bondar, Y. L. Slominsky and A. D. Kachkovski, Chem. Phys., 2004, 305, 259–270 CrossRef CAS PubMed.
- S. Pascal, A. Haefele, C. Monnereau, A. Charaf-Eddin, D. Jacquemin, B. Le Guennic, C. Andraud and O. Maury, J. Phys. Chem. A, 2014, 118, 4038–4047 CrossRef CAS PubMed.
- P. A. Bouit, C. Aronica, L. Toupet, B. Le Guennic, C. Andraud and O. Maury, J. Am. Chem. Soc., 2010, 132, 4328–4335 CrossRef CAS PubMed.
- K. T. Lin and J. W. Silzel, Spectrochim. Acta, Part A, 2015, 142, 210–219 CrossRef CAS PubMed.
- F. Terenziani, O. V. Przhonska, S. Webster, L. A. Padilha, Y. L. Slominsky, I. G. Davydenko, A. O. Gerasov, Y. P. Kovtun, M. P. Shandura, A. D. Kachkovski, D. J. Hagan, E. W. van Stryland and A. Painelli, J. Phys. Chem. Lett., 2010, 1, 1800–1804 CrossRef CAS.
- M. Matsumoto, H. Kobayashi, J. Matsubara, N. Watanabe, S. Yamashita, D. Oguma, Y. Kitano and H. Ikawa, Tetrahedron Lett., 1996, 37, 397–400 CrossRef CAS.
- J. Sivaguru, M. R. Solomon, T. Poon, S. Jockusch, S. G. Bosio, W. Adam and N. J. Turro, Acc. Chem. Res., 2008, 41, 387–400 CrossRef CAS PubMed.
- A. A. Frimer, Chem. Rev., 1979, 79, 359–387 CrossRef CAS.
- E. L. Clennan and K. Nagraba, J. Am. Chem. Soc., 1988, 110, 4312–4318 CrossRef CAS.
- A. Maranzana, G. Ghigo and G. Tonachini, J. Am. Chem. Soc., 2000, 122, 1414–1423 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS PubMed.
- J. A. Pople, J. S. Binkley and R. Seeger, Int. J. Quantum Chem., 1976, 1–19 CAS.
- G. D. Fletcher, M. W. Schmidt and M. S. Gordon, Adv. Chem. Phys., 1999, 110, 267–294 CrossRef CAS PubMed.
- C. M. Aikens, S. P. Webb, R. L. Bell, G. D. Fletcher, M. W. Schmidt and M. S. Gordon, Theor. Chem. Acc., 2003, 110, 233–253 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133–1138 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. J. Lee and D. Jayatilaka, Chem. Phys. Lett., 1993, 201, 1–10 CrossRef CAS.
- T. J. Lee, A. P. Rendell, K. G. Dyall and D. Jayatilaka, J. Chem. Phys., 1994, 100, 7400–7409 CrossRef CAS PubMed.
- C. M. Aikens, G. D. Fletcher, M. W. Schmidt and M. S. Gordon, J. Chem. Phys., 2006, 124, 014107 CrossRef PubMed.
- P. C. Harihara and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS PubMed.
- G. Herzberg and K. P. Huber, Molecular spectra and molecular structure. IV. Constants and diatomic molecules, Van Nostrand Reinhold, New York, London, 1979 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental protocols, HRMS spectra, representative HPLC chromatograms, full details regarding evaluation of B3LYP and MP2 methods against benchmark and experiment for the hypothetical model reaction: 3O2 + H2CCH2 → 2H2CO, geometrical conformations considered in comprehensive minima searches of all Cy7-O2 intermediates, and optimized geometries and energies of lowest-energy (electronic and free (G298), when determined) species. See DOI: 10.1039/c5sc02396c |
| This journal is © The Royal Society of Chemistry 2015 |