
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoelectronic source of the anomalous stability of bis-peroxides†
Gabriel dos Passos
Gomes
a,
Vera
Vil'
b,
Alexander
Terent'ev
b and
Igor V.
Alabugin
*a
aDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, Florida 32306-4390, USA. E-mail: ggomes@chem.fsu.edu; alabugin@chem.fsu.edu
bN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, 47 Leninsky Prospekt, Moscow 119991, Russian Federation. E-mail: vera_vill@mail.ru; alterex@yandex.ru
First published on 7th September 2015
Abstract
The unusual stability of bis- and tris-peroxides contradicts the conventional wisdom – some of them can melt without decomposition at temperatures exceeding 100 °C. In this work, we disclose a stabilizing stereoelectronic effect that two peroxide groups can exert on each other. This stabilization originates from strong anomeric nO → σ*CO interactions that are absent in mono-peroxides but reintroduced in molecules where two peroxide moieties are separated by a CH2 group. Furthermore, such effects can be induced by other σ-acceptors and amplified by structural constraints imposed by cyclic and bicyclic frameworks.
Introduction
Organic peroxides have a deserved reputation as explosive high-energy functionalities that should be avoided.1 However, in addition to classic applications of peroxides as radical initiators, explosives and oxidative reagents, more recently this functionality started to play an important role in the design of medicinal agents2 (Fig. 1). These new practical applications demand better understanding of the fundamental factors responsible for the instability of peroxides. In this context, the discovery of remarkably stable organic bis-peroxides that can melt without decomposition at temperatures as high as 142 °C (ref. 3) calls for a critical evaluation of the electronic properties responsible for the unusual stability of these structures. It is particularly counterintuitive that these surprisingly stable peroxides can contain not only one but multiple O–O bonds. | ||
| Fig. 1 A selection of biologically active peroxides. | ||
In this work, we investigate why combining in one molecule several O–O bonds, the very element responsible for the instability, can lead to a counterintuitive increase in thermodynamic stability in comparison to analogous mono-peroxides.
In the first part, we will analyze electronic structure of peroxides and related molecules with Natural Bond Orbital (NBO) analysis, the method of choice for analysis of stereoelectronic interactions.4 We will show that, among other factors, peroxides lack anomeric stabilization that strongly stabilizes their structurally related cousins, acetals. In the second part, we identify a variety of structural effects that bring the anomeric stabilization back to life and increase the overall thermodynamic stability of peroxides.
Computational details and methods
Calculations were carried with the Gaussian 09 software package,5 using the M06-2X DFT functional6 or the MP2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 method, both with the 6-311++G(d,p) basis set. For the relaxed PES scans, we also used double hybrid B2PLYPD functional.8 Delocalizing interactions were evaluated from M06-2X data with NBO method, using NBO 3.0 software. NBO analysis transforms the canonical delocalized molecular orbitals from DFT calculations into localized orbitals that are closely tied to the chemical bonding concepts. Each of the localized NBO sets is complete and orthonormal. The filled NBOs describe the hypothetical, strictly localized Lewis structure. The interactions between filled and antibonding orbitals represent the deviation from the Lewis structure and can be used to measure delocalization. For example, delocalizing interaction can be treated via the 2nd order perturbation energy approach as E(2) = ni|Fij|2/ΔE, where ni is the population of a donor orbitals, Fij is the Fock matrix element for the interacting orbitals i and j, and ΔE is the energy gap between these orbitals. Chemcraft 1.79 and CYLView10 were used to render the molecules and orbitals. Crystal structures were obtained from the Cambridge crystal structure database (CSD)11 using ConQuest12 and post-analyzed as histograms and heat plots with both Mercury13 and MATLAB.14 The latter software was also used to build the tridimensional PES with two dihedral scans.
7 method, both with the 6-311++G(d,p) basis set. For the relaxed PES scans, we also used double hybrid B2PLYPD functional.8 Delocalizing interactions were evaluated from M06-2X data with NBO method, using NBO 3.0 software. NBO analysis transforms the canonical delocalized molecular orbitals from DFT calculations into localized orbitals that are closely tied to the chemical bonding concepts. Each of the localized NBO sets is complete and orthonormal. The filled NBOs describe the hypothetical, strictly localized Lewis structure. The interactions between filled and antibonding orbitals represent the deviation from the Lewis structure and can be used to measure delocalization. For example, delocalizing interaction can be treated via the 2nd order perturbation energy approach as E(2) = ni|Fij|2/ΔE, where ni is the population of a donor orbitals, Fij is the Fock matrix element for the interacting orbitals i and j, and ΔE is the energy gap between these orbitals. Chemcraft 1.79 and CYLView10 were used to render the molecules and orbitals. Crystal structures were obtained from the Cambridge crystal structure database (CSD)11 using ConQuest12 and post-analyzed as histograms and heat plots with both Mercury13 and MATLAB.14 The latter software was also used to build the tridimensional PES with two dihedral scans.
Results and discussion
The high energy of the peroxide functionality
The high energy stored in the peroxide functionality is readily revealed by the exergonicity of reactions that decrease the number of O–O bonds. For example, 1,2,3-trioxolanes (i.e., the primary ozonides formed in ozonolysis of alkenes) are 52.4 kcal mol−1 less stable than 1,2,4-trioxolanes (i.e., the secondary ozonides). This energy difference is similar to the 51.3 kcal mol−1 difference in the energies of 1,2 and 1,3-dioxolanes because in both cases an O–O bond is converted into a stronger C–O bond (Scheme 1).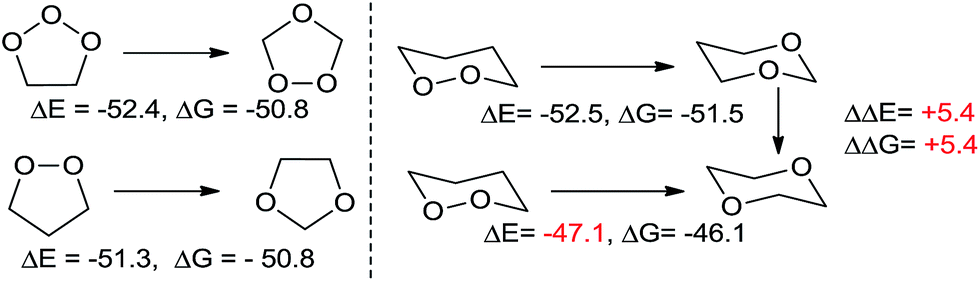 | ||
| Scheme 1 Left: ΔE and ΔG, in kcal mol−1, for translocation of oxygen in tri- and di-oxapentanes. Right: ΔE and ΔG for translocation of oxygen in dioxacyclohexanes. | ||
Clearly, the greater strength of C–O bonds is the largest contributor to the high exothermicity of such oxygen translocations. These isodesmic equations show the value of peroxides as a high energy functionality in the design of reactions where conversion of weak O–O bonds into stronger functionalities can serve as driving force for a chemical process. However, reactions in Scheme 1 also indicate the presence of an additional structural effect. The transformation of 1,2-dioxane into 1,4-dioxane is 5.4 kcal mol−1 less exothermic than the transformation into 1,3-dioxane (ΔE = −47.1 vs. −52.5 kcal mol−1, respectively). This difference indicates the presence of an additional stabilizing effect, specific for 1,3-dioxane. We will show below that this stabilization is associated with the activation of anomeric effect and that it provides a hint on how to stabilize peroxides without losing the O–O bonds, the very structural units that are responsible for the unique chemical and medicinal activity of peroxides.
The anomeric effect – the key stabilizing force in oxygen containing compounds
The anomeric effect, a well-known axial preference of acceptor substituents at the anomeric carbons of pyranoses and related heterocycles with an endocyclic lone pair, is one of the oldest and most-studied stereoelectronic effects15 (Scheme 1). It is a manifestation of the more general stereoelectronic preference for a lone pair nX at heteroatom X and C–Y bond in a YCH2X moiety to be aligned in an antiperiplanar geometry,16 maximizing the hyperconjugative nX → σ*C–Y interaction. The stabilizing effect of such interactions is considerable – for example, the gauche, gauche conformation of dimethoxymethane is 5.5 kcal more stable than the all-anti conformation in the gas phase. Solvents of high polarity provide more stabilization to the more polar conformations, weakening the stereoelectronic preference imposed by orbital interactions. Anomeric interactions are not limited to acetals. For example, n → σ*C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O interactions are responsible for the greater stability of Z-isomers of esters and related functionalities.17 NBO analysis provides a stereoelectronic rationale for the gauche preference in MeOCH2OMe by identifying two strong nO → σ*C–O interactions (with the NBO energies of 14.9 kcal each). These interactions are much stronger than other vicinal interactions in this molecule (e.g., the pairs of σC–H → σ*O–C and σC–H → σ*C–O interactions contribute 3.4 and 4.5 kcal mol−1, respectively, per interaction) (Scheme 2).
O interactions are responsible for the greater stability of Z-isomers of esters and related functionalities.17 NBO analysis provides a stereoelectronic rationale for the gauche preference in MeOCH2OMe by identifying two strong nO → σ*C–O interactions (with the NBO energies of 14.9 kcal each). These interactions are much stronger than other vicinal interactions in this molecule (e.g., the pairs of σC–H → σ*O–C and σC–H → σ*C–O interactions contribute 3.4 and 4.5 kcal mol−1, respectively, per interaction) (Scheme 2).
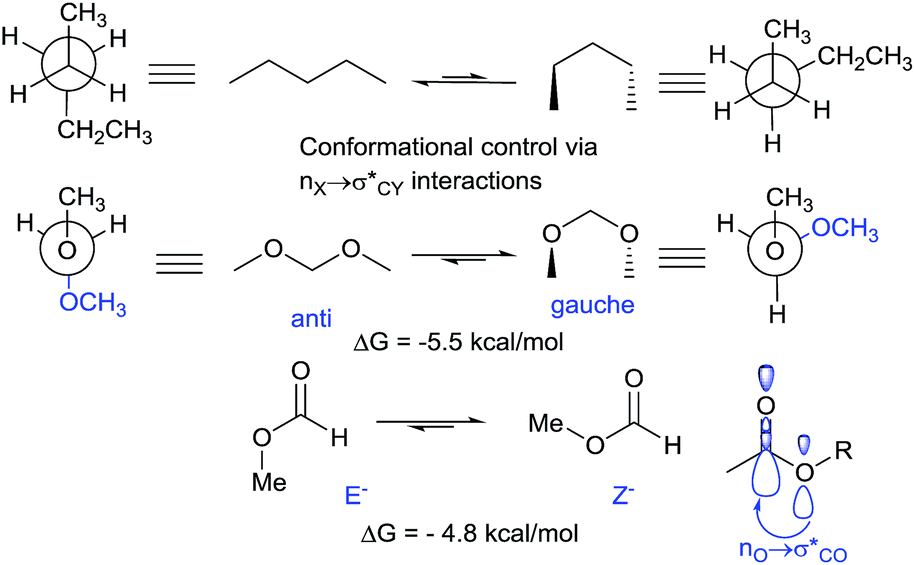 | ||
| Scheme 2 Generalized anomeric effect in dimethoxymethane and the role of anomeric hyperconjugation in esters, energies are in kcal mol−1. | ||
Lack of anomeric stabilization in peroxides
Conformational preferences of peroxides are drastically different from ketals. In particular, the most stable rotamer of dimethyl peroxide corresponds to the COOC dihedral angle of ∼150°.18 Furthermore, the adjacent region of the potential energy surface is rather flat and corresponds to an essentially isoenergetic ensemble of conformations with dihedral angles from 110 to 180° (all within 1 kcal mol−1 at MP2/6-311++G(d,p) level). This is in good agreement with the experimental values of the COOC dihedrals in crystal structures for acyclic peroxides, displayed as a histogram in Fig. 2. The gauche geometry analogous to the most stable geometry of dimethyl acetal (60°, PES scanned for both COCO dihedrals shown in depth on ESI†) is ∼5 kcal mol−1 higher in energy than the minimum (Fig. 2). Overall, these conformational trends point to dramatically different stereoelectronics of acetals compared to peroxides. This finding is noteworthy since the donor ability of lone pairs at the two adjacent heteroatoms is often considered to be enhanced, for example peroxide anions are better nucleophiles than hydroxides.19 In order to rationalize the conformational and structural peculiarities of peroxides, let us analyze the electronic structure of this functionality in more detail. | ||
| Fig. 2 The COOC and COCO dihedral PES scans in dimethylperoxide (a) and dimethoxy-methane (b), respectively. On right top, histogram displaying the frequency of COOC dihedral angles in crystal structures for acyclic peroxides.20 | ||
The anatomy of the peroxide moiety
Oxygen is well-known to form strong bonds to other elements, including some of the strongest bonds in the chemical universe (i.e., the O–H and Si–O bonds) and yet the O–O bond is weak.21One of oxygen's less-known idiosyncrasies is that this element does not efficiently use hybridization in non-polar covalent bonds. In particular, the O–O bond of the peroxide moiety forms via the overlap of nearly pure p-orbitals (∼sp8, Scheme 3). The inefficiency of hybridization decreases the covalent overlap between the bond forming orbitals and contributes to the weakness of the O–O bond.22,23
 | ||
| Scheme 3 Top: Polarization of σ*C–O explains why C–O bond is ∼40% stronger acceptor at the carbon end. The orbital interactions are quantified by NBO analysis and given in kcal mol−1. Middle and bottom: The difference between nO → σ*C–O interactions in acetals and nO → σ*O–C interactions in peroxides is amplified >16-fold (>1600% difference!). | ||
Furthermore, the apparent Bond Dissociation Energy (BDE) for an X–Y bond reflects not only the strength of direct X/Y overlap but also the presence of secondary delocalizing interactions between bonds, antibonds and lone pairs present at X and Y. For example, BDE for C–C bond in ethane also includes penalty for the loss of all hyperconjugative σC–H → σ*C–H interactions24 in addition to the direct cost of breaking overlap between the two ∼sp3 carbon hybrids in the C–C bond. Even without engaging in the debate of whether the lone pair/lone pair four-electron interactions are repulsive or simply non-stabilizing,25 it is clear that interactions between vicinal lone pairs do not contribute to stabilization of the O–O moiety in the same way as the σC–H → σ*C–H interactions contribute to the stabilization of CH2–CH2 moiety in an alkane.
In addition, unusual hybridization leads to unusual geometric features. For example, the OOC angle in dimethyl peroxide is ∼10° smaller than the OCO angle in dimethyl acetal, leading to a noticeably different alignment of the p-type lone pair of oxygen with the vicinal σ-bond in these two oxygen-containing organic functionalities (Fig. 3).
 | ||
| Fig. 3 The simplest organic functional groups with two oxygen atoms are dramatically different electronically and geometrically. | ||
The disappearing anomeric effect in peroxides
The effect of this combination of structural features is further amplified by anisotropic character of nO → σ*X–Y interactions. In our earlier work, we had identified the strong directionality of hyperconjugative interactions in ethers26 where C–O bonds are ∼40% better acceptors than O–C bonds in σC–H → σ*X–Y interactions (X, YO, C), as a consequence of polarization of σ*CO orbital towards carbon (Scheme 3). In the present work, we found that the directionality of nO → σ*X–Y interactions is pronounced even further.
As shown in Scheme 3, the NBO energy for nO → σ*C–O interaction in the acetal is 14.9 kcal mol−1, whereas the energy nO → σ*O–C interaction in the peroxide is ∼0.9 kcal mol−1 at the same level of theory. In other words, the stereoelectronic difference increased from 40% to >1600% in the σ* interactions with a stronger donor (σC–Hvs. nO).
The reason for the greatly increased hyperconjugative anisotropy lies in the intricate combination of effects that control the resulting orbital overlap. When a σ-orbital (e.g., the C–H bond in Scheme 3, top) serves as a donor, most of the stabilizing orbital overlap in the σ → σ* interaction originates from overlap of this σ-orbital with the back lobe of an antiperiplanar σ*-acceptor. In contrast, when a p-orbital serves as a donor in an anomeric interaction, the notions of syn- and antiperiplanarity disappear. In such systems, the np/σ* overlap is significant with both the back lobe of the σ*orbital (e.g., the O–C bond in peroxide in Scheme 3) and the antibonding region between the two atoms (e.g., O and C). In peroxides, the unusually small OOC angle brings the σ*OC node closer to the p-orbital. The destabilizing interaction with the out-of-phase hybrid at carbon largely offsets the in-phase stabilizing interaction of the p-donor with the oxygen part of the σ*O–C orbital (Scheme 3).
The >16-fold decrease in the magnitude of nO → σ*O–C interactions in peroxides in comparison to nO → σ*C–O interactions in acetals is striking. Taken together with the above-mentioned structural effects, the non-symmetric nature of σ-acceptors explains why the anomeric effect is dramatically diminished in peroxides in comparison with acetals. This stereoelectronic analysis reveals one more source of thermodynamic instability of dialkyl peroxides – the weakening of anomeric hyperconjugative interactions.
In the next part, we will show how to bring back the anomeric stabilization. This reactivation will also illustrate how combining multiple O–O moieties in one molecule can provide additional thermodynamic stability.
Converting peroxides into more stable molecules without removing O–O bonds
The paradoxical ∼4 kcal mol−1 stabilization due to the presence of an additional O–O group in the same molecule is illustrated by the comparison of 1,2,4,5-tetraoxane with 1,2-dioxacyclohexane (Scheme 4, top). Introduction of only a single oxygen (1,2,4-trioxane) decreases this stabilization by the factor of ∼2 whereas introduction of an additional carbon in the bridge that separates the two peroxides obliterates the stabilizing effect (Scheme 4, bottom). | ||
| Scheme 4 Top left: Stabilization energies, orbital interactions, and NBO plots of O1 lone pair, σ*O2–C3 and σ*C5–O6 in 1,2,4,5-tetraoxane. Top right: Reactivation of anomeric effect in 1,2,4,5-tetraoxane. Bottom left: Comparison with 1,2,4-trioxane. Bottom right: Comparison with 1,2,5,6-tetraoxacyclooctane. The NBO interaction energies as well as ΔE and ΔG for the isodesmic reactions are in kcal mol−1. | ||
NBO analysis illustrates that the origin of the observed stabilization is stereoelectronic. It is associated with the activation of the anomeric nO → σ*C–O interactions by the addition of σ-acceptors. Based on the prevalent hyperconjugative stabilization patterns, these bis-peroxides are stereoelectronically equivalent to bis-acetals. Indeed, each of the four nO → σ*C–O interactions in the significantly puckered 1,2,4,5-tetraoxane ring is almost as large as such interactions in 1,3-dimethoxymethane (14.1 and 14.9 kcal, respectively). The large magnitude of these interactions is noteworthy because the p-type lone pair at the endocyclic oxygen atom in a non-distorted chair geometry is usually aligned better with the axial substituents than with the vicinal endocyclic bond.
Taking advantage of stereoelectronic stabilization provided by anomeric effect opens an opportunity to increase thermodynamic stability of peroxides without decrease in the number of O–O bonds. Such stabilization should be general and incorporation of additional O–O units should be stabilizing as well as long as they are separated by a single carbon and the donor and acceptor orbitals are sufficiently well aligned with each other. This finding supports multiple directions for the design of peroxides with increased stability.
In search of the optimal stereoelectronics for the peroxide/peroxide motifs
In this section, we expand our analysis to other experimentally known cyclic bis- and tris-peroxides: the [2.2.1] bicyclic systems where the tetraoxacyclohexane moiety is constrained in a boat conformation and to nine-membered cycles with three O–O moieties, Scheme 5. The [2.2.1] bicyclic bis-peroxide is stabilized (∼4–5 kcal mol−1) in comparison to the monocyclic system Scheme 4. There, the stabilizing effect of each of the four symmetry-equivalent nO → σ*C–O interactions reaches 16.4 kcal, suggesting an even better arrangement of the donor and acceptor orbitals when the boat conformation in enforced in the 1,2,3,4-tetraoxane.The thermodynamic stabilization grows further in the 9-membered tris-peroxide. The overall ∼12 kcal mol−1 energy corresponds to ∼4 kcal mol−1 enthalpy lowering per each O–O moiety. Note, however, that this very large enthalpic stabilization is partially offset by entropy. The interplay between enthalpy and entropy depends on substitution. For example, whereas conversion of the parent six-membered bis-peroxide into nine-membered tris-peroxide still provides more than 5 kcal mol−1 decrease in the overall free energy per each formed tris-peroxide molecule, the analogous conversion of diacetone diperoxide (DADP) into triacetone triperoxide (TATP) is calculated to be slightly endergonic (Scheme 5).
 | ||
| Scheme 5 Structural modifications can further increase the stabilizing effect of multiple peroxides moieties. | ||
The large increase in stabilization for the 9-membered cycle is consistent with the increased NBO energies for the anomeric interactions in this more flexible cyclic system. We have not performed the exhaustive conformational search for this system but each of the found nO → σ*C–O interactions (∼16.5 kcal mol−1) is greater than the analogous interactions in the puckered chair conformation of 1,2,4,5-tetraoxane (ESI† part).
The intricate influence of molecular geometry on orbital interactions is further illustrated by Scheme 6 that shows that the additional stabilizing effect imposed by the bicyclic [2.2.1] system disappears in the analogous [2.2.2] bis-peroxide. This finding indicates that geometry of the larger bicycle decreases anomeric interactions in comparison to the [2.2.1] framework. According to NBO analysis, the 16.4 kcal mol−1 average energy for the nO → σ*C–O interactions in the [2.2.1] system is decreased to 12.5 kcal mol−1 in the [2.2.2] system.
 | ||
| Scheme 6 The bicyclic [2.2.2] system does not show the same large enhancement in the stabilization as the bicyclic [2.2.1] system. All energies in kcal mol−1. | ||
New stabilizing patterns
Indeed, NBO finds a strong anomeric interaction of 15.1 kcal mol−1 in the acyclic peroxide with two –CH2OMe substituents. The increased value of this effect in comparison to similar interaction in the acetals (14.9 kcal in 1,3-dimethoxymethane) can be attributed to the increased donor ability of peroxide lone pairs as a manifestation of α-effect.19 Much of the increased stabilization is retained in the relatively flexible 7-membered bis-peroxide with two embedded acetal functions (1,2,4,6-tetraoxacycloheptane) that exhibits >9 kcal mol−1 of the stabilization relative to tetraoxane. Nature uses this design to stabilize medicinally important peroxides such as artemisinin (Scheme 7). Furthermore, survey of stabilized peroxides provided by commercial sources also reveals increased kinetic and thermodynamic stabilities for a number of peroxides adjacent to a C–O bond (i.e., Trigonox 311).27
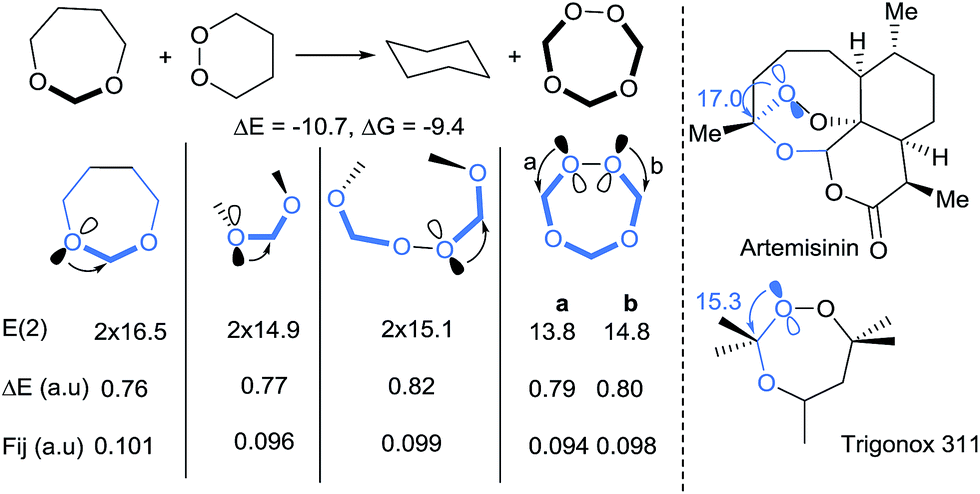 | ||
| Scheme 7 Top: Isodesmic equation showing the greater stabilization of a peroxide by a ketal group in 7-membered ketal. Bottom: E(2) for nO → σ*C–O interactions on ketals and peroxides that are stabilized by ketals; Right: Selected NBO interactions and its E(2) for nO → σ*C–O interactions in artemisinin and Trigonox 311. | ||
 | ||
| Scheme 8 Substituents can either stabilize or destabilize peroxides. | ||
In contrast, the presence of S–C–S, Si–C–Si and S–S moieties in cyclic structures shown in Scheme 8 has either a very small effect or becomes destabilizing. The mutually destabilizing effect of peroxide and disulfide groups within the same six-membered cycle is particularly interesting. Even a single S-atom in the cycle imposed a destabilizing influence on peroxides and the observed destabilization persisted even when S atom was oxidized to the respective sulfoxide and sulfone in order to increase the acceptor ability of the respective σ*C–S orbitals (see the ESI†).
On the other hand, C–F bonds impose a larger stabilizing effect than C–O bonds, as expected from the greater electronegativity of fluorine.29 The calculated conformational effects in these systems are large if oxygen p-lone pairs and the C–F bonds are properly aligned. In acyclic systems, such stabilization can reach ∼11 kcal mol−1 (Scheme 8 explored the stabilizing effect of C–F bonds in the greater detail and performed relaxed potential energy scan for the COOC and OOCF dihedrals in bis(fluoromethyl) peroxide (Fig. 4, see the ESI† for additional information). Interestingly, we have found several energy minima where the compromise between stereoelectronic, steric and electrostatic interactions led to similar stability.
 | ||
| Fig. 4 Multidimensional PES for scanning simultaneously both OOCF and COOC dihedrals of bis(fluoromethyl) peroxide. Three minima (deep blue pointed with arrows showing the OOCF dihedrals, boxed Newman projection conformation) and one global maximum (deep red) are located. | ||
The effect of fluorine on stability of cyclic peroxides further reflects the general requirements for the stereoelectronic stabilizing effects. Cyclic constraints can prevent the system from the optimal alignment of the donor and acceptor orbitals. However, when the acceptor σ*C–F orbitals are axial,30 the alignment is still favorable as illustrated by the 4.9 kcal mol−1 stabilization by the two axial C–F bonds (comparable with the 4.2 kcal mol−1 effect of an endocyclic O–O moiety in Scheme 4).
In contrast, the equatorial C–F groups are better aligned with the O–O bond than with the lone pairs. Such systems cannot enjoy strong anomeric nO → σ*C–F interactions and the stabilizing effect almost disappears (∼0.3 kcal mol−1).
We have also investigated the competition between endo- and exo-anomeric stabilization (Scheme 9). Introduction of two axial fluorines to the tetraoxane moiety adds 1.8 kcal mol−1 of stabilization. This value is significantly lower than 4.5 kcal mol−1 stabilization in the diaxial difluoro dioxane, demonstrating the saturation of donor acceptor interactions. The lack of cooperativity stems from the difficulty in simultaneously achieving efficient overlap of the oxygen p-lone pairs with the endocyclic C–O bond and the exocyclic C–F bond. Furthermore, addition of equatorial C–F groups to 1,2,4,5-tetraoxane is destabilizing due to the unfavorable antiperiplanar arrangement between the acceptor C–F and O–O orbitals.
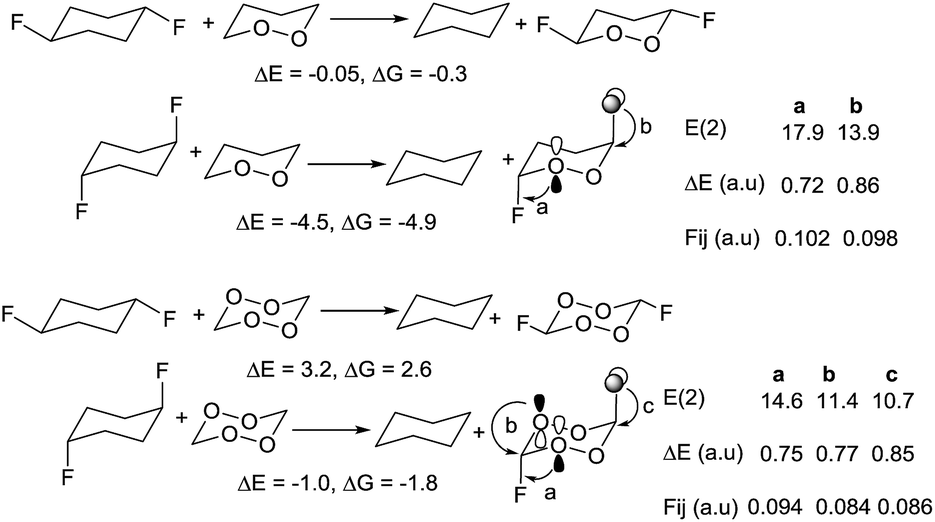 | ||
| Scheme 9 Effects of C–F bonds on stability of peroxides in cyclic systems. ΔE, ΔG and E(2) in kcal mol−1. | ||
We have compared these computational predictions by analyzing preferred OOCX dihedrals (X = O, N, halogens) using X-ray crystallographic data available for organic peroxides with an acceptor group X at the α-carbon (Fig. 5). The calculated optimal dihedral for OOCF is ∼72° (at the ∼128° COOC dihedral) is in a good agreement with the preferred dihedrals in the histogram (Fig. 5). The OOCX dihedral optimizes the nO → σ*C–X interaction that stabilizes the peroxide group by better aligning the best donor and acceptor orbitals. Since no structural restrains was imposed on the analyzed set, the range of observed dihedrals was relatively broad and another set of values close to 150–170° was found as well, mostly for exocyclic substituents in cyclic structures. We will analyze stereoelectronic features of the 2nd set of compounds in the future work.
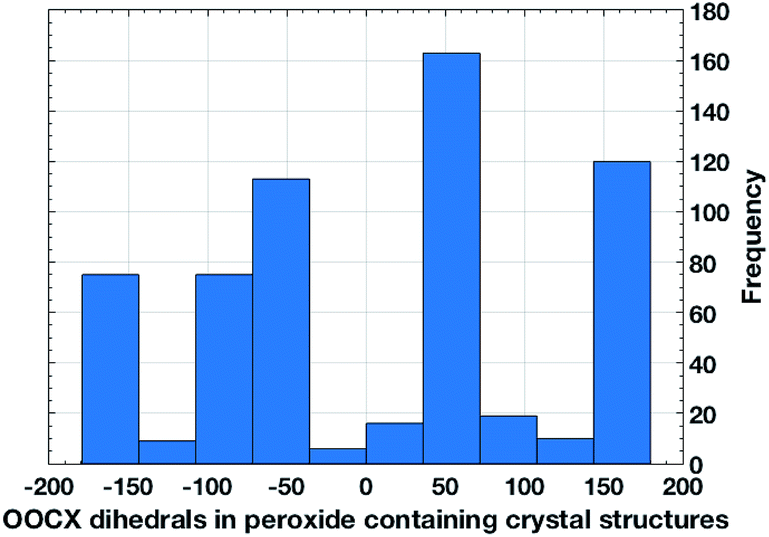 | ||
| Fig. 5 The distribution of OOCX dihedrals in peroxide-containing crystal structures. | ||
To test the effect of nO → pX interactions (corresponding to the formation of dative O = X bonds), we calculated stabilization energies associated with the presence of boron atoms (X = B).31 Since an empty a p-orbital can be an even better acceptor than a σ*C–X orbital, the effects associated with the OB bond formation are very large and strongly stabilizing. Although presence of two O–O moieties creates an antiaromatic system that partially offsets the stabilization, such systems can be stabilized via the formation of the anionic ate-complexes. For example, sodium perborate forms crystalline dimeric hydrates where two peroxide bridges are separated by two boron atoms.
The chair conformations of these six-membered cycles display exo-anomeric nO → σ*B–O interactions with the exocyclic B–O bonds. Even despite the lower electronegativity of boron, NBO finds these interactions to be quite strong (∼9 kcal mol−1, Scheme 10). The significant hyperconjugative stabilization can be one of the reasons for the increasing popularity of this oxidizing agent in synthesis.
 | ||
| Scheme 10 Left: The empty p-orbital of tricoordinated boron can stabilize peroxides but antiaromaticity decreases efficiency of the cyclic conjugation. Right: The dimeric perborate dianion is stabilized by anomeric n → σ*B–O interactions. ΔE, ΔG and E(2) in kcal mol−1. | ||
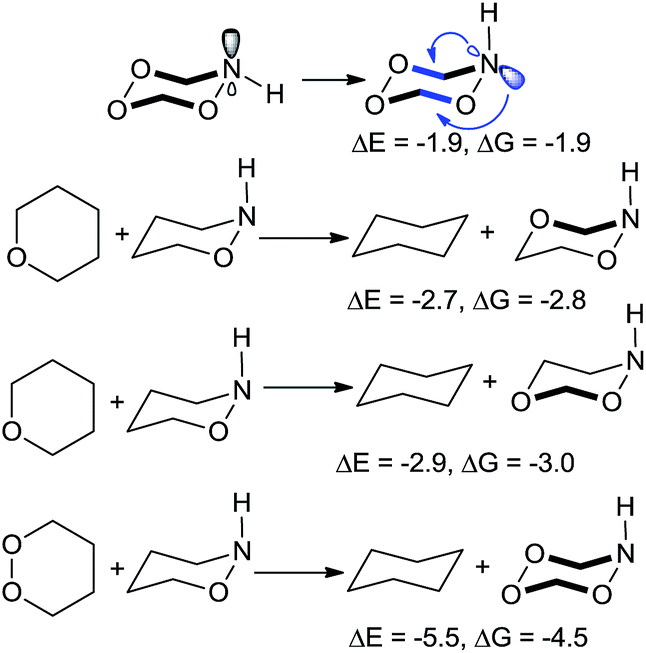 | ||
| Scheme 11 Different weak links can be stabilized through stereoelectronic effects. ΔE and ΔG in kcal mol−1. | ||
Furthermore, one can put the stabilizing C–O moiety antiperiplanar to the lone pairs of either nitrogen or oxygen atoms of the N–O bond: ∼3 kcal mol−1 of stabilization is found in the either case.32 As expected from the stereoelectronic origin of these effects, greater stabilization is observed when lone pair of an endocyclic nitrogen electron donor is equatorial. In this conformation, the two “weak links”, the O–O bond and the O–N bond, reinforce each other through four anomeric interactions.
Conclusions
Stereoelectronic analysis resolves the apparent paradox of why combining two peroxides in one molecule can lead to thermodynamic stabilization. The combination of hybridization and polarization effects leads to dramatic (>10-fold) weakening of anomeric effect in peroxides in comparison to their structural cousins, acetals. However, the anomeric stabilization in bis-peroxides is resurrected in molecules where two peroxides are separated by a one-atom bridge. Due to reactivation of strong anomeric interactions, such structures are better described as bis-acetals rather than bis-peroxides. Although these stabilizing effects cannot fully compensate for the weakness of O–O bond, this analysis opens new ways for design of more stable peroxides via the introduction of appropriate sigma acceptors (C–F, C–O or C–N bonds).But what does this stabilization mean from the practical point of view? “Stability” can have a different meaning when applied to peroxides. How far does thermodynamics translate into kinetic stability? How important is anomeric stabilization for reactivity of peroxides? Those are complex questions. Although the excessive heat of formation (high energy content) is one of the key properties of explosive materials, other factors can be important as well. For example, even though explosion of triacetone triperoxide (TATP) is not highly favored thermochemically, TATP is still a powerful explosive due to “entropy burst” associated with formation of one ozone and three acetone molecules from every molecule of TATP.33 The dramatic difference in sensitivities of diacetone diperoxide (DADP) cocrystals was shown to originate from subtle differences in non-covalent interactions in each cocrystal structure.1c In future work, it is important to investigate how the additional thermodynamic stabilization influences kinetic stability and reactivity of the hybrid peroxides described in this work.
Acknowledgements
We are grateful to the National Science Foundation (CHE-1465142) for support of this research and to the Research Computing Center of the Florida State University for the allocation of computational resources. Synthesis of stable organic peroxides was financially supported by the Russian Science Foundation (No. 14-23-00150). G. P. G. thanks Vinícius Ferrão and Prof. Pierre Esteves for their support, Dr Brian Gold and Rana Mohamed for helpful discussions.Notes and references
- (a) T. M. Klapötke and T. Wloka, Peroxide Explosives, John Wiley & Sons, Ltd, Chichester, UK, 2009, p. 1 Search PubMed; (b) H. Klenk, P. H. Götz, R. Siegmeier and W. Mayr, Peroxy Compounds, Organic, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, recent examples Search PubMed; (c) K. B. Landenberger, O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2013, 52, 6468 CrossRef CAS PubMed; (d) K. B. Landenberger, O. Bolton and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 5074 CrossRef CAS PubMed.
- (a) C. W. Jefford, Adv. Drug Res., 1997, 29, 271 CAS; (b) P. M. O'Neil and G. H. Posner, J. Med. Chem., 2004, 47, 2945 CrossRef PubMed; (c) A. Chung, M. R. Miner, K. J. Richert, C. J. Rieder and K. A. Woerpel, J. Org. Chem., 2015, 80, 266 CrossRef CAS PubMed; (d) K. Gilmore, D. Kopetzki, J. W. Lee, Z. Horváth, D. T. McQuade, A. Seidel-Morgenstern and P. H. Seeberger, Chem. Commun., 2014, 50, 12652 RSC; (e) K. Borstnik, I. K.-H. Paik, T. A. Shapiro and G. H. Posner, Int. J. Parasitol., 2002, 32, 1661 CrossRef CAS PubMed; (f) J. L. Vennerstrom, H.-N. Fu, W. Y. Ellis, A. L. Ager, J. K. Wood, S. L. Andersen, L. Gerena and W. K. Milhous, J. Med. Chem., 1992, 35, 3023 CrossRef CAS PubMed; (g) Y. Dong, H. Matile, J. Chollet, R. Kaminsky, J. K. Wood and J. L. Vennerstrom, J. Med. Chem., 1999, 42, 1477 CrossRef CAS PubMed; (h) M. H. Gelb, Curr. Opin. Chem. Biol., 2007, 11, 440 CrossRef CAS PubMed; (i) H.-S. Kim, Y. Shibata, Y. Wataya, K. Tsuchiya, A. Masuyama and M. Nojima, J. Med. Chem., 1999, 42, 2604 CrossRef CAS PubMed; (j) D. A. Casteel, Nat. Prod. Rep., 1999, 16, 55 RSC; (k) K. J. McCullough, J. K. Wood, A. K. Bhattacharjee, Y. Dong, D. E. Kyle, W. K. Milhous and J. L. Vennerstrom, J. Med. Chem., 2000, 43, 1246 CrossRef CAS PubMed; (l) F. Najjar, L. Gorrichon, M. Baltas, C. André-Barrès and H. Vial, Org. Biomol. Chem., 2005, 3, 1612 RSC; (m) G. L. Ellis, R. Amewu, C. Hall, K. Rimmer, S. A. Ward and P. M. O'Neill, Bioorg. Med. Chem. Lett., 2008, 18, 1720 CrossRef CAS PubMed; (n) D. C. Martyn, A. P. Ramirez, M. J. Beattie, J. F. Cortese, V. Patel, M. A. Rush, K. A. Woerpel and J. Clardy, Bioorg. Med. Chem. Lett., 2008, 18, 6521 CrossRef CAS PubMed; (o) S.-H. Lau, A. Galván, R. R. Merchant, C. Battilocchio, J. A. Souto, M. B. Berry and S. V. Ley, Org. Lett., 2015, 17, 3218 CrossRef CAS PubMed.
- (a) A. O. Terent'ev, D. A. Borisov, V. V. Chernyshev and G. I. Nikishin, J. Org. Chem., 2009, 74, 3335 CrossRef PubMed; (b) A. O. Terent'ev, I. A. Yaremenko, V. A. Vil', I. K. Moiseev, S. A. Kon'kov, V. M. Dembitsky, D. O. Levitsky and G. I. Nikishin, Org. Biomol. Chem., 2013, 11, 2613 RSC.
- A. E. Reed and F. J. Weinhold, Chem. Phys., 1985, 83, 1736 Search PubMed; A. E. Reed and F. Weinhold, Isr. J. Chem., 1991, 31, 277 CrossRef CAS; A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef; F. Weinhold, in Encyclopedia of Computational Chemistry, ed. Schleyer P.v.R., Wiley, New-York, 1998, vol. 3, p. 1792 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision B.01, Gaussian, Wallingford, CT, 2009, complete reference in the ESI Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed; S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed; T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2007, 9, 3397 RSC.
- ChemCraft 1.7 build number 405, http://www.chemcraftprog.com, accessed in February 2015 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- (a) F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef; (b) F. H. Allen and W. D. S. Motherwell, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 407 CrossRef.
- ConQuest 1.17, http://www.ccdc.cam.ac.uk/Solutions/CSDSystem/Pages/ConQuest.aspx, accessed on August 8th, 2015 Search PubMed.
- Mercury 3.5, http://www.ccdc.cam.ac.uk/Solutions/FreeSoftware/Pages/ConQuest.aspx, accessed on August 8th, 2015 Search PubMed.
- MATLAB R2015a, The MathWorks Inc., Natick, MA, http://www.mathworks.com/products/matlab/ Search PubMed.
- (a) R. U. Lemieux, Pure Appl. Chem., 1971, 25, 527 CrossRef CAS; (b) C. Altona, C. Romers, H. R. Buys and E. Havinga, Top. Stereochem., 1969, 4, 39 Search PubMed; (c) N. S. Zefirov and N. M. Shekhman, Russ. Chem. Rev., 1971, 40, 315 CrossRef; (d) I. Tvaroška and T. Bleha, Adv. Carbohydr. Chem. Biochem., 1989, 47, 45 CrossRef; (e) E. Juaristi and G. Cuevas, Tetrahedron, 1992, 48, 5019 CrossRef CAS; (f) P. P. Graczyk and M. Mikolajczyk, Anomeric effect: Origin and consequences, Top. Stereochem., 1994, 21, 159 CAS; (g) The Anomeric Effect and Associated Stereoelectronic Effects, ed. Thatcher G. R. J., ACS, Washington, 1993 Search PubMed; (h) E. Juaristi and G. Cuevas, The Anomeric Effect, CRC Press, Boca Raton, 1995 Search PubMedC. L. Perrin, Tetrahedron, 1995, 51, 11901 CrossRef CAS; (i) E. Juaristi and Y. Bandala, Adv. Heterocycl. Chem., 2012, 105, 189 CrossRef CAS.
- (a) C. Wang, Z. Chen, W. Wu and Y. Mo, Chem.–Eur. J., 2013, 19, 1436 CrossRef CAS PubMed; (b) S. David, O. Eisenstein, W. J. Hehre, L. Salem and R. Hoffmann, J. Am. Chem. Soc., 1973, 95, 3806 CrossRef CAS; (c) A. E. Reed and P. V. R. Schleyer, Inorg. Chem., 1988, 27, 3969 CrossRef CAS.
- I. V. Alabugin, K. M. Gilmore and P. W. Peterson, WIREs Computational Molecular Science, 2011, 1, 109 CrossRef CAS.
- Schwarz and coworkers found the same conformational preference: C. A. Schalley, J. N. Harvey, D. Schröder and H. Schwarz, J. Phys. Chem. A, 1998, 102, 1021 CrossRef CAS See also: S. L. Khursan and V. L. Antonovskii, Dokl. Phys. Chem., 2002, 382, 657 CrossRef . The preferred COOC dihedral was found to be 180° and 171° at the M06-2X and B2PLYPD levels respectively, both with 6-311++G(d,p) basis set.
- (a) J. O. Edwards and R. G. Pearson, J. Am. Chem. Soc., 1962, 82, 16 CrossRef; (b) S. Hoz and E. Buncel, Isr. J. Chem., 1985, 26, 313 CrossRef CAS.
- See ESI† for full analysis with cyclic peroxides included. Naturally, cyclic structures disfavor the larger COOC dihedral angles.
- Bond Dissociation Energy (BDE) ∼39 kcal mol−1 for CH3OOCH3: R. Bach, A. P. Ayala and H. B. Schlegel, J. Am. Chem. Soc., 1996, 118, 12758 CrossRef CAS.
- This is due to the increase in the s, p-gap that imposes penalty on hybridization by increasing promotion energy. For a more detailed discussion of hybridization effects on reactivity, see: (a) I. V. Alabugin, S. Bresch and M. Manoharan, J. Phys. Chem. A, 2014, 118, 3663 CrossRef CAS PubMedI. V. Alabugin and M. J. Manoharan, Comput. Chem., 2007, 28, 373 CrossRef CAS PubMed; (b) I. V. Alabugin, S. Bresch and G. Passos Gomes, J. Phys. Org. Chem., 2014, 28, 147 CrossRef.
- Not only is the σ-overlap of p-orbitals inefficient, but the unusual hybridization in the O–O bond has further consequences. For example, the increased allocation of p-character to an σ-bond leaves the relatively low amount of p- character in the two lone pairs of peroxide oxygen atoms. Although the p-type lone pairs in peroxides and acetals are almost the same, the hybridized σ-lone pairs are noticeably different: ∼sp0.7 for the peroxide and ∼sp1.4 for the acetal. Because p-character of lone pairs correlates with their donor ability, decreased p-character in the oxygen lone pair further decreases importance of the few remaining vicinal nO → σ* interactions.
- Combined energy of these interactions is estimated to be ∼15 kcal mol−1 by NBO analysis. V. Pophristic and L. Goodman, Nature, 2001, 411, 565 CrossRef CAS PubMed.
- F. Weinhold, Angew. Chem., Int. Ed., 2003, 42, 4188 CrossRef CAS.
- I. V. Alabugin and T. A. Zeidan, J. Am. Chem. Soc., 2002, 124, 3175 CrossRef CAS PubMed.
- For more information on the Trigonox 311®, visit: https://www.akzonobel.com/polymer/our_products/trigonox_311/, accessed in April 2015.
- Donation from lone pair of X to the σ*C–O of the peroxide also contributes to the overall isodesmic stabilization.
- For the effect of remote F-substitution on metabolic stability of artemisins, see: J.-P. Bégué and D. Bonnet-Delpon, ChemMedChem, 2007, 2, 608 CrossRef PubMed.
- Other examples of stabilizing axial substituents: C.-Y. Chiang, W. Butler and R. L. Kuczkowski, J. Chem. Soc., Chem. Commun., 1988, 465 RSC; N. Jorge, M. E. Gómez-Vara, L. F. R. Cafferata and E. A. Castro, Acta Chim. Slov., 2002, 49, 111 Search PubMed.
- J. A. Plumley and J. D. Evanseck, J. Phys. Chem. A, 2009, 113, 5985 CrossRef CAS PubMed.
- This is consistent with the similar donor ability of p-lone pair of O and hybrid lone pair of nitrogen. See ref. 26 for further discussion.
- F. Dubnikova, R. Kosloff, J. Almog, Y. Zeiri, R. Boese, H. Itzhaky, A. Alt and E. Keinan, J. Am. Chem. Soc., 2005, 127, 1146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates and energies of all calculated structures. See DOI: 10.1039/c5sc02402a |
| This journal is © The Royal Society of Chemistry 2015 |