
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphine and carbene azido-cations: [(L)N3]+ and [(L)2N3]+†
Daniel
Winkelhaus‡
a,
Michael H.
Holthausen‡
a,
Roman
Dobrovetsky
b and
Douglas W.
Stephan
*a
aDepartment of Chemistry, University of Toronto, 80 St. George St, Toronto, Ontario M5S3H6, Canada. E-mail: dstephan@chem.utoronto.ca
bSchool of Chemistry, Raymond and Beverly Sackler Faculty of Exact Sciences, Tel Aviv University, Tel Aviv 69978, Israel
First published on 21st July 2015
Abstract
The cationic N3-species [(p-HC6F4)3PN3]+ (1) featuring a perfluoro-arene phosphonium group serves as a N3+-source in stoichiometric reactions with several Lewis bases (L) allowing for the stepwise formation of [(L)N3]+ and [(L)2N3]+ cations (L = phosphine, carbene) with liberation of (p-HC6F4)3P. X-Ray diffraction analysis and computational studies provide insight into the bonding in these remarkably stable azido-cations.
Introduction
A major component of the recent renaissance in p-block chemistry1 has been the use of neutral, two electron donors like carbenes or phosphines for the stabilization of homoatomic, low valent main group element fragments.2 Most recently Cummins used an anthracene stabilized P2 for the synthesis of an aromatic [P2N3].3 Species of the form (L)2E2 (E = B,4 C,5 Si,6 Ge,7 Sn,8 N,9 P,10 As,11 Sb;12 L = carbene, phosphine) have been prepared affording ligand stabilization of unique diatomic main group fragments. In addition, the groups of Robinson, Bertrand and Roesky have exploited these donors for isolation of dications and radical cations of type [(carbene)2E2]n+ (E = C,5 P,13 As;14n = 1, 2) while Burford, Weigand, Jones and Grützmacher prepared [(carbene)2P3]+15 or bicyclic [(L)2P4]2+ species (L = carbene, AsPh3, PPh3).16 The latter compound is of special interest since L can be exchanged for more basic donors. A similar donor exchange was reported for [(Ph3P)PPh2]+ affording [(carbene)PPh2]+.17 The nature of these and related systems, especially the L–E and E–E bonding, has sparked vigorous debate.18Our recent studies of highly electrophilic phosphonium cations (EPCs) have demonstrated that species such as the fluorophosphonium cation [(C6F5)3PF]+19 exhibit remarkable Lewis acidity and thus act as effective catalysts in a range of Lewis acid mediated transformations.20 At the same time, we were motivated to probe the utility of these EPCs as synthons for cationic azides. Herein we describe the synthesis of [(p-HC6F4)3PN3]+ (1) and its use as a synthon to species of the form [(L)N3]+ and [(L)2N3]+ (L = phosphine, carbene).
Results and discussion
Compound [(p-HC6F4)3PN3][B(C6F5)4] (1) was readily prepared in quantitative yield by the reaction of [(p-HC6F4)3PF][B(C6F5)4] with Me3SiN3 in CH2Cl2 (Scheme 1).§ The 31P{1H} NMR spectrum of 1 in CD2Cl2 gives rise to a broad singlet resonance (δ(31P) = 17.5 ppm, Δν1/2 = 30 Hz).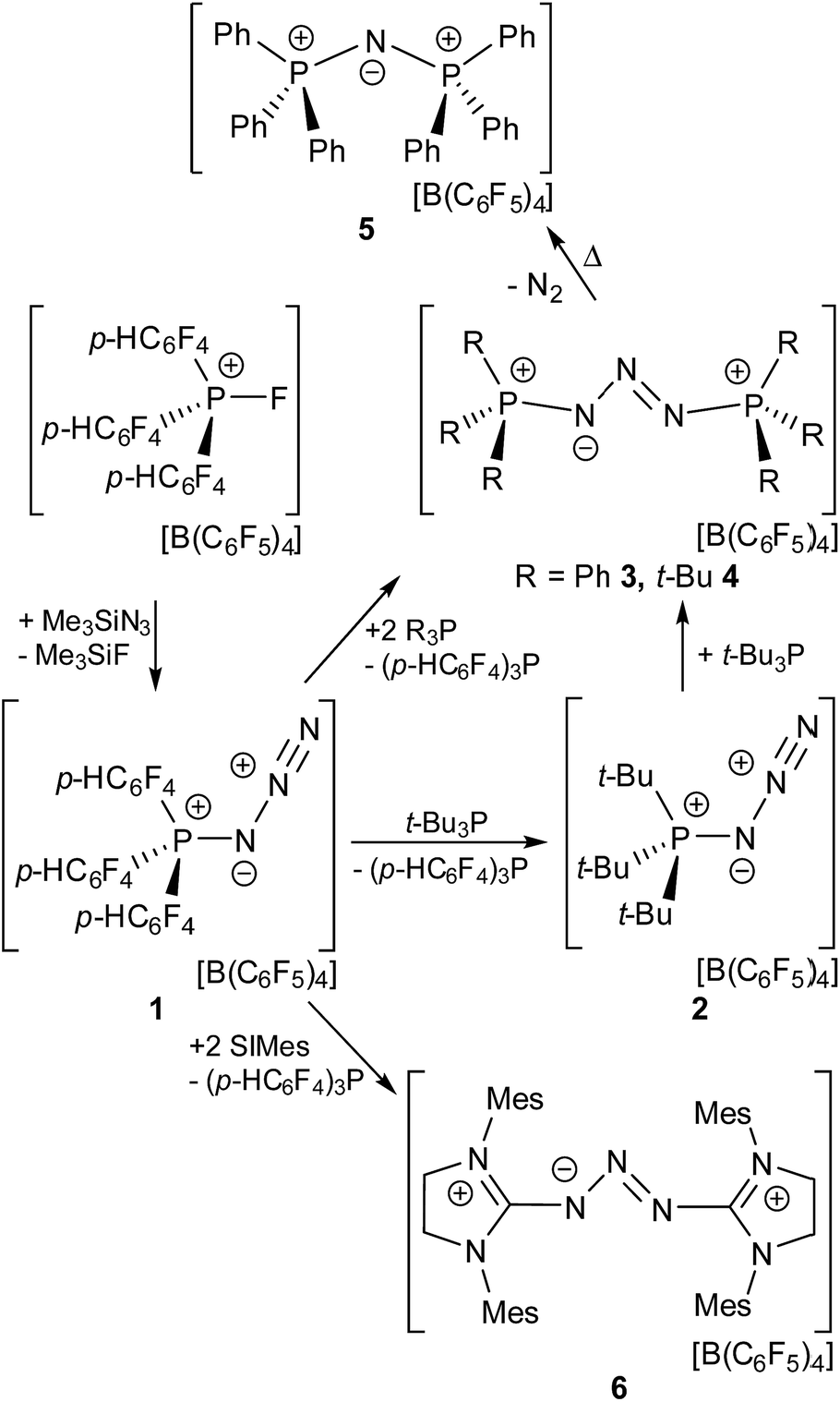 | ||
| Scheme 1 Preparation of 1–6. | ||
The presence of the para hydrogen substituents in 1 is crucial as the fully fluorinated derivative [(C6F5)3PF]+ is prone to nucleophilic attack at this position.21 The formulation of compound 1 was subsequently confirmed by an X-ray crystallographic study (Fig. 1).
 | ||
| Fig. 1 POV-ray depiction of the cation in 1 (top). Hydrogen atoms are omitted for clarity. Selected bond distances and angles: P(1)–N(1) 1.651(2), P(1)–C(13) 1.783(2), P(1)–C(1) 1.789(2), P(1)–C(7) 1.789(2), N(1)–N(2) 1.260(2), N(2)–N(3) 1.114(2), N(1)–P(1)–C(13) 111.6(2), N(1)–P(1)–C(1) 102.3(2), C(13)–P(1)–C(1) 112.6(2), N(1)–P(1)–C(7) 109.8(2), C(13)–P(1)–C(7) 110.5(2), C(1)–P(1)–C(7) 109.7(2), N(2)–N(1)–P(1) 120.9(2), N(3)–N(2)–N(1) 172.4(2). Graphical presentation of the HOMO and LUMO+10 of 1 calculated at the wB97XD/def2-TZV level of theory (surface iso-value = 0.05). | ||
The molecular structure of 1 shows a distorted tetrahedral environment about the phosphorus atom with angles at P ranging from 102.3(1)–112.6(1)°. The P(1)–N(1) bond length of 1.651(2) Å is shorter than those observed in azido-substituted phosphenium cations (1.67 Å)22 and azido-phosphines (1.73 Å),23 consistent with the electron deficient nature of the phosphonium center and the strongly polarized nature of the P–N bond. The azido moiety shows an N(1)–N(2)–N(3) angle that slightly deviates from the ideal 180° (172.4(2)°) while the N(1)–N(2) (1.260(2) Å) and N(2)–N(3) bond length (1.114(2) Å) are in the expected range for azide substituents. Compound 1 is a rare example of a crystallographically characterized salt of a cationic azido-phosphonium species24 while several neutral, azido-substituted P(III) and P(V) derivatives have been reported to date.25 Other examples of monocationic nitrogen species, reported in the literature, include diazonium ions,26 the homoleptic [N5]+ of Christe,27 [N2F]+,28 Hünig's stable triazenium ions [R2N3R2]+29 and aminodiazonium ions of the form [(R2N)N2]+30 (R = H, silyl, alkyl, aryl).
To gain further insights into the nature of 1, the geometry of the cation was optimized at the wB97XD/def2-TZV level of theory (see ESI†). The p-type HOMO of 1 is located at the P-bonded N atom while the LUMO is part of two sets of degenerated π*-type orbitals involving the p-HC6F4-groups (LUMO to LUMO+9). The first accessible acceptor orbital is a π*-type orbital located at the terminal N2 moiety (LUMO+10). This stands in contrast to other EPCs where the accessible acceptor orbital involves the P–F σ* orbital at the P center.19 The corresponding P–N σ* orbital in 1 is much higher in energy (LUMO+11). NBO analysis reveals that the P–N single bond in 1 is occupied by 1.92 electrons, while the P-bound N atom features two lone pairs of p and sp0.69 type. Donation of electron density from these lone pairs into σ* orbitals of the adjacent P–C bonds occurs (LP(p)N1 → σ*P–C7 11.2 kcal mol−1, LP(p)N1 → σ*P–C7 7.4 kcal mol−1, LP(sp0.69)N1 → σ*P–C7 3.7 kcal mol−1). The secondary interactions of these lone pairs may account for the observed shortening of the P–N bond in 1 in comparison to other P-azido species.22,23
Compound 1 reacts with the Lewis base t-Bu3P in CH2Cl2 solution (Scheme 1).§ The 31P{1H} NMR spectrum of the reaction mixture showed a septet resonance at δ(31P) = −72.3 ppm (3JPF = 36.4 Hz)19b which corresponds to (p-HC6F4)3P and a new singlet resonance at low field (δ(31P) = −85.9 ppm) which is in the typical chemical shift range for an N-substituted trialkyl-phosphonium derivative.31 Collectively, these observations indicate the formal transfer of the N3+-moiety in 1 to t-Bu3P yielding [t-Bu3PN3][B(C6F5)4] (2) which was isolated in 98% yield. The nature of 2 was confirmed by X-ray single crystallography and the metrics were found similar to 1 (see ESI†). Interestingly, 2 and 1 are very stable and do not show any signs of degradation on heating in C6D5Br solution to 100 °C for 24 h. Attempts to independently synthesize 2 from the reaction of [t-Bu3PF][B(C6F5)4]19 with Me3SiN3 failed even after prolonged reaction times at 100 °C in C6D5Br solution. This reflects the increased steric demand of the [t-Bu3PF]+ cation.
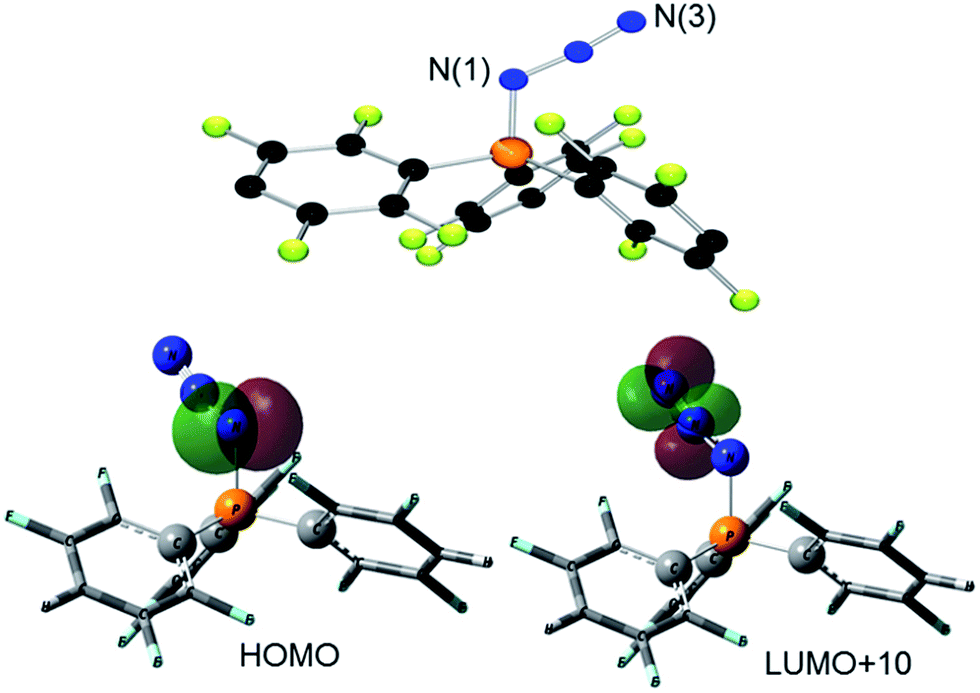
The corresponding reaction of 1 with Ph3P in CH2Cl2 was monitored by 31P{1H} NMR spectroscopy revealing the complete consumption of Ph3P and the liberation of only 0.5 equivalents of (p-HC6F4)3P, leaving approximately 50% of 1 unreacted. Addition of another equivalent of Ph3P resulted in its complete consumption. Collectively, this indicates the formation of a bis-adduct [(Ph3P)2(N3)][B(C6F5)4] (3, Scheme 1). While the proposed intermediate [Ph3PN3]+ was independently synthesized by the reaction of [Ph3PF][B(C6F5)4]20c and Me3SiN3 (see ESI†), this species was not observed in the reaction of 1 with Ph3P. This indicates that reaction of [Ph3PN3]+ with a second equivalent of Ph3P is rapid, in agreement with Wiberg who described the [(Ph3P)2(N3)]+ cation in 1967.24c Compound 3 was isolated in high yields (97%). The t-Bu-substituted analog 4 was obtained from 1 with two equivalents of t-Bu3P or by reaction of 2 with one equivalent of t-Bu3P (Scheme 1). Species 4 was also obtained by reaction of 3 with two equivalents of t-Bu3P with concurrent release of Ph3P. All methods furnish 4 in high yields (76–99%). Compounds 3 and 4 show 31P{1H} NMR resonances at 30.6 and 56.5 ppm that are between the chemical shift ranges of phosphinimine and N-substituted phosphonium derivatives.31 X-Ray structure determination confirmed the formulations (Fig. 2). The P2N3-moiety adopts a W-shaped geometry with all five atoms almost located within one plane (largest deviation: 3: 0.013(2) Å for N2, 4: 0.039(1) Å for N2). The acute N–N–N angles (3: 113.2(4)°, 4: 110.8(1)°) are in the typical range for phosphazides.32 The two P–N–N angles in each compound are 3: 114.5(3)°/114.7(3)°; 4: 118.1(1)°/118.9(1)° with the larger angles in 4 reflecting the increased steric congestion. Similarly, the P–N bond lengths of 1.648(3) Å and 1.643(3) Å and 1.675(2) Å and 1.673(2) Å seen in 3 and 4, respectively, range between those of phosphinimine and N-substituted phosphonium derivatives.31 The N–N bonds in 3 and 4 were found to be 1.298(4) Å, 1.309(4) Å and 1.309(2) Å, 1.300(2) Å,28 which are intermediate between single and double bond distances. A similar conformation is observed for several crystallographically characterized organo-phosphazides R3PN3R,32 formed in the initial step of a Staudinger reaction.33
 | ||
| Fig. 2 POV-ray depiction of the cations in 3 (top) and 4 (bottom). Hydrogen atoms are omitted for clarity. Selected bond distances and angles for 3: N(1)–P(1) 1.648(3), N(3)–P(2) 1.643(3), N(1)–N(2) 1.298(4), N(2)–N(3) 1.309(4), N(2)–N(1)–P(1) 114.5(3), N(1)–N(2)–N(3) 113.2(4), N(2)–N(3)–P(2) 114.7(3), for 4: P(1)–N(3) 1.675(2), P(2)–N(1) 1.673(2), N(1)–N(2) 1.309(2), N(3)–N(2) 1.230(2), N(2)–N(1)–P(2) 118.1(2), N(2)–N(3)–P(1) 118.9(2), N(3)–N(2)–N(1) 110.8(2). | ||
It is of interest to note that, similar to 1 and 2, 4 is thermally stable and can be heated to 120 °C in C6D5Br solution over several days without decomposition. In contrast, compound 3 is not temperature stable, decomposing quantitatively with release of N2 within 3 h at 100 °C.24c The 31P{1H} NMR spectrum shows only one resonance at δ(31P) = 21.1 ppm assignable to [(Ph3P)2N][B(C6F5)4] (5) (Scheme 1).34 N2 elimination is thought to follow isomerization of the W-shaped 3 to a U-shaped isomer. It is noteworthy that compound 3 was isolated as a mixture of both isomers in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The 31P{1H} NMR spectrum of the minor isomer gave two resonances at δ(31P) = 28.4 and 11.0 ppm (4JPP = 5.5 Hz). At elevated temperatures, rapid conversion of the cation 3 to [(Ph3P)2N]+5 was observed. Nonetheless, the ratio of the isomers of 3 is unchanged inferring the rate of N2-loss is comparable to the rate of isomerization of 3. Computations at the wB97XD/def2-TZV level of theory with addition of the conductor-like polarizable continuum solvation model (CPCM)35 showed only a small energy difference between the W and U-shaped isomers of 3 (ΔGR298 = −2.6 kcal mol−1).
:1 ratio. The 31P{1H} NMR spectrum of the minor isomer gave two resonances at δ(31P) = 28.4 and 11.0 ppm (4JPP = 5.5 Hz). At elevated temperatures, rapid conversion of the cation 3 to [(Ph3P)2N]+5 was observed. Nonetheless, the ratio of the isomers of 3 is unchanged inferring the rate of N2-loss is comparable to the rate of isomerization of 3. Computations at the wB97XD/def2-TZV level of theory with addition of the conductor-like polarizable continuum solvation model (CPCM)35 showed only a small energy difference between the W and U-shaped isomers of 3 (ΔGR298 = −2.6 kcal mol−1).
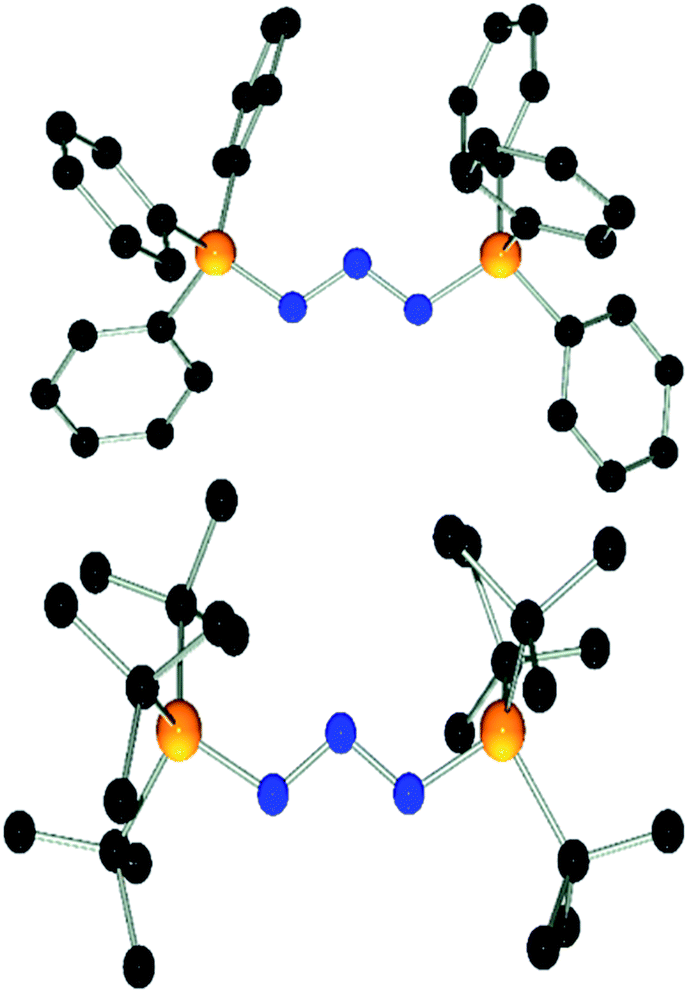
Compound 1 also reacts with two equivalents of 1,3-dimesitylimidazolidin-2-ylidene (SIMes) in C6D5Br solution with liberation of (p-HC6F4)3P as evidenced by the 31P{1H} NMR spectrum. The 13C{1H} NMR spectrum shows a new resonance for the C-2 carbon atom at δ(13C) = 162.2 ppm.36 Collectively, the NMR data indicate the formation of [(SIMes)2N3][B(C6F5)4] (6) which was isolated in 70% yield. Interestingly, 6 was the only product obtained from the reaction using a 1:1 stoichiometry. This contrasts with the reactivity observed for t-Bu3P and is likely a result of the low solubility of 1 in bromobenzene. Interestingly, compound 6 was also obtained in high yields by reaction of 3 or [Ph3PN3][B(C6F5)4] with SIMes in 1:2 stoichiometry (see ESI†). Compound 6 is thermally stable even under prolonged heating to 120 °C for several days in C6D5Br solution. The nature of compound 6 was further confirmed by single crystal X-ray diffraction (Fig. 3). The structure features a W-shaped N3-chain terminated by imidazolidiniumyl-groups with a N–N–N angle (110.4(2)°) comparable to that seen for 3 and 4. The planes of the imidazole-rings are skewed with respect to the N3-plane (38.3(7)°/41.2(5)°) and the torsion angles involving the C–N bonds (N5–C1–N1–N2: 27.6°, N6–C22–N3–N2: 35.0°) deviate from those expected for a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond. This indicates that electron delocalization across the imidazole and azide moiety may be hampered by the steric demands of the Mes substituents. The N–N (1.309(2) Å/1.311(2) Å) and C–N (1.359(2) Å/1.369(2) Å) bond distances involving the W-type fragment of 6 fall in the range between those expected for single and double bonds.37
N double bond. This indicates that electron delocalization across the imidazole and azide moiety may be hampered by the steric demands of the Mes substituents. The N–N (1.309(2) Å/1.311(2) Å) and C–N (1.359(2) Å/1.369(2) Å) bond distances involving the W-type fragment of 6 fall in the range between those expected for single and double bonds.37
 | ||
| Fig. 3 POV-ray depiction of the cation in 6. Hydrogen atoms are omitted for clarity. Selected bond distances and angles: C(1)–N(4) 1.332(3), C(1)–N(5) 1.346(2), C(1)–N(1) 1.359(2), C(22)–N(7) 1.335(2), C(22)–N(6) 1.337(2), C(22)–N(3) 1.369(2), N(1)–N(2) 1.309(2), N(2)–N(3) 1.311(2), N(4)–C(1)–N(5) 111.1(2), N(4)–C(1)–N(1) 119.3(2), N(5)–C(1)–N(1) 129.4(2), N(7)–C(22)–N(6) 111.7(2), N(7)–C(22)–N(3) 119.9(2), N(6)–C(22)–N(3) 128.2(2), N(2)–N(1)–C(1) 113.6(2), N(1)–N(2)–N(3)110.4(2), N(2)–N(3)–C(22) 111.9(2). | ||
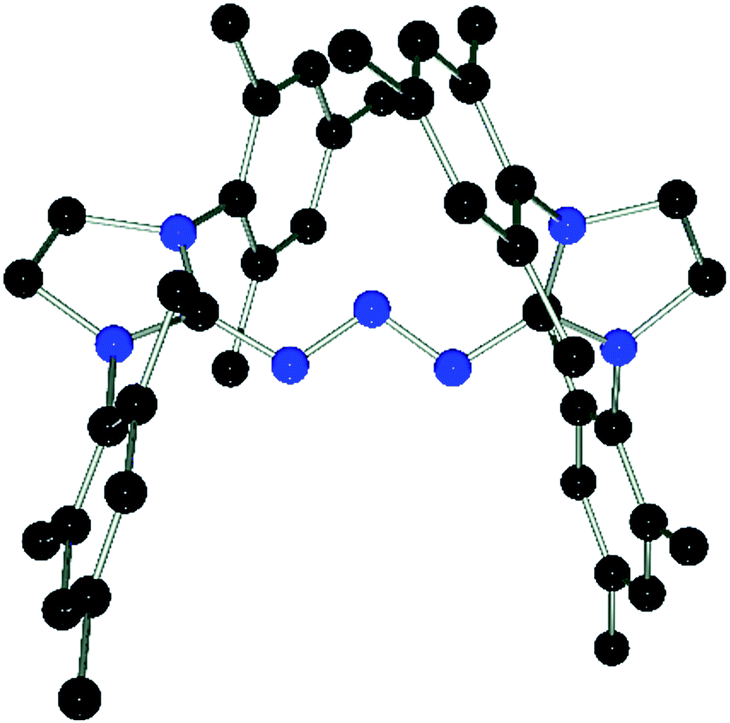
DFT calculations (wB97XD/def2-TZV, see ESI†) carried out on 3 and the model compound 6-Me, (mesityl-substituents replaced by methyl groups) showed similar molecular orbitals for both compounds (Fig. 4). The HOMOs exhibit strong n character comprised primarily of the lone pairs of the N-atoms. The LUMOs are π-type orbitals delocalized across the N3-linkage and the L donors. This is in contrast to the isolated P3-allyl anion frontier orbitals reported for [(carbene)2P3]+.15 Interestingly these frontier orbitals are also reminiscent of the HOMO and HOMO−1 of neutral triazenes (NHC)N3R.38
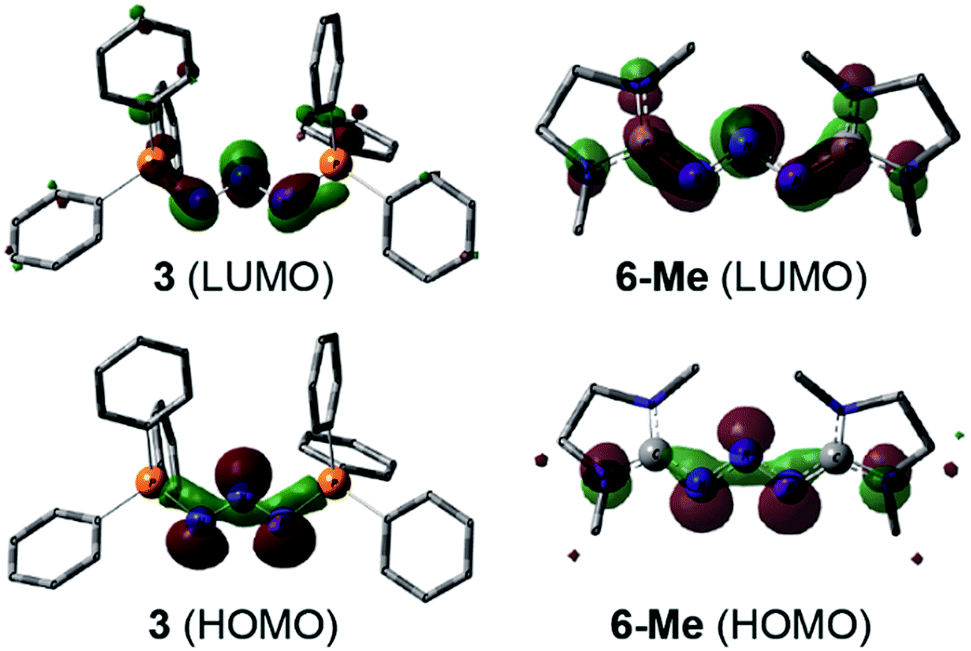 | ||
| Fig. 4 Graphical presentation of the LUMO (top) and HOMO (bottom) of the cations in 3 and 6-Me calculated at the wB97XD/def2-TZVPP level of theory (surface iso-value = 0.05). | ||
Conclusions
In summary, the EPC [(p-C6F4H)3PF][B(C6F5)4] is used to prepare the phosphonium ion salt 1 which serves as a precursor for the formal transfer of [N3]+ to other donors affording stable and isolable mono- and bis-adducts of the form [(L)N3]+ and [(L)2N3]+. The reactivity of these species containing terminal and bridging azide-fragments is the subject of continuing studies. In addition, the exploration of the reactivity of EPCs as synthetic building blocks for other unusual main group cations is ongoing.Acknowledgements
D. W. S. gratefully acknowledges the financial support of the NSERC of Canada and the award of a Canada Research Chair. M. H. H. thanks the Alexander von Humboldt Foundation for a Feodor Lynen Research Fellowship.Notes and references
- (a) Y. Z. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326–12337 CrossRef CAS PubMed; (b) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (c) C. A. Dyker and G. Bertrand, Science, 2008, 321, 1050–1051 CrossRef CAS PubMed; (d) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815–11832 CrossRef CAS PubMed.
- D. J. D. Wilson, S. A. Couchman and J. L. Dutton, Inorg. Chem., 2012, 51, 7657–7668 CrossRef CAS PubMed.
- A. Velian and C. C. Cummins, Science, 2015, 348, 1001–1004 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed.
- (a) Y. Li, K. C. Mondal, P. P. Samuel, H. Zhu, C. M. Orben, S. Panneerselvam, B. Dittrich, B. Schwederski, W. Kaim, T. Mondal, D. Koley and H. W. Roesky, Angew. Chem., Int. Ed., 2014, 53, 4168–4172 ( Angew. Chem. , 2014 , 126 , 4252–4256 ) CrossRef CAS PubMed; (b) L. Jin, M. Melaimi, L. Liu and G. Bertrand, Org. Chem. Front., 2014, 1, 351–354 RSC.
- Y. Z. Wang, Y. M. Xie, P. R. Wei, R. B. King, H. F. Schaefer, P. V. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed.
- A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701–9704 ( Angew. Chem. , 2009 , 121 , 9881–9884 ) CrossRef CAS PubMed.
- C. Jones, A. Sidiropoulos, N. Holzmann, G. Frenking and A. Stasch, Chem. Commun., 2012, 48, 9855–9857 RSC.
- N. Holzmann, D. Dange, C. Jones and G. Frenking, Angew. Chem., Int. Ed., 2013, 52, 3004–3008 ( Angew. Chem. , 2013 , 125 , 3078–3082 ) CrossRef CAS PubMed.
- (a) Y. Z. Wang, Y. M. Xie, P. R. Wei, R. B. King, H. F. Schaefer, P. V. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS PubMed; (b) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530–5533 ( Angew. Chem. , 2009 , 121 , 5638–5641 ) CrossRef CAS PubMed; (c) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180–14181 CrossRef CAS PubMed; (d) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052–7055 ( Angew. Chem. , 2007 , 119 , 7182–7185 ) CrossRef CAS PubMed.
- M. Y. Abraham, Y. Z. Wang, Y. M. Xie, P. R. Wei, H. F. Schaefer, P. V. Schleyer and G. H. Robinson, Chem.–Eur. J., 2010, 16, 432–435 CrossRef CAS PubMed.
- R. Kretschmer, D. A. Ruiz, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 8176–8179 ( Angew. Chem. , 2014 , 126 , 8315–8318 ) CrossRef CAS PubMed.
- O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 CrossRef CAS PubMed.
- M. Y. Abraham, Y. Wang, Y. Xie, R. J. Gilliard Jr, P. Wei, B. J. Vaccaro, M. K. Johnson, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2013, 135, 2486–2488 CrossRef CAS PubMed.
- (a) M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078–11082 ( Angew. Chem. , 2013 , 125 , 11284–11288 ) CrossRef CAS PubMed; (b) A. M. Tondreau, Z. Benko, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC.
- (a) M. Donath, E. Conrad, P. Jerabek, G. Frenking, R. Fröhlich, N. Burford and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 2964–2967 ( Angew. Chem. , 2012 , 124 , 3018–3021 ) CrossRef CAS PubMed; (b) A. Sidiropoulos, B. Osborne, A. N. Simonov, D. Dange, A. M. Bond, A. Stasch and C. Jones, Dalton Trans., 2014, 43, 14858–14864 RSC.
- N. Burford, T. S. Cameron and P. J. Ragogna, J. Am. Chem. Soc., 2001, 123, 7947–7948 CrossRef CAS.
- (a) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 370–374 ( Angew. Chem. , 2014 , 126 , 378–382 ) CrossRef CAS PubMed; (b) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 6047–6048 ( Angew. Chem. , 2014 , 126 , 6159–6160 ) CrossRef CAS PubMed; (c) G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040–6046 ( Angew. Chem. , 2014 , 126 , 6152–6158 ) CrossRef CAS PubMed.
- (a) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed; (b) C. B. Caputo, D. Winkelhaus, R. Dobrovetsky, L. J. Hounjet and D. W. Stephan, Dalton Trans., 2015, 44, 12256–12264 RSC; (c) M. Perez, Z. W. Qu, C. B. Caputo, V. Podgorny, L. J. Hounjet, A. Hansen, R. Dobrovetsky, S. Grimme and D. W. Stephan, Chem.–Eur. J., 2015, 21, 6491–6500 CrossRef CAS PubMed.
- (a) M. Perez, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10917–10921 CrossRef CAS PubMed; (b) M. Perez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308–18310 CrossRef CAS PubMed; (c) M. H. Holthausen, R. R. Hiranandani and D. W. Stephan, Chem. Sci., 2015, 6, 2016–2021 RSC; (d) M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 ( Angew. Chem. , 2014 , 126 , 6656–6659 ) CrossRef CAS PubMed.
- L. J. Hounjet, C. B. Caputo and D. W. Stephan, Dalton Trans., 2013, 42, 2629–2635 RSC.
- C. Hering, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 6241–6245 ( Angew. Chem. , 2012 , 124 , 6345–6349 ) CrossRef CAS PubMed.
- (a) J. J. Weigand, K.-O. Feldmann and F. D. Henne, J. Am. Chem. Soc., 2010, 132, 16321–16323 CrossRef CAS PubMed; (b) D. Michalik, A. Schulz, A. Villinger and N. Weding, Angew. Chem., Int. Ed., 2008, 47, 6465–6468 ( Angew. Chem. , 2008 , 120 , 6565–6568 ) CrossRef CAS PubMed.
- Only two examples were crystallographically characterized: (a) C. Hering, A. Schulz and A. Villinger, Inorg. Chem., 2013, 52, 5214–5225 CrossRef CAS PubMed; for derivatives that were not characterized by X-ray see: (b) K. Bott, Angew. Chem., Int. Ed. Engl., 1965, 4, 695 ( Angew. Chem. , 1965 , 77 , 683 ) CrossRef PubMed; (c) N. Wiberg and K. H. Schmid, Angew. Chem., Int. Ed. Engl., 1967, 6, 953–954 ( Angew. Chem. , 1967 , 79 , 938 ) CrossRef CAS PubMed; (d) A. Schmidt, Chem. Ber., 1968, 101, 4015–4021 CrossRef CAS PubMed; (e) A. Schmidt, Chem. Ber., 1970, 103, 3923–3927 CrossRef CAS PubMed; (f) W. Buder and A. Schmidt, Chem. Ber., 1973, 106, 3812–3816 CrossRef CAS PubMed; (g) C. Hering, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 6241–6245 CrossRef CAS PubMed.
- (a) U. Müller, Chem. Ber., 1977, 110, 788–791 CrossRef PubMed; (b) H. W. Roesky, M. Noltemeyer and G. M. Sheldrick, Z. Naturforsch., B: Chem. Sci., 1986, 41, 803–807 Search PubMed; (c) D. Schomburg, U. Wermuth and R. Schmutzler, Chem. Ber., 1987, 120, 1713–1718 CrossRef CAS PubMed; (d) A. H. Cowley, F. Gabbaï, R. Schluter and D. Atwood, J. Am. Chem. Soc., 1992, 114, 3142–3144 CrossRef CAS; (e) U. Englert, P. Paetzold and E. Eversheim, Z. Kristallogr., 1993, 208, 307–309 CrossRef CAS; (f) J. R. Goerlich, M. Farkens, A. Fischer, P. G. Jones and R. Schmutzler, Z. Anorg. Allg. Chem., 1994, 620, 707–715 CrossRef CAS PubMed; (g) A. H. Cowley, F. P. Gabbaï, G. Bertrand, C. J. Carrano and M. R. Bond, J. Organomet. Chem., 1995, 493, 95–99 CrossRef CAS; (h) M. Larbig, M. Nieger, V. von der Gönna, A. V. Ruban and E. Niecke, Angew. Chem., Int. Ed. Engl., 1995, 34, 460–462 ( Angew. Chem. , 1995 , 107 , 505 ) CrossRef CAS PubMed; (i) C. Aubauer, T. M. Klapötke, H. Noth, A. Schulz, M. Suter and J. Weigand, Chem. Commun., 2000, 2491–2492 RSC; (j) N. Götz, S. Herler, P. Mayer, A. Schulz, A. Villinger and J. J. Weigand, Eur. J. Inorg. Chem., 2006, 2051–2057 CrossRef PubMed; (k) A. Schulz and A. Villinger, Eur. J. Inorg. Chem., 2008, 4199–4203 CrossRef CAS PubMed; (l) X. Zeng, M. Gerken, H. Beckers and H. Willner, Inorg. Chem., 2010, 49, 3002–3010 CrossRef CAS PubMed; (m) X. Zeng, E. Bernhardt, H. Beckers and H. Willner, Inorg. Chem., 2011, 50, 11235–11241 CrossRef CAS PubMed; (n) C. Hering, M. Hertrich, A. Schulz and A. Villinger, Inorg. Chem., 2014, 53, 3880–3892 CrossRef CAS PubMed.
- (a) A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed; (b) S. Mahouche-Chergui, S. Gam-Derouich, C. Mangeney and M. M. Chehimi, Chem. Soc. Rev., 2011, 40, 4143–4166 RSC.
- (a) K. O. Christe, W. W. Wilson, J. A. Sheehy and J. A. Boatz, Angew. Chem., Int. Ed., 1999, 38, 2004–2009 CrossRef CAS; (b) K. O. Christe, W. W Wilson, J. A. Sheehy and J. A. Boatz, Angew. Chem., Int. Ed., 1999, 111, 2112–2118 CrossRef.
- D. Moy and A. R. Young, J. Am. Chem. Soc., 1965, 87, 1889–1892 CrossRef CAS.
- H. Hansen, S. Hünig and K.-I. Kishi, Chem. Ber., 1979, 112, 445–461 CrossRef CAS PubMed.
- (a) A. Mertens, K. Lammertsma, A. Massoud and G. A. Olah, J. Am. Chem. Soc., 1983, 105, 5657–5660 CrossRef; (b) K. O. Christe, W. W. Wilson, D. A. Dixon, S. I. Khan, R. Bau, T. Metzenthin and R. Lu, J. Am. Chem. Soc., 1993, 115, 1836–1842 CrossRef CAS; (c) A. Schulz and A. Villinger, Chem.–Eur. J., 2010, 16, 7276–7281 CrossRef CAS PubMed; (d) C. W. Huh and J. Aube, Chem. Sci., 2014, 5, 699–706 RSC.
- M. H. Holthausen, I. Mallov and D. W. Stephan, Dalton Trans., 2014, 43, 15201–15211 RSC.
- (a) L. LePichon and D. W. Stephan, Inorg. Chem., 2001, 40, 3827–3829 CrossRef CAS; (b) G. C. Fortman, B. Captain and C. D. Hoff, Inorg. Chem., 2009, 48, 1808–1810 CrossRef CAS PubMed; (c) P. Molina, C. López-Leonardo, J. Llamas-Botía, C. Foces-Foces and C. Fernández-Castaño, Tetrahedron, 1996, 52, 9629–9642 CrossRef CAS; (d) R. D. Kennedy, Chem. Commun., 2010, 46, 4782–4784 RSC; (e) C. G. Chidester, J. Szmuszkovicz, D. J. Duchamp, L. G. Laurian and J. P. Freeman, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1080–1083 CrossRef; (f) C. Reddy, V. Reddy, B. M. Fetterly and J. G. Verkade, Energy Fuels, 2007, 21, 2466–2472 CrossRef CAS; (g) M. W. P. Bebbington and D. Bourissou, Coord. Chem. Rev., 2009, 253, 1248–1261 CrossRef CAS PubMed.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635 CrossRef CAS PubMed.
- S. Cameron and M. Peach, J. Chem. Crystallogr., 1998, 28, 919–924 CrossRef.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed; (c) J. Andzelm, C. Kölmel and A. Klamt, J. Chem. Phys., 1995, 103, 9312–9320 CrossRef CAS PubMed; (d) A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- M. Iglesias, D. J. Beetstra, J. C. Knight, L.-L. Ooi, A. Stasch, S. Coles, L. Male, M. B. Hursthouse, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2008, 27, 3279–3289 CrossRef CAS.
- A. F. Holleman and E. Wiberg, Lehrbuch der Anorganischen Chemie, ed. von Nils Wiberg, de Gruyter, Berlin, 2007 Search PubMed.
- (a) J. E. Leffer and R. D. Temple, J. Am. Chem. Soc., 1967, 89, 5235–5246 CrossRef; (b) J. G. Gololobov, L. F. Kasukhin and U. S. Petrenko, Phosphorus Sulfur Relat. Elem., 1987, 30, 393–396 CrossRef PubMed; (c) H. Bock and M. Schnöller, Angew. Chem., Int. Ed. Engl., 1968, 7, 636–637 ( Angew. Chem. , 1968 , 80 , 667 ) CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on VT NMR experiments, preparation of 5, quantum chemical calculations and X-ray crystallography. CCDC 1403531–1403535. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02336j |
| ‡ These authors contributed equally. |
| § Extra precaution must be taken when working with azides in CH2Cl2 solution as they are potentially dangerous and can result in the possible formation of dangerous and explosive diazidomethane. |
| This journal is © The Royal Society of Chemistry 2015 |