
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of the RTH-type layer: the first small-pore, two dimensional layered zeolite precursor†
Joel E.
Schmidt
 a,
Dan
Xie
b and
Mark E.
Davis
*a
a,
Dan
Xie
b and
Mark E.
Davis
*a
aChemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: mdavis@cheme.caltech.edu
bChevron Energy Technology Company, Richmond, CA 94802, USA
First published on 27th July 2015
Abstract
There are several distinct two-dimensional zeolite building layers that can condense through different stacking arrangements of the layers to form various three-dimensional framework materials. All known building layers are dense layers in that they do not contain 8-membered ring (MR) or larger pores perpendicular to the two-dimensional layers. Herein, we report a new material (CIT-10) that consists of a two-dimensional layer (termed “rth” layer) that contains an 8 MR perpendicular to the layer. Calcination of CIT-10 forms pure-silica RTH (SSZ-50). CIT-10 can be pillared to form a new framework material with a three-dimensional pore system of 8 and 10 MRs, denoted CIT-11, that can be calcined to form a new microporous material, denoted CIT-12.
1. Introduction
The rate of discovery of new microporous materials has accelerated in recent years due to factors including new organic structure directing agents, the use of fluoride as a mineralizing agent as well as germanium as a heteroatom in syntheses.1–6 Much of this discovery is motivated by the fact that a single framework and composition are normally found to achieve optimal performance in a process.7 Another synthesis strategy that has received increased attention is that of synthesizing layered silicates that can directly form microporous materials via topotactic condensation, or can be pillared using silyating agents, forming structures with larger pores (generally larger by 2 tetrahedral atoms) than would be formed via topotactic condensation.8–14In a topotactic condensation, a three-dimensional (3D) framework structure is formed from a two-dimensional (2D) layered silicate by condensation of surface silanol groups (Si-OH), releasing water. Some of the initial framework materials prepared via topotactic condensation were FER, formed from the layered precursor denoted PREFER,15 and MWW, formed from the layered precursor MCM-22(P).16 After these pioneering efforts, several additional frameworks have been prepared using topotactic condensation, and they include (layered precursor given in parentheses): AST (β-helix-layered silicate),17CAS-NSI intermediate (EU-19, NU-6(1)),18–20CDO (PLS-1, RUB-36),19,21FER (PREFER),15MTF (HPM-2),22MWW (MCM-22),16PCR (IPC-4 prepared by disassembly of UTL),23RRO (RUB-39),24RWR (RUB-18)25 and SOD (RUB-15).26 The layered materials that form 3D frameworks can be built by different stacking arrangements of a few 2D building layers (such as CAS, FER, HEU, MWW and RWR).8,9 Additionally, methods have been developed to prepare MFI nanosheets that are a single unit cell thick,27,28 however, this material generally is not considered to be a building layer.29
The silanol groups of the layered zeolite precursors can also be used to prepare larger pore materials through a pillaring process. This process normally uses dichlorodimethylsilane or diethoxydimethylsilane to react with the silanol groups to form pillars that are coordinated to two methyl groups (or two hydroxyl groups after calcination). This process is typically carried out in acidic media under hydrothermal conditions. Some of the layered materials that have been pillared include PREFER, MWW(P), PLS-1, MCM-47, RUB-36, RUB-39 and Nu-6(1).8,30,31 Additionally, related strategies to prepare porous materials include delamination or exfoliation. Recently, it has also been shown to be possible to introduce catalytic activity in the pillars.32,33 Excellent reviews of the above strategies are available.8,9
All of the previously reported layered zeolite precursors are dense layers, that is, they contain no pores (8-membered ring (MR) or larger) that are perpendicular to the layers. The MWW layer contains a sinusoidal 10 MR channel parallel to the ab-plane, but it is still dense as this channel is not perpendicular to the layer. However, the nanosheets of MFI that are single unit cell thick do contain a 10 MR perpendicular to the layer, a medium size pore.27 We recently reported a method to prepared high-silica heulandite (denoted CIT-8) via topotactic condensation from a layered precursor (denoted CIT-8P), that was prepared using a diquaternary organic structure directing agent (OSDA) in fluoride-mediated syntheses.34 In that case, the building layer that forms CIT-8 (HEU) is the same as that of RUB-41 (RRO), but is formed from a different stacking of the HEU building layer: AA stacking gives the RRO structure while AA′ stacking (where the A′-layer is related to the A-layer by a 180 degree rotation) gives the HEU structure.25 CIT-8 was prepared from fluoride-mediated, aluminosilicate inorganic conditions across a relatively narrow composition range. It is interesting to note with this material that the OSDA used was considerably larger than what is normally found in preparing topotactic materials such as piperdines,15 as well as methyl, ethyl and propyl substituted ammoniums.31
In the absence of aluminium, we have found conditions to prepare the layered precursor to pure-silica RTH, denoted CIT-10. This layered material can be directly calcined to prepare pure-silica RTH (SSZ-50 (ref. 35)) or can be pillared, leading to a new microporous material (denoted CIT-11), that is stable to calcination (calcined material denoted CIT-12). CIT-10 is a layered material composed of a new type of 2D building layer (termed “rth”, according to the nomenclature defined in ref. 9) containing an 8 MR channel going through the layer (8 MR pore dimensions of 5.6 × 2.5 Å). The discovery of CIT-10 adds a new group of layered zeolite precursors and is the first to contain an 8 MR through the 2D layer. Pure-silica RTH is now the sixth known microporous material that can be obtained by both direct synthesis and topotactic condensation.
2. Experimental
2.1. OSDA synthesis
The diquaternary OSDA used in this work (shown in Fig. 1) was synthesized by reacting 200 mmol of 1,2,4,5-tetramethylimidazole (TCI Chemicals) with 100 mmol of 1,5-dibromopentane (Aldrich) at reflux in methanol overnight. The solvent was then removed using rotary evaporation and the product washed with ether. The product was verified using 13C NMR in D2O with methanol added as an internal standard. 13C-NMR (125 MHz, D2O): δ 7.76, 7.82, 9.61, 22.82, 28.58, 31.42, 44.72, 124.84, 126.03, 141.95. The product was ion exchanged to hydroxide form using Dowex Marathon A exchange resin and the final product concentration was determined using a Mettler-Toledo DL22 autotitrator using 0.01 M HCl as the titrant.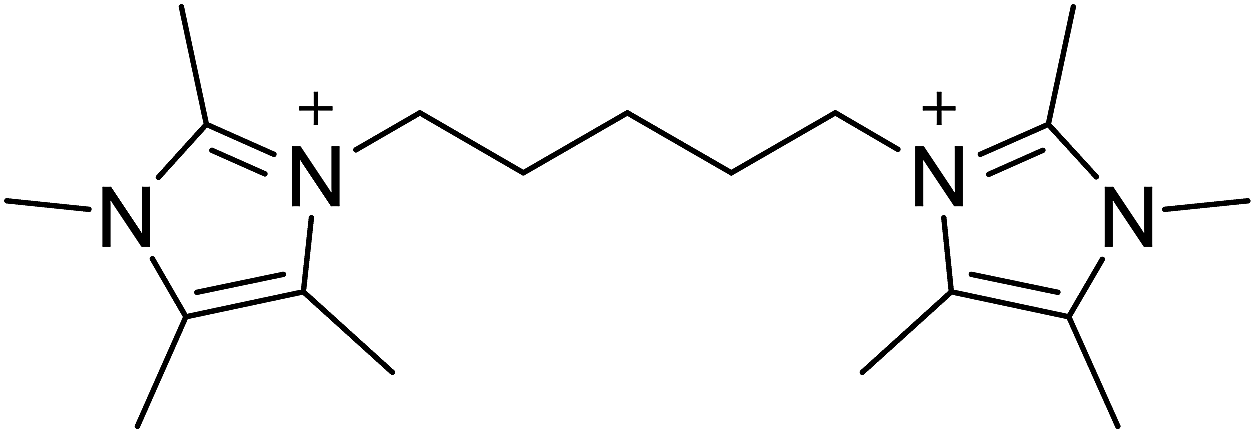 | ||
| Fig. 1 OSDA used to prepare CIT-10. | ||
2.2. Synthesis of CIT-10
Tetraethylorthosilicate was added to the OSDA in its hydroxide form in a Teflon Parr Reactor. The container was closed and stirred overnight to allow for complete hydrolysis. The lid was then removed, and the ethanol and some water were allowed to evaporate under a stream of air. Once the gel was dry, a small amount of water was added to obtain a homogenous liquid. Then aqueous HF was added and the mixture was stirred by hand. A second evaporation step was then used to give a final gel molar ratio of 1SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5R1/2(OH):0.5HF:4H2O. Seeds of CIT-10 were then added and the autoclave was sealed and placed in a rotating oven (43 rpm) at 175 °C. Aliquots of the material were taken periodically by first quenching the reactor in water and then removing enough material for powder X-ray diffraction (PXRD). Synthesis times for pure silica RTH were on the order of 20 days when no seeds were added and 10 days when seeds were added. The product was recovered via centrifugation and was washed with water 3 times, a final time with acetone and dried in air at 100 °C.
:0.5R1/2(OH):0.5HF:4H2O. Seeds of CIT-10 were then added and the autoclave was sealed and placed in a rotating oven (43 rpm) at 175 °C. Aliquots of the material were taken periodically by first quenching the reactor in water and then removing enough material for powder X-ray diffraction (PXRD). Synthesis times for pure silica RTH were on the order of 20 days when no seeds were added and 10 days when seeds were added. The product was recovered via centrifugation and was washed with water 3 times, a final time with acetone and dried in air at 100 °C.
2.3. Pure-silica RTH
The as-made material prepared in Section 2.2 was calcined in breathing grade air. The material was heated to 150 °C at 1 °C min−1, held for three hours, then heated to 580 °C at 1 °C min−1 and held for six hours to assure complete combustion of the organic.2.4. Pillaring of CIT-10 to obtain CIT-11
The procedure that led to the pillared product with the highest crystallinity (judged using PXRD) was found to be as follows. 500 mg of CIT-10 were placed in a 45 mL Teflon Parr Reactor, then 20 g of a 1.25 M solution of HCl in ethanol were added. Finally 500 mg of silyating agent (dichlorodimethylsilane or diethoxydimethylsilane, both were found to produce a similar product) were added. The reactor was sealed and place in a rotating oven at 175 °C for 24 hours. The product was recovered via centrifugation and was washed one time with absolute ethanol, three times with water and finally one time with acetone and then dried in air at 100 °C.2.5. CIT-12
CIT-12 was obtained by the calcination of CIT-11 using the procedure described in Section 2.3.2.6. Characterizations
Liquid NMR spectra were recorded with a 500 MHz spectrometer. 13C and 29Si solid-state NMR were performed using a Bruker DSX-500 spectrometer (11.7 T) and a Bruker 4 mm MAS probe. The spectral operating frequencies were 500.2 MHz, 125.721 MHz and 99.325 MHz for 1H, 13C and 29Si nuclei, respectively. Spectra were referenced to external standards as follows: tetramethylsilane (TMS) for 1H and 29Si and adamantane for 13C as a secondary external standard relative to tetramethylsilane. Samples were spun at 8 kHz for 13C and 29Si MAS and CPMAS NMR experiments. Thermogravimetric analysis was performed on a Perkin-Elmer STA 6000 with a ramp of 1 °C min−1 to 900 °C under air atmosphere. Argon physical adsorption isotherms were performed at 87 K using a Quantachrome Autosorb iQ and were conducted using a quasi-equilibrium, volumetric technique.36 PXRD data were collected on a Rigaku MiniFlex II with Cu Kα radiation. Variable temperature Powder X-ray diffraction (PXRD) patterns were collected from 30 °C to 580 °C at increments of 50 °C under ambient conditions, using a PANalytical Empyrean powder diffractometer (Cu Kα radiation) equipped with an Anton Paar HTK 1200N high-temperature chamber. The sample was stabilized at each measurement temperature for 15 min before starting each measurement. The temperature ramp between two consecutive temperatures was 5 °C min−1. Scanning electron microscope (SEM) images were acquired on a ZEISS 1550 VP FESEM, equipped with in-lens SE. Energy-dispersive X-ray spectroscopy (EDS) spectra were acquired with an Oxford X-Max SDD X-ray Energy Dispersive Spectrometer system. Three-dimensional electron diffraction data were collected using the rotation electron diffraction (RED) technique.37,38 The RED software was installed on a JEOL 2010 microscope operating at 200 kV, and data were collected over a tilt range of ±50° with a tilt step of 0.50°, the exposure time is 3 seconds per tilt step.3. Results and discussions
3.1. Synthesis of CIT-10 and calcination to produce pure-silica RTH
We have recently been investigating imidazolium OSDAs in the synthesis of microporous materials, and have found that they are able to produce a wide range of phases including LTA,39RTH,40,41STW,42CSV43 and HEU34 in addition to a number of additional phases discussed in the previous references. While the majority of these products are microporous materials that were made with OSDAs intact inside the framework, the high-silica HEU (CIT-8) could be prepared from a layered precursor (CIT-8P). CIT-8P was synthesized in fluoride-media from a gel containing a relatively high amount of aluminium (gel Si/Al = 15 or 20). The result of finding a layered material in these conditions led us to continue to explore similar inorganic conditions. In aluminum-free syntheses, we have reported that diquats formed from tetramethylimidazole can be used to prepare pure-silica CSV (CIT-7).43 However, under similar conditions, we found that the diquat containing a five-carbon chain linker length led to a phase that could not be identified (shown in Fig. 2). Upon calcination, this material yielded a phase that was easily identified as pure-silica RTH (Fig. 2). This is the second reported method to synthesize pure-silica RTH, and may broaden its evaluation in applications as previously, the synthesis of this type of material required a difficult to prepare OSDA.35 SEM images of CIT-10 and pure-silica RTH are shown in Fig. 3. These images do not show solids with regular morphologies that are commonly observed in highly crystalline materials, but instead show morphologies resembling thick plates. Plate-like morphology is common in layered materials, however, the thickness of the plates in these samples is unusual.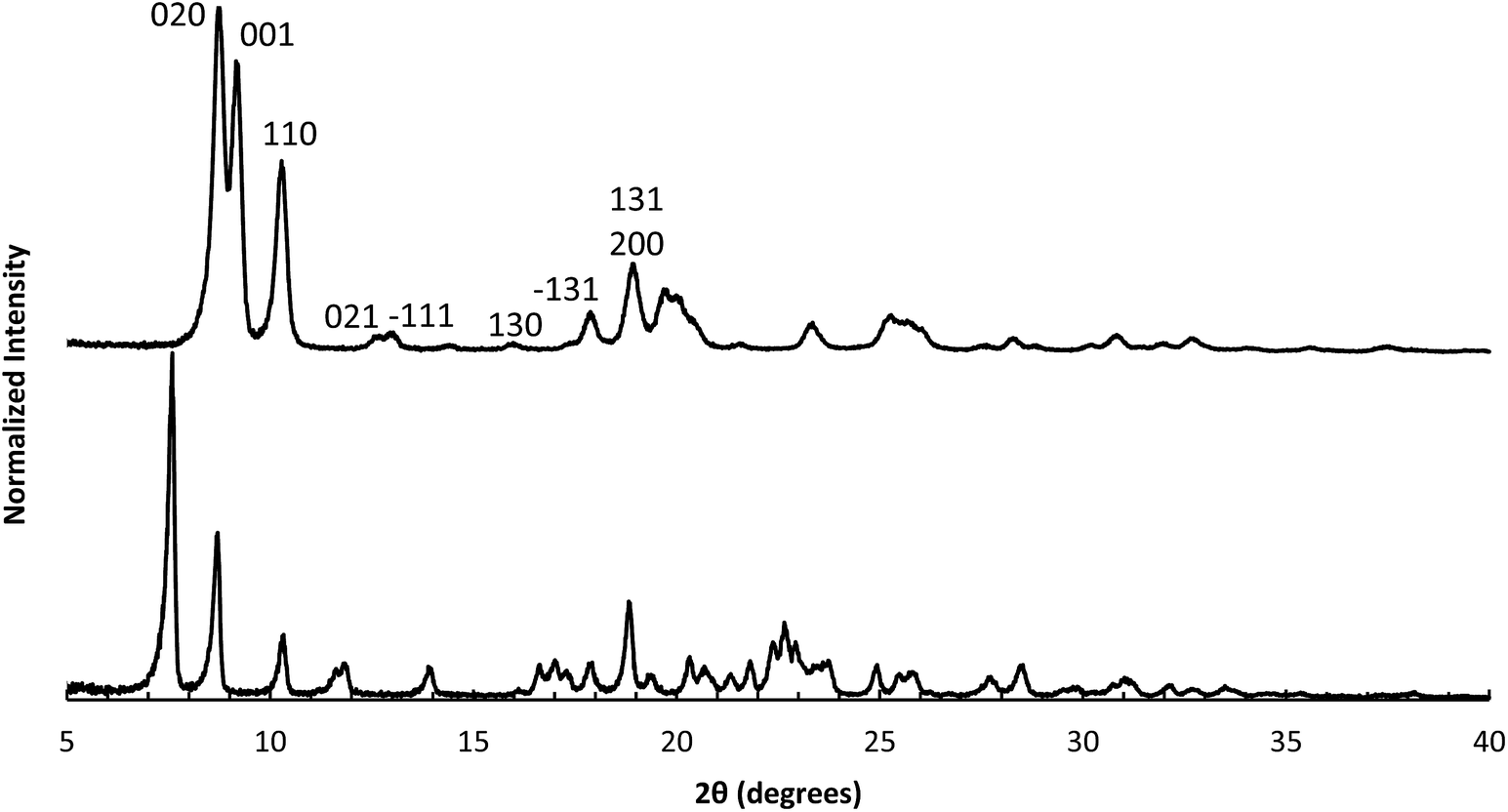 | ||
| Fig. 2 PXRD of CIT-10 (lower) and calcined CIT-10 (pure silica RTH, upper) with selected crystallographic indicies. | ||
 | ||
| Fig. 3 SEM images of (a) CIT-10, (b) Si-RTH, (c) CIT-11 and (d) CIT-12. | ||
To determine the mechanism of formation of pure-silica RTH, the materials were studied using 13C CPMAS NMR, 29Si MAS and CPMAS NMR and variable temperature PXRD. The 13C CPMAS NMR of CIT-10 (Fig. 4) shows that the diquat OSDA was occluded intact in the material. It is interesting to note that many of the peaks in this spectrum are split, indicating that otherwise equivalent carbon atoms are present in non-equivalent environments; this has been previously reported in layered materials.10 The 29Si MAS and CPMAS NMRs of CIT-10 are shown in Fig. 5. CIT-10 was studied using CPMAS NMR in addition to MAS NMR to confirm the resonances (organic-containing materials often exhibit a poor signal-to-noise ratio). In the as-made material there are three resonances at −113, −107 and −102 ppm with approximate area ratios of 8:5:3. The signals at −113 and −107 ppm are assigned to Q4 silicon, Si(OSi)4 coordination, while the signal at −102 ppm is assigned to Q3 silicon, Si(OSi)3(OH) coordination. The presence of Q3 silicon is expected in a layered material. The ratio of Q3/(Q3 + Q4) silicon in the as made material is 0.23, which is very close to the theoretical value of 0.25 (see the ESI Fig. S1† for additional details). Upon calcination, the 29Si MAS NMR spectrum no longer shows the presence of any Q3 silicon and instead shows 3 resonances in the Q4 region at −116, −114 and −109 ppm, with area ratios of 1:2:1. These area ratios agree with the crystal structure of RTH, as it contains 4 independent T-sites.
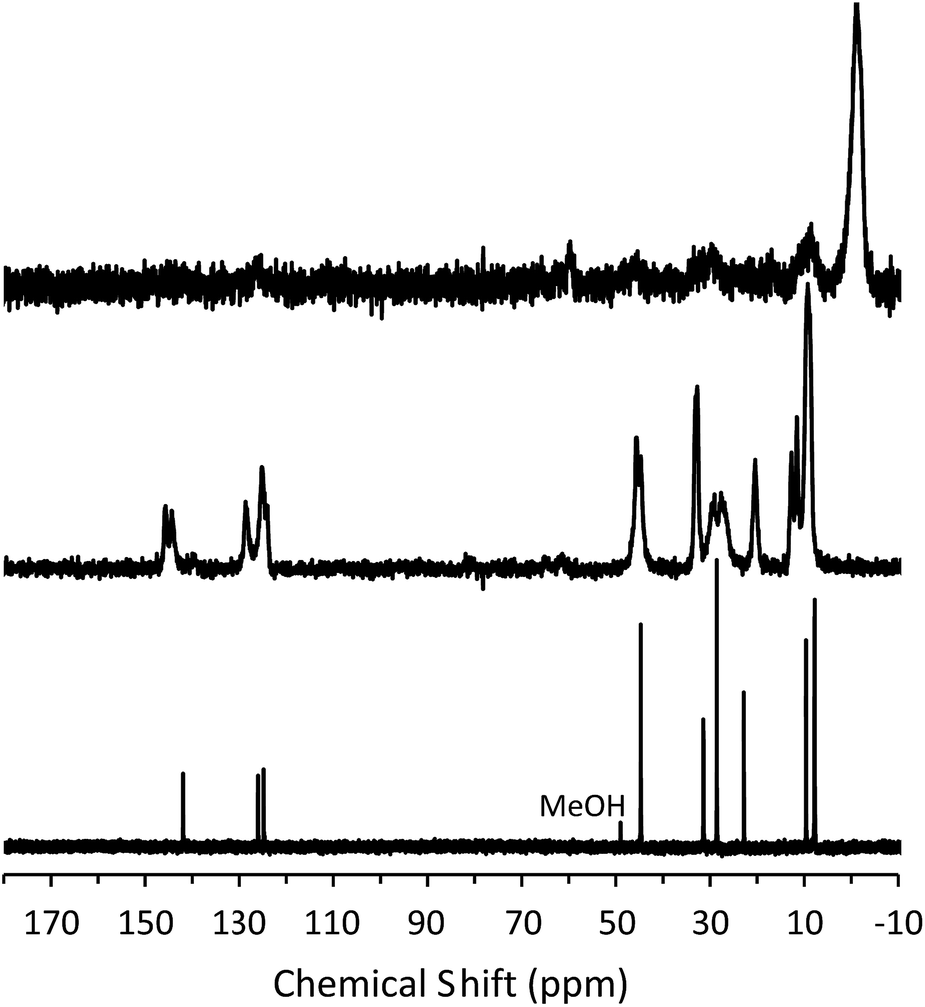 | ||
| Fig. 4 13C NMR of the diquat in D2O (lower, methanol added as an internal standard), 13C CPMAS NMR of CIT-10 showing the occluded diquat (middle) and 13C CPMAS NMR of CIT-11 (upper). | ||
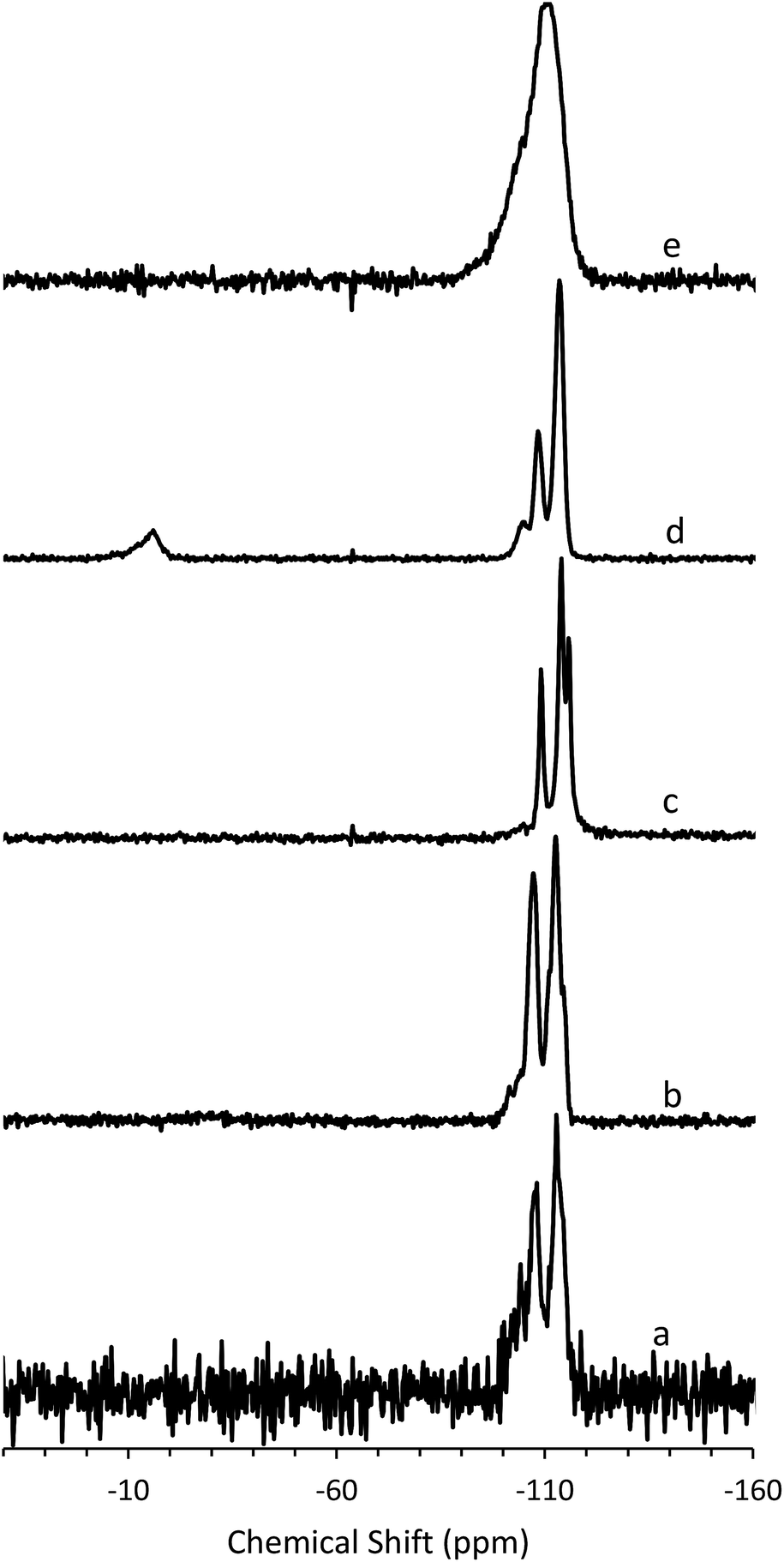 | ||
| Fig. 5 29Si (a) MAS NMR of CIT-10, (b) CPMAS NMR of CIT-10, (c) MAS NMR of pure-silica RTH prepared by calcination of CIT-10, (d) MAS NMR of CIT-11 and (e) MAS NMR of CIT-12. | ||
The structural mechanism of condensation was determined by using variable temperature PXRD as well as RED. The variable temperature PXRD of CIT-10 is shown in Fig. 7. When compared with the PXRD patterns of RTH in Fig. 2 (labelled with the crystallographic indices), it is apparent that peak positions for hk0 reflections remain during heating, while the peak positions for the hkl (l ≠ 0) reflections are shifted to higher 2θ angles (i.e., lower d-spacing). This result indicates that the 3D RTH structure forms via topotactic condensation along the c-axis, and that the a and b axes are intact in the layered material. The structural change was further confirmed by studying CIT-10 using RED (Fig. 6). The RED clearly shows that CIT-10 is ordered in the a and b directions (indicated by clearly defined diffraction spots), but that some disorder is present in the c direction (indicated by diffraction streaks between diffraction spots). Thus, results from using both techniques confirm that CIT-10 contains 2D sheets in the a and b directions that are separated by a disordered organic in the c direction.
 | ||
| Fig. 6 RED structure analysis of CIT-10. | ||
 | ||
| Fig. 7 Variable temperature PXRD of CIT-10. | ||
The TGA data are consistent with the condensation temperature observed in the variable temperature PXRD. In the variable temperature PXRD, the structure of the layered material is intact until 330 °C, then the low angle peak corresponding to the 001 direction abruptly disappears. This reflection is absent at the PXRD pattern taken at 380 °C, then begins to emerge around 430 °C. From the TGA trace in Fig. 8, a sharp mass loss occurring around 375 °C is seen, and is in the same temperature range where the low angle peak disappeared in the variable temperature PXRD. The rapid change observed with RTH is in contrast to the TGA trace and structural changes observed with CIT-8P (layered precursor to HEU) where a gradual shift in position of the low-angle peak was observed along with the gradual decrease in mass.34
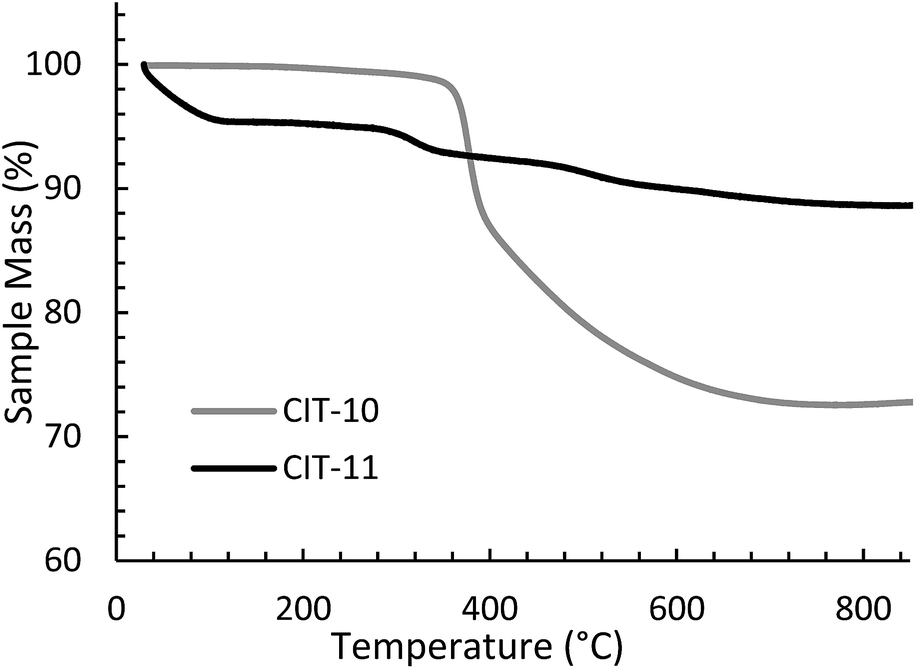 | ||
| Fig. 8 TGA of CIT-10 and CIT-11. | ||
A detailed analysis of the structural changes occurring during the topotactic condensation process for RUB-39 have been reported as a series interconnected endothermic (OSDA decomposition and condensation of silicate layers) and exothermic (OSDA combustion) processes that are dominated by combustion of the OSDA.44 It was found that as soon as the OSDA was removed, the condensation occurred. Attempts to stabilize the intermediate material were not successful. The sharp mass loss over a narrow temperature range observed with CIT-10 is similar to what is reported with RUB-39, and for the latter the loss was ascribed to a fast series of connected processes that include OSDA decomposition and combustion as well as condensation of the silicate layers.
CIT-10 has an 8 MR channel running through the layer along the c axis, with dimensions of 2.5 × 5.6 Å. As the structure condenses along the c-axis, a second 8 MR channel system running through the a-axis is formed, and a cavity is created at the intersection of the two 8 MR channels, forming the RTH framework structure. The condensation process is shown schematically in Fig. 9. As the schemes in the figure depict, the RTH layer is actually a half unit cell thick compared to the final RTH framework unit cell.
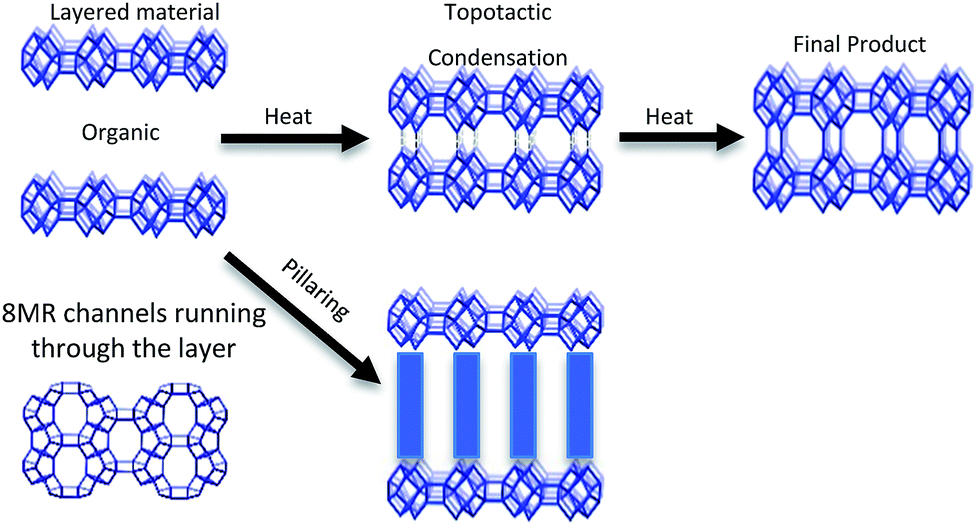 | ||
| Fig. 9 Depiction of the topotactic condensation and pillaring of CIT-10. | ||
Table 1 shows the comparisons of the d-spacing corresponding to the first and most intense PXRD peak for known 2D layered materials, and the corresponding d-spacing shrinkage after topotactic condensation to form 3D framework materials. In most of the cases (including CIT-10) the d-spacing shrinkage due to topotactic condensation is around 2 Å, an observation that has been discussed by Roth et. al.46 It is also interesting to note that although the OSDAs used to make CIT-10 and CIT-8P are very similar, the latter demonstrated a d-spacing contraction nearly twice that of the former.
| 2D zeolite | d-spacing (Å) | 3D zeolite | Corresponding d-spacing (Å) | d-spacing shrinkage (Å) | Ref. |
|---|---|---|---|---|---|
| CIT-10 | 11.8 | Siliceous RTH | 9.8 | 2.0 | This work |
| RUB-36 | 11.1 | RUB-37 (CDO) | 9.2 | 1.9 | 45 |
| MCM-22P | 26.9 | MCM-22 (MWW) | 24.9 | 2.0 | 16, 46 |
| HMP-2 | 17.5 | MCM-35 (MTF) | 15.4 | 2.1 | 22 |
| RUB-39 | 10.8 | RUB-41 (RRO) | 8.7 | 2.1 | 24 |
| R-RUB-18 | 9.1 | RUB-24 (RWR) | 6.8 | 2.3 | 25 |
| EU-19 | 11.5 | EU-20 (CAS-NSI) | 8.3 | 3.2 | 19 |
| PREFER | 13.1 | FER | 9.4 | 3.7 | 15 |
| CIT-8P | 12.8 | CIT-8 (HEU) | 8.9 | 3.9 | 34 |
3.2. Pillaring of CIT-10
In some cases it is possible to pillar layered materials using a monomeric silane in order to prepare materials with pores larger than would have been formed by topotactic condensation. These materials are commonly referred to as interlayer expanded zeolites (IEZ), and they have been prepared from precursors such as PREFER, MWW(P), PLS-1, MCM-47, RUB-36, RUB-39 and Nu-6(1).8,30,31 Pillaring is normally carried out in acidic media, under hydrothermal conditions, and two of the most common pillaring agents are dichlorodimethylsilane and diethoxydimethylsilane.In attempting to pillar CIT-10, a wide range of conditions were explored including acid type and concentration, aqueous versus ethanolic acid, silane source, and reaction temperature and time. The optimal conditions to pillar CIT-10 were found to be 1.25 M HCl in ethanol with either dichlorodimethylsilane or diethoxydimethylsilane at 175 °C for 24 hours. Other conditions lead to what appeared to be pillared materials (based on PXRD), but these materials exhibited very weak X-ray reflections. We interpret these results as indicating framework destruction, and these solids were often not stable to calcination. This phenomenon has been observed before, i.e., that acidic ethanol is the only effective medium to carry out pillaring. It has been postulated that the reason for this is that effective pillaring takes place when the rate of removal of OSDA is well matched by the rate of silylation.30 It should be noted that while we made no special efforts to preclude trace amounts of water in these syntheses (such as working in a glovebox or using a Schlenk line), the water content was likely very low.
The X-ray diffraction results of pillared CIT-10 are shown in Fig. 10. As can be observed from the PXRD patterns, pillaring causes a shift in the most intense reflection from 7.5° 2θ in CIT-10 to 6.8° 2θ in CIT-11 (i.e., 1.1 Å expansion). This peak continues to shift to 7.7° 2θ in CIT-12 (after calcination). The 13C CPMAS NMR of CIT-11 (Fig. 4) shows that the majority of the organic was removed under acidic conditions (while CPMAS NMR is not quantitative this was confirmed by TGA, shown in Fig. 8). This result was expected as a change in color of the acidic medium was observed. The strong resonance observed near −1 ppm is consistent with (CH3)2Si carbon that is expected from the pillaring. (Prior to NMR analysis it was necessary to degas the material under vacuum at 150 °C to remove any residual ethanol or acetone.) The TGA analysis of CIT-11 (Fig. 8) shows several distinct mass loss regions. The first mass loss of 5% is attributed to the loss of water and possibly residual ethanol or acetone (material was dried in air at 100 °C prior to analysis but not under vacuum as was done with the NMR sample). The second sharp mass loss begins around 300 °C, and is attributed to removal of residual organic (present in 13C CPMAS NMR). There is a third, distinct region of mass loss that begins around 500 °C and is attributed to combustion of the Si–CH3 groups to form hydroxyl groups.
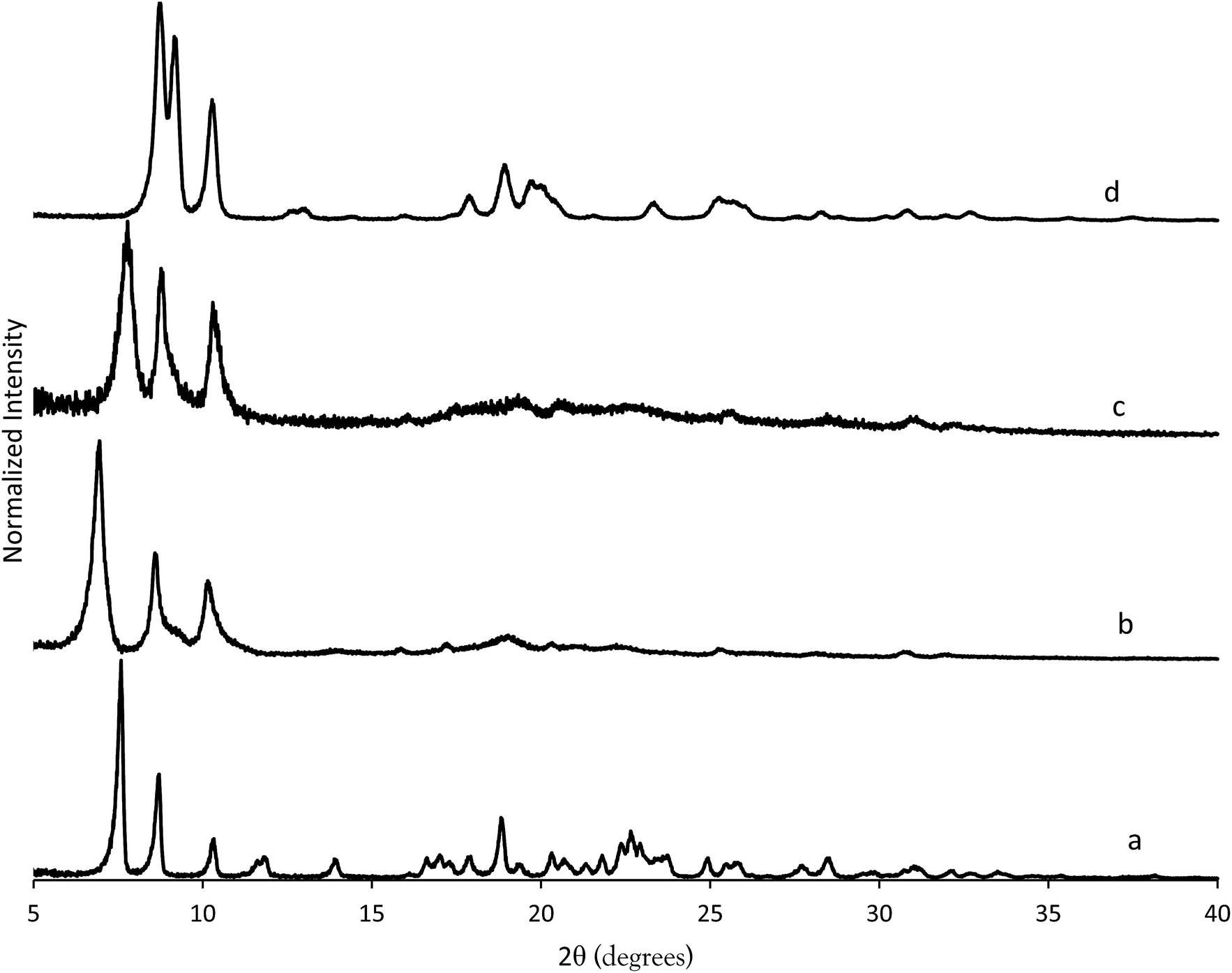 | ||
| Fig. 10 PXRD patterns of (a) CIT-10, (b) CIT-11, (c) CIT-12 and (d) pure-silica RTH. | ||
The 29Si NMR spectra are consistent with a pillared material (Fig. 5). In CIT-10, both Q4 and Q3 silicon environments are observed, consistent with a layered material (vide supra). In the pillared material, CIT-11, 29Si NMR resonances are observed at −113.5, −108.4, −104.5 and −15.3 ppm with approximate area ratios of 20:8:2:5. The resonances at −113.5 and −108.4 are assigned to Q4 silicon and the resonance at −104.5 is assigned to residual Q3 silicon. The Q3/(Q3 + Q4) ratio in the pillared material is 0.07, a significant decrease from 0.23 in CIT-10, indicating that a substantial amount of Q3 species have been consumed in linking the layers of the material. The resonance at −15.3 is assigned to bridging silanol groups bonded to two methyl groups, that is Si(CH3)2(OSi)2 coordination. The ratio of (Q2 + Q3)/(Q2 + Q3 + Q4) is 0.25, consistent with the expected value from the RTH layer (see ESI†). Upon calcination the material exhibits a broad resonance around −110 ppm. A single broad resonance has been observed in other pillared, calcined materials such as PLS-4.47 No obvious peak for Q2 silicon can be seen near −90 ppm, but it is likely obscured by the much broader resonance.
The structure of CIT-11/12 is a 3D pore system consisting of 8 and 10 MRs, shown in Fig. 9. The 8 MR running the in the c-direction perpendicular to the RTH layer remains intact. The pillars form two new ring sizes as the previous 8 MR along the a-direction expands to a 10 MR and the 6 MR along the b-direction expands to an 8 MR. This means that the previous 2D ring system in RTH expands be a 3D ring system in CIT-11/12.
The pore system of CIT-12 was confirmed using argon adsorption. The results from this analysis are shown in Fig. S2† compared to pure-silica RTH as well as zeolite 5A and pure-silica BEA. We chose to plot all of these isotherms on the same graph as the shape of the isotherm in the low pressure regime is an indication of the pore size distribution. The comparison of the low pressure region of the argon adsorption isotherms indicates that this material has a pore system that has expanded compared to pure-silica RTH (8 MRs) but is smaller than pure-silica BEA (12 MRs), consistent with the structure solution.
4. Conclusions
A new 2D building layer has been synthesized, the rth type layer, denoted CIT-10. This is the first reported 2D building layer that contains small pores that are perpendicular to the layer. Upon calcination, this material forms pure-silica RTH, making pure-silica RTH accessible without using a difficult to synthesize organic. CIT-10 can be pillared, forming CIT-11, that can then be calcined, forming CIT-12. CIT-12 contains a 3D pore system of 8 and 10 MRs. As CIT-10 contains a new 2D layer there are many further studies to be conducted with this material such as swelling, exfoliation and delamination as well as heteroatom introduction to introduce sites for ion exchange and catalysis. CIT-10 is the first layered material to contain small pores running through the layer; it is possible this material could find use in separations, especially of small molecules. Such possibilities have already been explored with other microporous material frameworks such as LTA and MFI. However, the RTH layers are only half a unit cell thick compared to a full unit cell with MFI27 (which has medium pores) and multiple unit cells with LTA48–50 (small pores). Additionally, the RTH layers have elliptical pores running through them, which may offer additional size discrimination compared to other small pore materials with circular pores, such as LTA.Acknowledgements
We would like to thank Dr Stacey Zones (Chevron Energy Technology Company) for insightful discussions and Thomas Rea (Chevron Energy Technology Company) for collecting the RED data. We would also like to thank Dr Sonjong Hwang (Caltech) for assistance with collecting the NMR spectra. We would like to thank Chevron Energy Technology Company for providing funding for this work. J.E.S. would like to thank the NDSEG for their support through a fellowship.Notes and references
- M. Moliner, C. Martínez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 3560–3579 CrossRef CAS PubMed.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 CrossRef CAS PubMed.
- Z. Wang, J. Yu and R. Xu, Chem. Soc. Rev., 2012, 41, 1729–1741 RSC.
- a. Corma, J. Catal., 2003, 216, 298–312 CrossRef CAS.
- M. E. Davis, Chem. Mater., 2014, 26, 239–245 CrossRef CAS.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120–3145 CrossRef CAS PubMed.
- R. Pophale, F. Daeyaert and M. W. Deem, J. Mater. Chem. A, 2013, 1, 6750–6760 CAS.
- U. Díaz and A. Corma, Dalton Trans., 2014, 43, 10292–10316 RSC.
- B. Marler and H. Gies, Eur. J. Mineral., 2012, 24, 405–428 CrossRef CAS PubMed.
- P. Wu, J. Ruan, L. Wang, L. Wu, Y. Wang, Y. Liu, W. Fan, M. He, O. Terasaki and T. Tatsumi, J. Am. Chem. Soc., 2008, 130, 8178–8187 CrossRef CAS PubMed.
- D. Mochizuki, A. Shimojima, T. Imagawa and K. Kuroda, J. Am. Chem. Soc., 2005, 127, 7183–7191 CrossRef CAS PubMed.
- S. L. Suib, Chem. Rev., 1993, 93, 803–826 CrossRef CAS.
- W. J. Roth, P. Nachtigall, R. E. Morris and J. Cejka, Chem. Rev., 2014, 114, 4807–4873 CrossRef CAS PubMed.
- U. Díaz, ISRN Chem. Eng., 2012, 2012, 1–35 CrossRef PubMed.
- L. Schreyeck, P. Caullet, J. C. Mougenel, J. L. Guth and B. Marler, Microporous Mater., 1996, 6, 259–271 CrossRef CAS.
- M. E. Leonowicz, J. a. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910–1913 CAS.
- Y. Asakura, R. Takayama, T. Shibue and K. Kuroda, Chemistry, 2014, 20, 1893–1900 CrossRef CAS PubMed.
- A. J. Blake, K. R. Franklin and B. M. Lowe, J. Chem. Soc., Dalton Trans., 1988, 2513 RSC.
- B. Marler, M. a. Camblor and H. Gies, Microporous Mesoporous Mater., 2006, 90, 87–101 CrossRef CAS PubMed.
- S. Zanardi, A. Alberti, G. Cruciani, A. Corma, V. Fornés and M. Brunelli, Angew. Chem., Int. Ed., 2004, 43, 4933–4937 CrossRef CAS PubMed.
- T. Ikeda, Y. Akiyama, Y. Oumi, A. Kawai and F. Mizukami, Angew. Chem., Int. Ed., 2004, 43, 4892–4896 CrossRef CAS PubMed.
- A. Rojas and M. A. Camblor, Chem. Mater., 2014, 26, 1161–1169 CrossRef CAS.
- W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubná, L. Grajciar, M. Položij, A. Zukal, O. Shvets and J. Cejka, Nat. Chem., 2013, 5, 628–633 CrossRef CAS PubMed.
- Y. X. Wang, H. Gies, B. Marler and U. Müller, Chem. Mater., 2005, 17, 43–49 CrossRef CAS.
- B. Marler, N. Ströter and H. Gies, Microporous Mesoporous Mater., 2005, 83, 201–211 CrossRef CAS PubMed.
- T. Moteki, W. Chaikittisilp, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2008, 130, 15780–15781 CrossRef CAS PubMed.
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef CAS PubMed.
- M. Tsapatsis, AIChE J., 2014, 60, 2374–2381 CrossRef CAS PubMed.
- U. Díaz and A. Corma, Dalton Trans., 2014, 10292–10316 RSC.
- J. Jiang, L. Jia, B. Yang, H. Xu and P. Wu, Chem. Mater., 2013, 25, 4710–4718 CrossRef CAS.
- H. Gies, U. Müller, B. Yilmaz, M. Feyen, T. Tatsumi, H. Imai, H. Zhang, B. Xie, F. S. Xiao, X. Bao, W. Zhang, T. De Baerdemaeker and D. de Vos, Chem. Mater., 2012, 24, 1536–1545 CrossRef CAS.
- T. de Baerdemaeker, H. Gies, B. Yilmaz, U. Müller, M. Feyen, F.-S. Xiao, W. Zhang, T. Yokoi, X. Bao and D. E. de Vos, J. Mater. Chem. A, 2014, 2, 9709 CAS.
- T. de Baerdemaeker, W. Vandebroeck, H. Gies, B. Yilmaz, U. Müller, M. Feyen and D. de Vos, Catal. Today, 2014, 235, 169–175 CrossRef CAS PubMed.
- J. E. Schmidt, D. Xie and M. E. Davis, J. Mater. Chem. A, 2015, 3, 12890–12897 CAS.
- G. S. Lee and S. I. Zones, J. Solid State Chem., 2002, 167, 289–298 CrossRef CAS.
- P. E. Hathaway and M. E. Davis, Catal. Lett., 1990, 5, 333–347 CrossRef CAS.
- D. Zhang, P. Oleynikov, S. Hovmöller and X. Zou, Z. Kristallogr., 2010, 225, 94–102 CrossRef CAS.
- W. Wan, J. Sun, J. Su, S. Hovmöller and X. Zou, J. Appl. Crystallogr., 2013, 46, 1863–1873 CAS.
- J. E. Schmidt, S. I. Zones, D. Xie and M. E. Davis, Microporous Mesoporous Mater., 2014, 200, 132–139 CrossRef CAS PubMed.
- J. E. Schmidt, M. A. Deimund and M. E. Davis, Chem. Mater., 2014, 26, 7099–7105 CrossRef CAS.
- J. E. Schmidt, M. a. Deimund, D. Xie and M. E. Davis, Chem. Mater., 2015, 27, 3756–3762 CrossRef CAS.
- J. E. Schmidt, M. W. Deem and M. E. Davis, Angew. Chem., Int. Ed., 2014, 53, 8372–8374 CrossRef CAS PubMed.
- J. E. Schmidt, D. Xie, T. Rea and M. E. Davis, Chem. Sci., 2015, 6, 1728–1734 RSC.
- Y. X. Wang, H. Gies and J. H. Lin, Chem. Mater., 2007, 19, 4181–4188 CrossRef CAS.
- J. Song and H. Gies, Stud. Surf. Sci. Catal., 2004, 154, 295–300 CrossRef.
- W. J. Roth, D. L. Dorset and G. J. Kennedy, Microporous Mesoporous Mater., 2011, 142, 168–177 CrossRef CAS PubMed.
- T. Ikeda, S. Kayamori and F. Mizukami, J. Mater. Chem., 2009, 19, 5518 RSC.
- A. Huang and J. Caro, Chem. Commun., 2011, 47, 4201–4203 RSC.
- I. Tiscornia, S. Valencia, A. Corma, C. Téllez, J. Coronas and J. Santamaría, Microporous Mesoporous Mater., 2008, 110, 303–309 CrossRef CAS PubMed.
- H. K. Hunt, PhD thesis, California Insititue of Technology, 2010.
Footnote |
| † Electronic supplementary information (ESI) available: Figure of the RTH-type layer with T-site labels and log-plot high resolution argon adsorption isotherms. See DOI: 10.1039/c5sc02325d |
| This journal is © The Royal Society of Chemistry 2015 |