DOI:
10.1039/C5SC02205C
(Edge Article)
Chem. Sci., 2015,
6, 5922-5927
Taming the beast: fluoromesityl groups induce a dramatic stability enhancement in boroles†
Received
18th June 2015
, Accepted 10th July 2015
First published on 13th July 2015
Abstract
The electron-deficient pentaarylborole 1-(2′,4′,6′-tris(trifluoromethyl)phenyl)-2,3,4,5-tetraphenylborole (1) has been synthesised with the long-term aim of developing borole-based optoelectronic materials. The bulky 2,4,6-tris(trifluoromethyl)phenyl (FMes) group on the boron atom of 1 significantly improves (>600 times) its air stability relative to its mesityl analogue. Moreover, 1 shows good thermal stability without undergoing the dimerisation or isomerisation reactions reported for some other boroles. A triarylborole analogue (2), belonging to a new class of borole with the 3- and 4-positions of the BC4 ring linked by a –(CH2)3– group, has also been synthesised to elucidate the influence of carbon-bonded substituents on the stability of boroles. Both boroles were prepared through the reaction of Li[FMesBF3] and divinyldilithium reagents, a new and general method for borole syntheses. Compound 2 was found to isomerise through a [1,3]-H shift and double-bond rearrangement to an s-trans-butadienylborane species under highly basic (NaOH) conditions. The increased steric crowding at the boron centre through incorporation of the FMes group does not preclude binding of Lewis bases to either 1 or 2, as demonstrated by their fully reversible binding of pyridine. Interestingly, 1 exhibits a blue-shifted absorption spectrum, as compared with its mesityl analogue, a result contrary to previous understanding of the influence of substituent electronics on the absorption spectra of boroles. Most importantly, these boroles exhibit much greater air-stability than previously reported analogues without sacrificing the strong electron-accepting ability that makes boroles so attractive; indeed, 1 and 2 have very low reduction potentials of −1.52 and −1.69 eV vs. Fc/Fc+, respectively.
Introduction
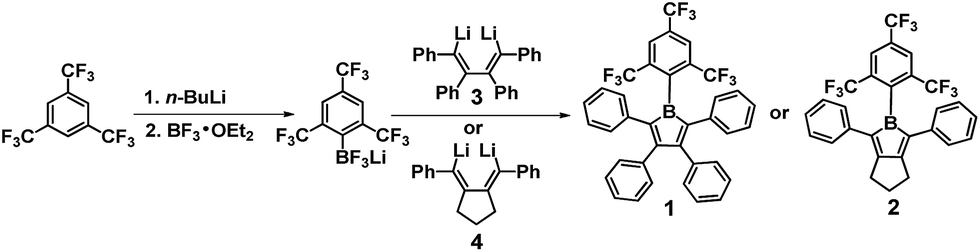
Boroles,1 a class of heterocyclopentadienes with an anti-aromatic 4π-electron BC4 ring, have attracted increasing interest due to their strong Lewis acidity, unique reactivity and facile reduction to radical-anions.2–7 The first crystallographic characterisation of a member of this class of compounds was reported in 2008,2a although the first borole was reported by Eisch in 1969.7a One general and very attractive feature of boroles is their strong electron-accepting ability and high electron-affinity (typical Ered1/2ca. −1.6 to −2.0 V vs. Fc/Fc+), which indicates their clear potential for applications as optoelectronic materials, such as electron-transporting and acceptor materials in organic light-emitting diodes (OLEDs) and photovoltaics (OPVs), respectively.8 However, boroles sometimes undergo Diels–Alder and related dimerisations2e,7g and, more importantly, are typically extremely sensitive to air2b,j,4b (both to O2 (ref. 7a and f) and water3a) due to the empty 2pz orbital of the boron atom and the antiaromaticity resulting from the 4π-electron structure. This instability has presented a major obstacle to their practical use in optoelectronics. One strategy to stabilise boroles for isolation and characterisation and to avoid dimerisation is the use of bulky groups as the exocyclic substituents1c and, thus, pentaarylboroles have become the predominant targets of recent studies.1–4 However, their air stability is still poor. Thus, improving the air stability of boroles is an essential but challenging issue for realising optoelectronic applications. ortho-Substituted aryl groups on boron can provide steric shielding above and below the BC4 plane, protecting both the boron atom and the B–C bonds. Therefore, they are expected to improve air stability as boroles undergo B–C bond cleavage when attacked by water3a and O2.7a,b However, it is synthetically challenging to introduce very bulky substituents at the boron centre because of the steric congestion caused by the use of large substituents on the carbon atoms adjacent to the boron centre at the 2,5-positions of the BC4 ring. This is different from ring-fused boroles, such as 9-borafluorenes (dibenzoboroles),9 wherein there is less congestion, allowing bulky substituents, e.g. 2,4,6-tris(tert-butyl)phenyl,9d to be used to construct air-stable compounds. Unfortunately, although more air stable, these compounds typically show significantly weakened electron-accepting ability due to the benzannulation, e.g. Ered1/2 = −2.28 eV vs. Fc/Fc+ for 2,4,6-tris(tert-butyl)phenyl dibenzoborole, which somewhat limits one of their key attractions.9d Exceptions to this are air-sensitive π-extended benzothiophene-fused ladder boroles (Ered1/2 = −1.72 eV)9f and pre-organised bis(borafluorene) compounds that are easily reduced (e.g., −1.49 eV for a 2,2-biphenylene-bridged example) due to cooperativity of the two boron centres and the generation of a 2c–1e bond.9i,j
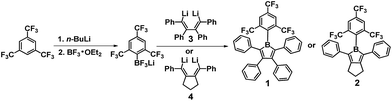
In line with our interest in constructing strongly electron-accepting boroles with much improved air-stability, we decided to utilise the bulky 2,4,6-tris(trifluoromethyl)phenyl (FMes) group, in which the CF3 unit has a similar volume to an ethyl group,10 as the boron-bonded substituent. We have recently shown that the introduction of two, or even just one, FMes groups on boron provides both the steric protection required for excellent air stability and much enhanced electron-accepting ability in three-coordinate boron optical materials.11 We have now succeeded in synthesising not only a pentaarylborole (1, in Scheme 1) with the B(FMes) moiety, but also a triarylborole that represents a new class of borole system (2, in Scheme 1).12 This has allowed us to examine the influence of substituents at the 3,4-positions of the BC4 ring on air-stability, as recent reports by the groups of Braunschweig, Piers and Yamaguchi indicate that both the B- and C-bonded substituents influence the properties of boroles.2–4 In addition to their air stability, the photophysical and electrochemical properties, thermal stability and Lewis acidity of these boroles have also been studied.
 |
| | Scheme 1 Syntheses of compounds 1 and 2. | |
Results and discussion
Synthesis
Salt-elimination reactions of ArBCl2 precursors with dilithium reagents, such as 1,4-dilithio-2,3,4,5-tetraphenylbuta-1,3-diene (3, Scheme 1), are known to be efficient for the synthesis of boroles with relatively bulky boron-bonded substituents, e.g. the Mes group.2h However, the preparation of FMesBCl2 by reaction of FMesLi with BCl3 is known to give an unacceptable yield (5%) and very low purity, because of significant Cl–F exchange.13 Therefore, we attempted to use BF3·Et2O as an alternative to BCl3 to avoid the Cl–F exchange and prepare FMesBF2 as the intermediate for the syntheses of our boroles (Scheme 1). Interestingly, the reaction of FMesLi and BF3·Et2O instead produced a mixture of Li[FMesBF3], (FMes)2BF and LiBF4 as the major products. After a simple workup, a mixture of Li[FMesBF3], LiBF4 and Et2O in a molar ratio of ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:0.8 was obtained according to NMR studies (Fig. S1–S7†), and the existence of Li[FMesBF3] and LiBF4 in the mixture was further confirmed by mass spectrometry and single-crystal X-ray diffraction (Fig. S8 and S9†). Isolation of pure Li[FMesBF3] failed due to unavoidable co-crystallisation with LiBF4. Nevertheless, when the crude mixture was stirred with dilithium reagent 3 or 4 in toluene, the desired borole compounds were observed by in situ19F NMR spectroscopy, with only very small amounts of by-products. No products were observed arising from reaction between the LiBF4 component and either of the dilithium reagents. Compounds 1 and 2 were isolated as black crystals in yields of 53 and 42%, respectively. Our experiments suggest that readily accessible and relatively stable aryltrifluoroborates14 could serve as good and more convenient alternatives to extremely air-sensitive arylborondihalides as precursors for the synthesis of boroles.
:1.5:0.8 was obtained according to NMR studies (Fig. S1–S7†), and the existence of Li[FMesBF3] and LiBF4 in the mixture was further confirmed by mass spectrometry and single-crystal X-ray diffraction (Fig. S8 and S9†). Isolation of pure Li[FMesBF3] failed due to unavoidable co-crystallisation with LiBF4. Nevertheless, when the crude mixture was stirred with dilithium reagent 3 or 4 in toluene, the desired borole compounds were observed by in situ19F NMR spectroscopy, with only very small amounts of by-products. No products were observed arising from reaction between the LiBF4 component and either of the dilithium reagents. Compounds 1 and 2 were isolated as black crystals in yields of 53 and 42%, respectively. Our experiments suggest that readily accessible and relatively stable aryltrifluoroborates14 could serve as good and more convenient alternatives to extremely air-sensitive arylborondihalides as precursors for the synthesis of boroles.
Crystal structures
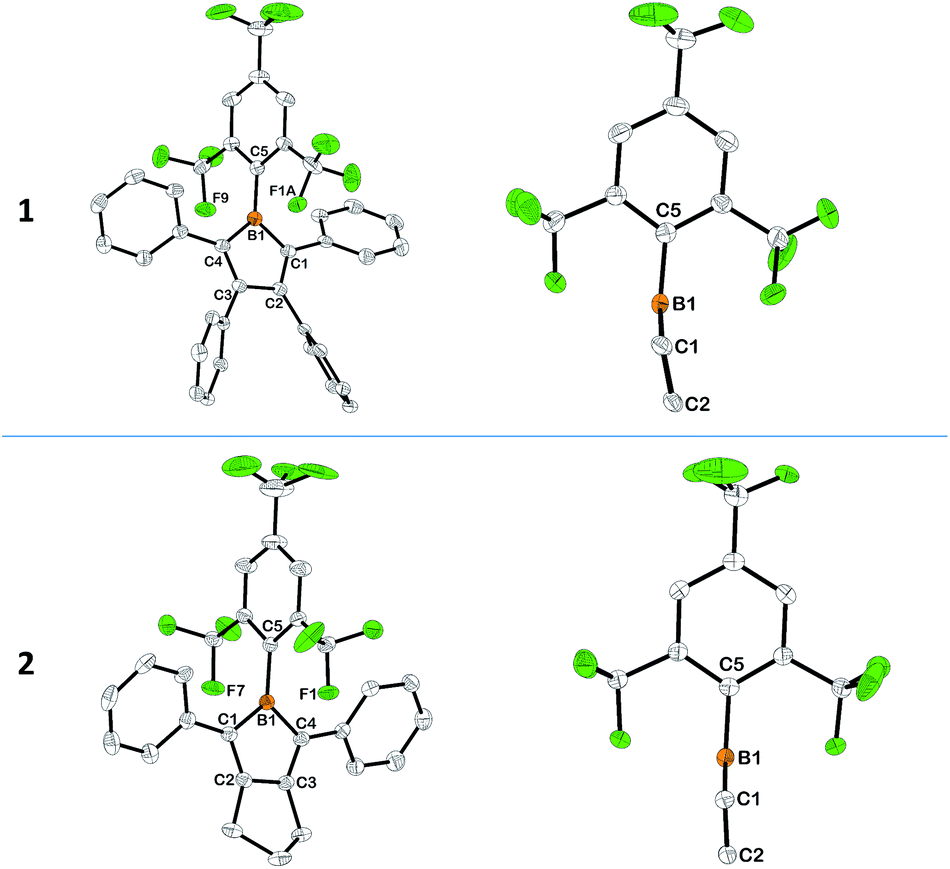
Single crystals of 1 and 2 were obtained by slow evaporation of their respective diethyl ether–hexane mixed-solvent solutions at room temp. (see ESI†). Within the BC4 rings of 1 and 2, corresponding B–C and C–C bonds have roughly similar lengths, which are also close to those observed in MesBC4Ph4 (Fig. 1, S10 and S11†). The near single-bond character of the B1–C1/B1–C4 (1.571(3)–1.592(3) Å) and C2–C3 (1.485(2)–1.526(3) Å) bonds and the clear double-bond character of the C1–C2/C3–C4 (1.350(2)–1.359(3) Å) bonds are consistent with the antiaromaticity of the BC4 ring, although B1–C1 is slightly shorter in 1 than 2, and C2–C3 is slightly shorter in 2 than 1. It is notable that the BC4 ring in 1 is not completely planar, the boron atom slightly departing from the C4 plane by a distance of 0.133(4) Å (Fig. 1). Consequently, the plane defined by B1–C1–C4 shows a dihedral angle of 8.1(3)° with respect to the C4 plane. Moreover, the B1–C5 bond is further bent out of the B1–C1–C4 plane by an angle of 7.6(3)°. The combination of these two deviations results in a distance of 0.561(6) Å from the C5 atom to the aforementioned C4 plane. The analogous distance is 0.496(4) Å in MesBC4Ph42h and 0.181(4) Å in BC4Ph5.2a The larger deviation in 1 is likely caused by the enhanced spatial congestion around the bulkier CF3 groups at the positions ortho to the boron atom, which also leads to short distances between the boron and the closest fluorine atoms of the two o-CF3 groups, ranging from 2.385(3) to 2.556(6) Å, close to those observed in (2-thienyl)2B–FMes and 2,5-[(2-thienyl)(FMes)B]2thiophene.11a Moreover, it causes a significantly larger dihedral angle of 82.4(3)° between the phenyl ring of the FMes group and the B1–C1–C4 plane than the corresponding angle of 68.76(6)° in MesBC4Ph4. These structural characteristics serve to make the boron atom well protected by the o-CF3 groups. The planes defined by the phenyl rings on C1 and C4 form dihedral angles of 41.1(1) and 45.0(1)°, respectively, with respect to the C4 plane, which are close to those in MesBC4Ph4 (47.05(8) and 42.41(9)°). However, the corresponding dihedral angles are much smaller (24.40(9)–28.44(9)°) in 2, allowing better conjugation between the two phenyl groups and the electron deficient BC4 ring. Such smaller dihedral angles result from decreased steric hindrance arising from the replacement of the two bulky phenyl groups at C2 and C3 with the –(CH2)3– moiety. Better coplanarity of the BC4 ring and C5 atoms is achieved in 2 (Fig. 1) than in 1, as evidenced by the smaller deviation of the C5 atom from the C4 plane (0.222(5), 0.267(5) Å). In 2, the FMes group is essentially perpendicular to the B1–C1–C4 plane (dihedral angles: 89.85(8), 90.58(7)°), and the closest B⋯F distances range from 2.400(2) to 2.498(2) Å (sum of B and F van der Waals atomic radii: 3.39 Å15).
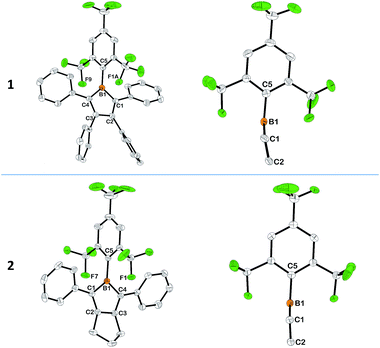 |
| | Fig. 1 Molecular structures of 1 and 2 obtained by X-ray diffraction (left) and views along the C1–C2–C3–C4 plane showing the deviation of the B1 and C5 atoms (right). Hydrogen atoms, the minor component of disordered CF3 groups and the second molecule of the asymmetric unit of 2 are omitted for clarity. Thermal ellipsoids correspond to 50% probability at 100 K. Selected bond lengths (Å) for 1: B1–C1 1.571(3), C1–C2 1.359(3), C2–C3 1.526(3), C3–C4 1.358(3), B1–C4 1.576(3), B1–C5 1.580(3); for 2 (values of the second molecule in [ ]): B1–C1 1.591(3) [1.592(3)], C1–C2 1.352(2) [1.355(2)], C2–C3 1.488(2) [1.485(2)], C3–C4 1.350(2) [1.354(2)], B1–C4 1.591(3) [1.589(3)], B1–C5 1.591(2) [1.591(2)]. | |
Photophysical properties and theoretical study
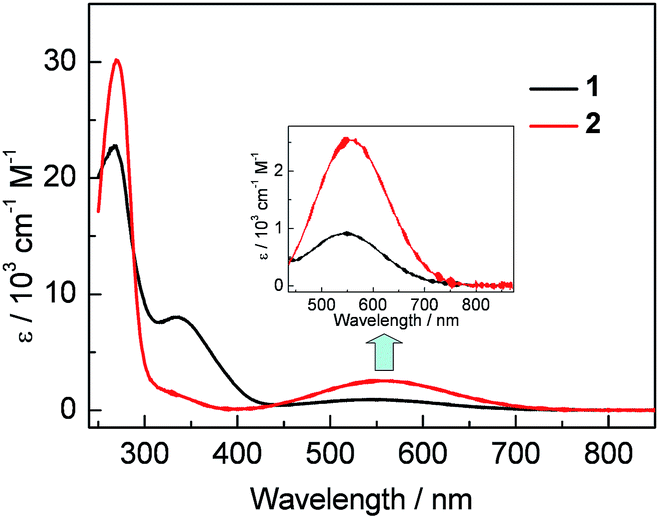
Compounds 1 and 2 form purple and blue CH2Cl2 solutions, respectively, displaying broad low-energy absorptions with maxima (λabs) located at 549 nm (ε = 900 cm−1 M−1) for 1 and slightly red shifted to 558 nm (ε = 2500 cm−1 M−1) for 2 (Fig. 2). These low-energy absorptions are dominated by electronic transitions from HOMO to LUMO, which are located mainly on the (2-Ph)-BC4-(5-Ph) moiety and the BC4 ring, respectively, according to DFT (PBE0/6-31+G(d,p)) and TD-DFT (CAM-B3LYP/6-31+G(d,p)) calculations (Fig. S12–S14†). The red shift in absorption and the increase in ε from 1 to 2, are related to better conjugation between the BC4 ring and the aryl groups at the 2- and 5-positions in the latter compound.4b Another notable phenomenon is that λabs of 1, with a strongly electron-withdrawing FMes group, is significantly blue-shifted from that of MesBC4Ph4 (λabs = 578 nm in CH2Cl2). This was not anticipated, as relatively electron-poor substituents on boron normally cause a red shift in λabs for boroles by significantly lowering the LUMO level,1c as concluded from the comparison between BC4Ph52a and a number of boroles with electron-rich heterocycles,2i amino2b or metal-containing groups2f on the boron atom. Interestingly, our DFT results indicate that replacing Mes with FMes stabilises the HOMO even more effectively than the LUMO, and thus 1 has a larger HOMO–LUMO gap than MesBC4Ph4. No fluorescence was observed from samples of 1 or 2 in solution or in the solid state.
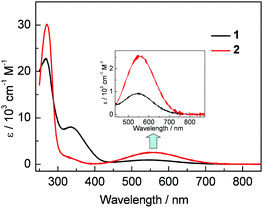 |
| | Fig. 2 UV-visible absorption spectra of 1 and 2 in CH2Cl2. Inset: expansion of the lowest-energy absorption band. | |
Nucleus-independent chemical shifts (NICS) at the geometric centres of the borole rings of 1 and 2, i.e. NICS(0), were calculated at the optimised geometries using the GIAO PBE0/6-31+G(d,p) level of theory. Both compounds have larger NICS(0) values (13.9 ppm for 1 and 14.8 ppm for 2) than MesBC4Ph4 (13.6 ppm), nominally indicating stronger anti-aromaticity. The interpretation of these result must be made with caution though; large changes in the dihedral angles at both the boron-bonded substituent and the phenyl rings in the 2,3,4,5-positions were found to result in very different values. For instance, when the rings of MesBC4Ph4 are constrained to have the same dihedral angles as those of 1, a significantly higher value of 14.9 ppm is obtained. Thus, it is important to note, the NICS(0) values reflect changes in both the geometry and electronics upon changing the substituent on the B atom.
Electrochemical properties
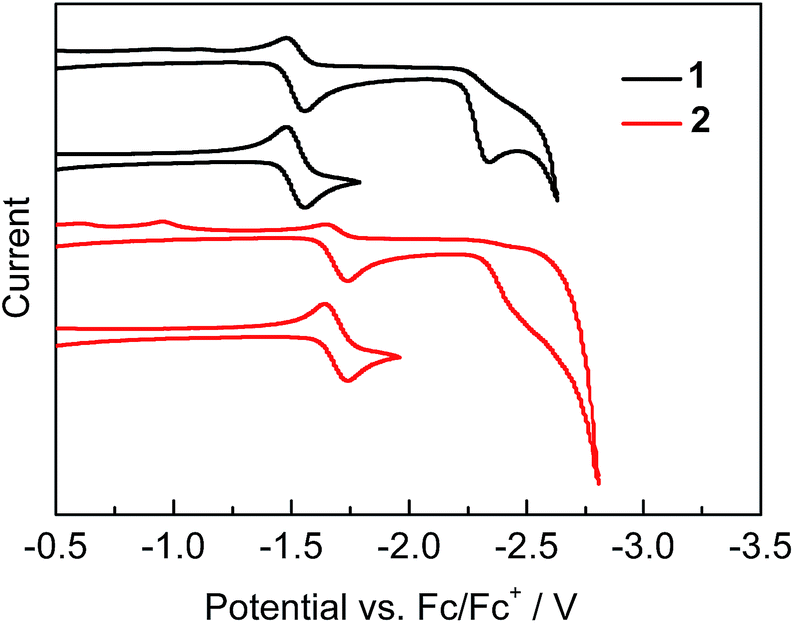
Cyclic voltammetry measurements on both 1 and 2, conducted to evaluate their electron-accepting properties, show a reversible first reduction wave and a subsequent irreversible second reduction wave corresponding to the formation of the radical anion and dianion (Fig. 3). It is notable that the first reduction of 1 occurs at only −1.52 V (Ered1/2), which is less negative than that of MesBC4Ph4 (Ered1/2 = −1.69 V)2h and all other reported first reduction potentials of boroles,2d,i,j,l,4b,16 indicating the very strong electron-accepting character of 1. Compound 2 displays a negatively shifted potential (Ered1/2 = −1.69 V) for the first reduction compared with 1 but equal to that of MesBC4Ph4, suggesting that its BC4 ring is more electron-rich than that of 1. The second reduction processes of 1 and 2 start at −2.23 and −2.32 V (Eonsetred), respectively, close to that of MesBC4Ph4 (ca. −2.3 V).
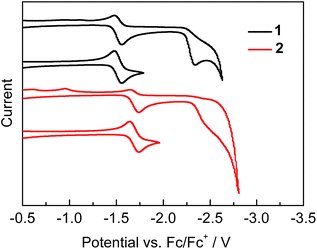 |
| | Fig. 3 Cyclic voltammograms of 1 and 2 measured in CH2Cl2. | |
Water and air stability
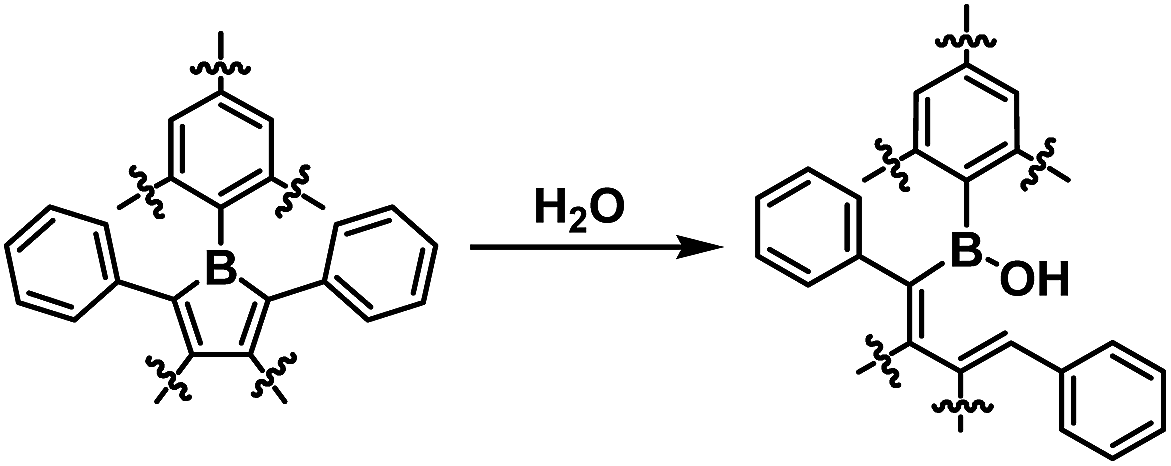
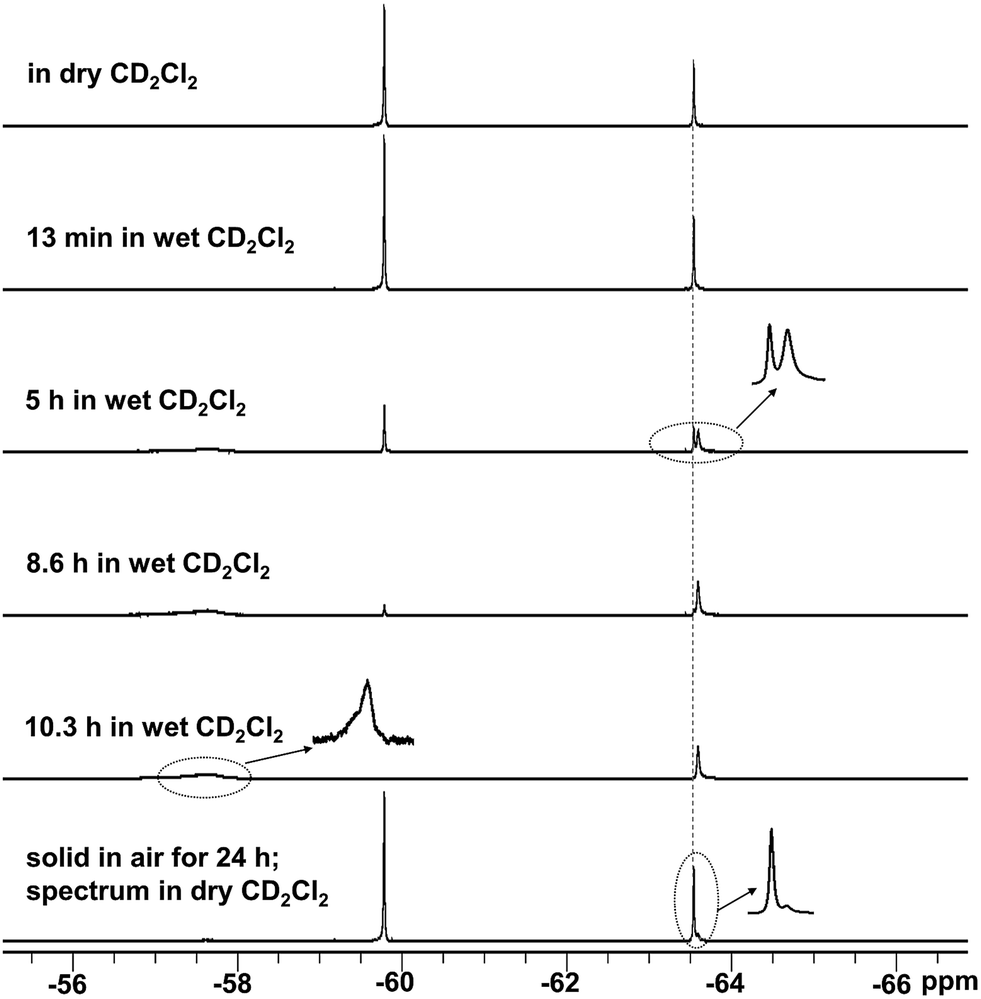
To determine the effectiveness of the FMes group at improving water and air stability, and to understand the influence of carbon-bonded groups on stability, 1, 2 and MesBC4Ph4, which also contains a relatively bulky boron-bonded aryl substituent, were compared. MesBC4Ph4 was found to be very unstable towards water; an NMR measurement of a freshly prepared sample (4.7 × 10−3 M) in wet CD2Cl2 (borole:H2O = ca. 1:1.7) showed total hydrolysis of the compound to generate a decomposition product containing a B–OH moiety through the cleavage of one B–C bond (Scheme 2 and Fig. S27–S29†). The hydrolysis process was so rapid that it was complete within one minute, as readily observed by the total disappearance of the deep green colour within this timescale to afford a colourless solution. In contrast, a reasonably clean NMR spectrum was obtained for a freshly prepared sample of 1 at a similar concentration using the same solvent, which clearly indicates the significantly improved stability of 1 towards water. Although this sample still showed gradual hydrolysis under these conditions, the full consumption of 1 required 10 h (Fig. 4 and S30–S33†), i.e. making it over 600 times less reactive. An NMR sample of 2, prepared under the same conditions, fully hydrolysed within ca. 1.5 h (Fig. S34–S38†), indicating that replacement of the phenyl groups at the 3- and 4-positions of 1 with a –(CH2)3– moiety leads to lower water stability, albeit still considerably greater (>90 times more) than that of MesBC4Ph4. The structure of the hydrolysis product of 2 was confirmed by X-ray diffraction (Fig. S39†).
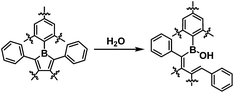 |
| | Scheme 2 The hydrolysis of 1, 2 and MesBC4Ph4. | |
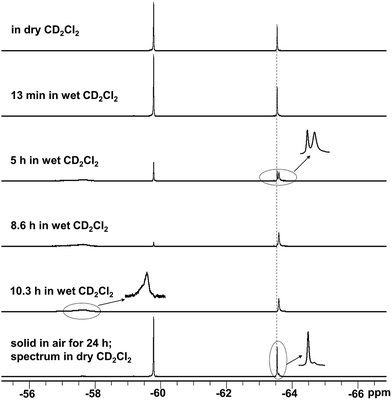 |
| | Fig. 4
19F{1H} NMR spectra showing the slow hydrolysis of 1 in wet CD2Cl2 and after exposure of a solid sample to air for 24 h. | |
In air, crystalline MesBC4Ph4 showed 83% hydrolysis within 24 h (Fig. S27†), while only 13% of crystalline 1 hydrolysed during the same time (Fig. 4 and S30†). The much higher air stability of 1 is consistent with its high tolerance toward water. For 2, 33% hydrolysis was detected under the same conditions in air (Fig. S34 and S35†), which is still noticeably better than that of MesBC4Ph4, despite the removal of the sterically protecting phenyl rings in the 3- and 4-positions. The relative stability in the solid state may also be affected partly by the size distribution or the morphology of the crystals, although efforts were made to minimise these effects. Compared with MesBC4Ph4, compounds 1 and 2 have a more (1) or an equally (2) electron-deficient BC4 ring, according to electrochemical measurements; however, their boron atoms and B–C bonds are much more resistant to attack by water molecules, as indicated by their higher water- and air-stability, presumably a result of the bulk of the FMes moiety.
It has been reported that oxidation of in situ generated BC4Ph5 with dry O2 followed by acidic hydrolysis in aqueous ethanol and subsequent column chromatography led to the isolation of tetraphenylfuran in good yield.7f However, in our air-stability measurements of solid 1, 2 and MesBC4Ph4, the same hydrolysis products as those observed in wet CD2Cl2, rather than oxidation products, were the predominant species observed by NMR spectroscopy and GC-MS. In aerated solutions, decomposition of 1 and 2 is more complicated and leads in both cases to an oxidation product with a mass of M + 16 as the major component of a complex mixture, as determined by GC-MS. The M + 16 mass may suggest the insertion of one oxygen atom into the borole, but the structure of the oxidation product has not yet been elucidated.
Thermal stability
For applications in organic electronic devices, good thermal stability is required. To assess the thermal stability of 1 and 2, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were conducted. Compound 1 has a high 5%-weight-loss temperature (Td5) of 271 °C and a sharp melting point (Tm) of 199 °C, indicating excellent thermal stability (Fig. S40 and S41†). This is impressive as boroles are typically very reactive species and some boroles are known to undergo isomerisation5a or dimerisation2e when heated at much lower temperatures. Compound 2 also has an excellent Td5 value of 262 °C (Fig. S42†), almost as high as that of 1. In the DSC curve (Fig. S43†), 2 displayed an exothermic process which occurred at ca. 140 °C before the melting of the sample at 190 °C, which may be related to a crystal–crystal phase transition or reactions within the sample. Importantly, both compounds can be sublimed under vacuum (160 °C at 3 mbar for 1, 100–110 °C at 100 mbar for 2), which make them suitable for vacuum deposition processes commonly used in device fabrication. Furthermore, the volatility, and therefore processability, of 1 and 2 can be inferred from their ability to pass through a 30 m GC column in a stream of helium carrier gas (Fig. S44 and S45†).
Reactivity towards base
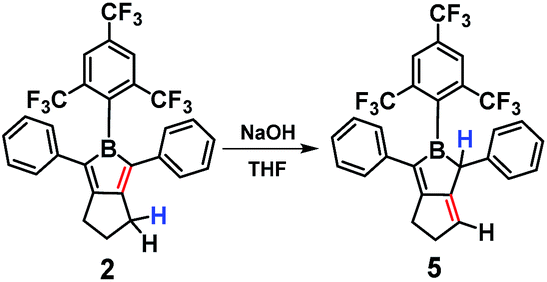
Although the FMes group is bulky, 1 and 2 are still Lewis acidic, e.g. they readily bind pyridine in solution to form an adduct that dissociates completely and cleanly upon addition of BF3·Et2O (Fig. S46 and S47†). Reaction of an excess of NaOH (99.99% purity, dry) with 2 in THF resulted in a colour change to light yellow, and the major reaction product was readily isolated in 77% yield after column chromatography on neutral alumina. Multinuclear NMR spectroscopy and GC-MS clearly indicated the product (5) to be an isomer of 2 in which a nominal [1,3]-H migration and a concomitant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond shift from endo to exo with respect to the borole ring have taken place, presumably via deprotonation at the allylic site (Scheme 3, Fig. S23–S26†). The CF3 groups of the FMes moiety ortho to the boron atom are equivalent in the 19F NMR spectrum of 2 but they are inequivalent in 5, as the two sides of the BC4 plane are now different. Furthermore, in the 1H NMR spectrum of 5, the presence of a vinylic proton (6.31 ppm, 1H), an allylic proton (3.79 ppm, 1H), the inequivalence of the two phenyl rings, and the two inequivalent CH2 resonances (2H each, reduced in integration from 4H and 2H in 2) all support the structural assignment of 5 as the thermodynamically more stable s-trans-butadienylborane.
C double bond shift from endo to exo with respect to the borole ring have taken place, presumably via deprotonation at the allylic site (Scheme 3, Fig. S23–S26†). The CF3 groups of the FMes moiety ortho to the boron atom are equivalent in the 19F NMR spectrum of 2 but they are inequivalent in 5, as the two sides of the BC4 plane are now different. Furthermore, in the 1H NMR spectrum of 5, the presence of a vinylic proton (6.31 ppm, 1H), an allylic proton (3.79 ppm, 1H), the inequivalence of the two phenyl rings, and the two inequivalent CH2 resonances (2H each, reduced in integration from 4H and 2H in 2) all support the structural assignment of 5 as the thermodynamically more stable s-trans-butadienylborane.
 |
| | Scheme 3 The isomerisation of 2 under strongly basic conditions to s-trans-butadienylborane 5. | |
Conclusions
In summary, the bulky FMes group has been successfully incorporated as the boron-bonded substituent in pentaarylboroles, as well as in novel triarylboroles via the reaction of Li[FMesBF3] with divinyldilithium reagents. A very congested environment around the two o-CF3 groups in our pentaarylborole compound leads to significantly improved water- and air-stability in comparison with MesBC4Ph, whilst simultaneously enhancing its electron-accepting ability. The triarylborole compound possesses better conjugation within the (2-Ph)-BC4-(5-Ph) moiety, but somewhat diminished water- and air-stability with respect to the pentaarylborole compound, although still notably better than the previously reported compound MesBC4Ph4. In addition, the pentaarylborole compound possesses good thermal stability, essential for potential applications in electronic devices. Interestingly, compared with MesBC4Ph, our pentaarylborole compound with the more electron-withdrawing FMes group exhibits a blue-shifted absorption, a result contrary to previous understanding. Our results represent a great stride towards making boroles available to the material science community for the full exploitation of their outstanding electronic properties. The design and synthesis of new boroles with enhanced stability and their incorporation into electronic devices are ongoing in our laboratory.
Acknowledgements
We are grateful for generous financial support by the DFG and the Bavarian State Ministry of Science, Research, and the Arts for the Collaborative Research Network “Solar Technologies go Hybrid”. Z. Z. and R. M. E. thank the Alexander von Humboldt Foundation for Postdoctoral Research Fellowships.
Notes and references
-
(a) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS;
(b) H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC;
(c) H. Braunschweig, I. Krummenacher and J. Wahler, Adv. Organomet. Chem., 2013, 61, 1–53 CrossRef CAS.
-
(a) H. Braunschweig, I. Fernández, G. Frenking and T. Kupfer, Angew. Chem., Int. Ed., 2008, 47, 1951–1954 CrossRef CAS PubMed;
(b) H. Braunschweig and T. Kupfer, Chem. Commun., 2008, 4487–4498 RSC;
(c) H. Braunschweig, C.-W. Chiu, K. Radacki and T. Kupfer, Angew. Chem., Int. Ed., 2010, 49, 2041–2044 CrossRef CAS PubMed;
(d) H. Braunschweig, F. Breher, C.-W. Chiu, D. Gamon, D. Nied and K. Radacki, Angew. Chem., Int. Ed., 2010, 49, 8975–8978 CrossRef CAS PubMed;
(e) H. Braunschweig, C.-W. Chiu, J. Wahler, K. Radacki and T. Kupfer, Chem.–Eur. J., 2010, 16, 12229–12233 CrossRef CAS PubMed;
(f) H. Braunschweig, C.-W. Chiu, K. Radacki and P. Brenner, Chem. Commun., 2010, 46, 916–918 RSC;
(g) K. Ansorg, H. Braunschweig, C.-W. Chiu, B. Engels, D. Gamon, M. Hügel, T. Kupfer and K. Radacki, Angew. Chem., Int. Ed., 2011, 50, 2833–2836 CrossRef CAS PubMed;
(h) H. Braunschweig, V. Dyakonov, J. O. C. Jimenez-Halla, K. Kraft, I. Krummenacher, K. Radacki, A. Sperlich and J. Wahler, Angew. Chem., Int. Ed., 2012, 51, 2977–2980 CrossRef CAS PubMed;
(i) H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, C. Hörl, I. Krummenacher, T. Kupfer, L. Mailänder and K. Radacki, J. Am. Chem. Soc., 2012, 134, 20169–20177 CrossRef CAS PubMed;
(j) H. Braunschweig, A. Damme, D. Gamon, H. Kelch, I. Krummenacher, T. Kupfer and K. Radacki, Chem.–Eur. J., 2012, 18, 8430–8436 CrossRef CAS PubMed;
(k) H. Braunschweig, C.-W. Chiu, A. Damme, B. Engels, D. Gamon, C. Hörl, T. Kupfer, I. Krummenacher, K. Radacki and C. Walter, Chem.–Eur. J., 2012, 18, 14292–14304 CrossRef CAS PubMed;
(l) H. Braunschweig, C. W. Chiu, D. Gamon, M. Kaupp, I. Krummenacher, T. Kupfer, R. Müller and K. Radacki, Chem.–Eur. J., 2012, 18, 11732–11746 CrossRef CAS PubMed;
(m) R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 CrossRef CAS PubMed;
(n) H. Braunschweig, C. Hörl, L. Mailänder, K. Radacki and J. Wahler, Chem.–Eur. J., 2014, 20, 9858–9861 CrossRef CAS PubMed.
-
(a) C. Fan, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2009, 48, 2955–2958 CrossRef CAS PubMed;
(b) C. Fan, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2010, 29, 5132–5139 CrossRef CAS;
(c) A. Fukazawa, J. L. Dutton, C. Fan, L. G. Mercier, A. Y. Houghton, Q. Wu, W. E. Piers and M. Parvez, Chem. Sci., 2012, 3, 1814–1818 RSC;
(d) A. Y. Houghton, V. A. Karttunen, C. Fan, W. E. Piers and H. M. Tuononen, J. Am. Chem. Soc., 2013, 135, 941–947 CrossRef CAS PubMed.
-
(a) C.-W. So, D. Watanabe, A. Wakamiya and S. Yamaguchi, Organometallics, 2008, 27, 3496–3501 CrossRef CAS;
(b) T. Araki, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 5484–5487 CrossRef CAS PubMed.
-
(a) F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2014, 136, 68–71 CrossRef CAS PubMed;
(b) F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, Organometallics, 2015, 34, 229–235 CrossRef CAS.
-
(a) K. Huynh, J. Vignolle and T. D. Tilley, Angew. Chem., Int. Ed., 2009, 48, 2835–2837 CrossRef CAS PubMed;
(b) K. Nozaki, Nature, 2010, 464, 1136–1137 CrossRef CAS PubMed;
(c) M. Yamashita, Angew. Chem., Int. Ed., 2010, 49, 2474–2475 CrossRef CAS PubMed.
-
(a) J. J. Eisch, N. K. Hota and S. Kozima, J. Am. Chem. Soc., 1969, 91, 4575–4577 CrossRef CAS;
(b) J. J. Eisch and J. E. Galle, J. Am. Chem. Soc., 1975, 97, 4436–4437 CrossRef CAS;
(c) G. E. Herberich, J. Hengesbach, U. Kölle and W. Oschmann, Angew. Chem., Int. Ed. Engl., 1977, 16, 42–43 CrossRef;
(d) L. Killian and B. Wrackmeyer, J. Organomet. Chem., 1978, 148, 137–146 CrossRef CAS;
(e) A. Sebald and B. Wrackmeyer, J. Organomet. Chem., 1986, 307, 157–165 CrossRef CAS;
(f) J. J. Eisch, J. E. Galle and S. Kozima, J. Am. Chem. Soc., 1986, 108, 379–385 CrossRef CAS PubMed;
(g) P. J. Fagan, E. G. Burns and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2979–2981 CrossRef CAS;
(h) G. E. Herberich, M. Hostalek, R. Laven and R. Boese, Angew. Chem., Int. Ed. Engl., 1990, 29, 317–318 CrossRef;
(i) J. J. Eisch, J. E. Galle, B. Shafii and A. L. Rheingold, Organometallics, 1990, 9, 2342–2349 CrossRef CAS.
-
(a) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS;
(b) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS;
(c) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed;
(d) Y. Sun, N. Ross, S.-B. Zhao, K. Huszarik, W.-L. Jia, R.-Y. Wang, D. Macartney and S. Wang, J. Am. Chem. Soc., 2007, 129, 7510–7511 CrossRef CAS PubMed;
(e) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed;
(f) P. Sonar, J. P. F. Lim and K. L. Chan, Energy Environ. Sci., 2011, 4, 1558–1574 RSC.
-
(a) P. A. Chase, W. E. Piers and B. O. Patrick, J. Am. Chem. Soc., 2000, 122, 12911–12922 CrossRef CAS;
(b) S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS PubMed;
(c) S. Kim, K. H. Song, S. O. Kang and J. Ko, Chem. Commun., 2004, 68–69 RSC;
(d) A. Wakamiya, K. Mishima, K. Ekawa and S. Yamaguchi, Chem. Commun., 2008, 579–581 RSC;
(e) A. Hübner, Z.-W. Qu, U. Englert, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2011, 133, 4596–4609 CrossRef PubMed;
(f) A. Iida and S. Yamaguchi, J. Am. Chem. Soc., 2011, 133, 6952–6955 CrossRef CAS PubMed;
(g) J. F. Araneda, B. Neue, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2012, 51, 8546–8550 CrossRef CAS PubMed;
(h) D. M. Chen, Q. Qin, Z.-B. Sun, Q. Peng and C.-H. Zhao, Chem. Commun., 2014, 50, 782–784 RSC;
(i) A. Hübner, A. M. Diehl, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2014, 53, 4832–4835 CrossRef PubMed;
(j) A. Hübner, T. Kaese, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2015, 137, 3705–3714 CrossRef PubMed.
- F. Leroux, ChemBioChem, 2004, 5, 644–649 CrossRef CAS PubMed.
-
(a) X. Yin, J. Chen, R. A. Lalancette, T. B. Marder and F. Jäkle, Angew. Chem., Int. Ed., 2014, 53, 9761–9765 CrossRef CAS PubMed;
(b) Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Steffen, L. E. Longobardi, D. W. Stephan, C. Lambert and T. B. Marder, Chem. Sci., 2015, 6, 308–321 RSC;
(c) Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem.–Eur. J., 2015, 21, 177–190 CrossRef CAS PubMed.
- Analogous heterocyclopentadienes with boron replaced by other atoms are known. For examples, see:
(a) T. Takahashi, F.-Y. Tsai and Y. Li, Chem. Lett., 1999, 1173–1174 CrossRef CAS;
(b) M. C. Suh, B. Jiang and T. D. Tilley, Angew. Chem., Int. Ed., 2000, 39, 2870–2873 CrossRef CAS.
- S. M. Cornet, K. B. Dillon, C. D. Entwistle, M. A. Fox, A. E. Goeta, H. P. Goodwin, T. B. Marder and A. L. Thompson, Dalton Trans., 2003, 4395–4405 RSC.
- G. A. Molander, S. L. J. Trice, S. M. Kennedy, S. D. Dreher and M. T. Tudge, J. Am. Chem. Soc., 2012, 134, 11667–11673 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- With the exception of the bis(borafluorene)s of Wagner and co-workers,9i,jvide supra.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1051711–1051713. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02205c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence