
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBillion-fold rate enhancement of urethane polymerization via the photothermal effect of plasmonic gold nanoparticles†
Kaitlin M.
Haas
and
Benjamin J.
Lear
*
The Pennsylvania State University, 102 Chemistry Building, University Park, PA 16802, USA. E-mail: bul14@psu.edu
First published on 31st July 2015
Abstract
We use the photothermal effect of gold nanoparticles (AuNPs) to provide billion-fold enhancement of on-demand bulk-scale curing of polyurethane. We follow the course of this polymerization using infrared spectroscopy, where we can observe the loss of both isocyanate and alcohol stretches, and the rise of the urethane modes. Application of 12.5 MW cm−2 of 532 nm light to a solution of isocyanate and alcohol with 0.08% w/v of 2 nm AuNPs results in the billion-fold enhancement of the rate of curing. This result is intriguing, as it demonstrates the ability of nanoscale heat to drive bulk transformations. In addition, the reaction is strongly exothermic and results in a relatively weak bond, both of which would preclude the use of bulk-scale heat, highlighting the unique utility of the photothermal effect for driving thermal reactions.
Introduction
The advance of chemistry requires continual development of new and improved means for driving the cleavage, formation, and isomerisation of chemical bonds. Recently, the photothermal effect of gold nanoparticles (AuNPs) emerged as a way in which to supply heat sufficient to cleave very strong chemical bonds,1–7 and to do so with unprecedented resolution in both time (picoseconds) and space (nanometres). The origin of the photothermal heat in these cases is the absorption of light by the surface plasmon resonance (SPR) of AuNPs, which excites a collective oscillation of electrons that quickly (femtoseconds) dephase, transferring energy into the phonon modes of the AuNP.8–11 The photothermal system reaches peak temperature on the picosecond timescale, and then transfers thermal energy away from the AuNP, elevating the temperature of the local environment, but leaving the bulk temperature largely unperturbed.12–15 Beyond bond cleavage,2,4,6,16–21 the intense localised heat produced by the photothermal effect of nanoparticles can be harnessed to drive other destructive processes including the melting of solids,8–11 the boiling of liquids,12–15 controlled release of molecules from materials,2,4,6,16–21 and killing of cancerous cells.1,3,5,7,22 Remarkably, all of these transformations are realised without damage to the surroundings – a result that highlights the fact that the transformations occur at localised ‘effective temperatures’ that are not experienced by the bulk material. This result is particularly intriguing for chemical bond cleavage, as it allows targeted destruction of chemicals with molecular scale precision. However, while the destructive power of the photothermal effect is undeniable, its general applicability for other chemical transformations – such as bond formation – remains to be established.In many respects, the application of extreme temperatures is ideal for bond cleavage. For even the strongest bond, a temperature can be identified that allows escape from its potential energy well. However, this same reasoning also suggests that the photothermal effect might not be useful for accomplishing constructive events (i.e. bond formation) that require occupation of these same potential energy wells. Nevertheless, the rapid cooling of the particles provides a possible means for trapping of species transiently generated at high temperatures. Below, we demonstrate the ability of the photothermal effect of gold nanoparticles to drive bond formation between isocyanates and alcohols, curing bulk-scale polyurethane films with unprecedented rates of reaction.
Results and discussion
Our work focuses on the polymerisation of polyurethane films (Fig. 1a) from hexamethylene diisocyanate (HDI – formulated as Desmodur N3600), and the diester polyol poly-bis(triethylol) heptanedioate (BTEH – formulated as K-FLEX-188). Both of the structures shown in Fig. 1a are approximate. Though HDI exists primarily as the trimer shown, both dimers and monomers of the basic HDI unit are present.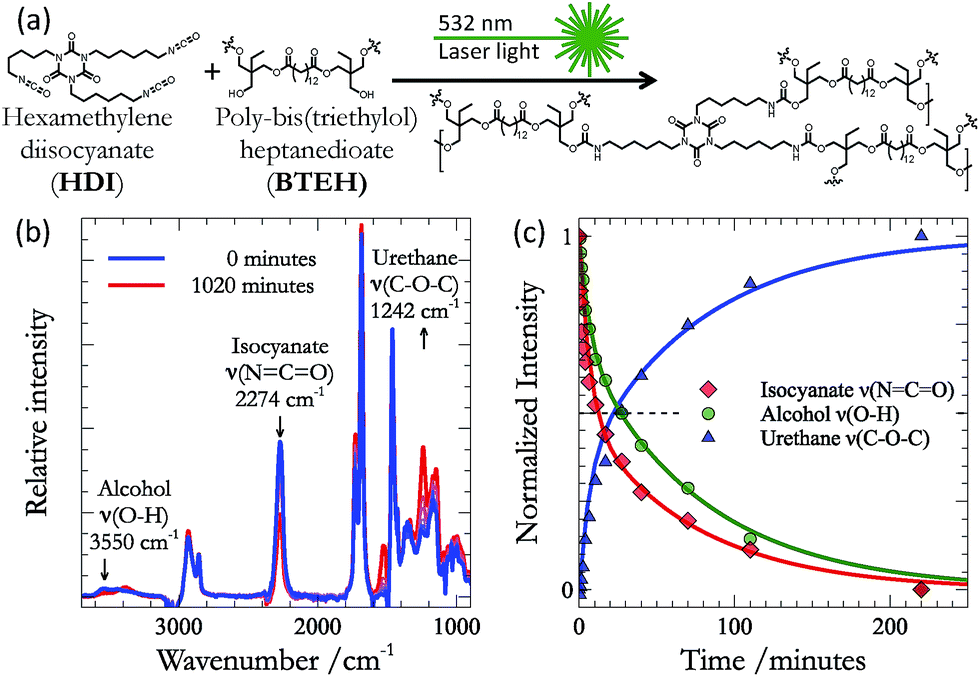 | ||
| Fig. 1 (a) The reaction between HDI and BTEH to form polyurethane can be followed using (b) infrared spectroscopy. In particular, formation of the urethane moiety results in the loss of bands associated with the isocyanate (2274 cm−1) and alcohol (3550 cm−1) modes, and the appearance of the C–O–C band (1242 cm−1) associated with the urethane. (c) The kinetics associated with these changes indicate direct conversion of isocyanates and alcohols to urethane. | ||
For a number of reasons, our selected transformation is an ideal probe of the efficacy of the photothermal effect for driving bond formation. First, the formation of urethane is spontaneous at room temperature,1,3,5,7,23,24 and this provides a baseline rate of bond formation to which we can compare the effect of photothermal heating. Second, there are many known chemical catalysts for this reaction – the most common being dibutyltin dilaurate (DBTDL)25,26 – to which we can also compare the efficacy of the photothermal effect. Third, the urethane bond is relatively weak (100–130 kJ mol−1), and can be cleaved at temperatures (125 °C to 250 °C)27,28 far below the peak temperatures produced by the photothermal effect of gold nanoparticles (which can reach thousands of degrees).29 Thus, if net bond formation is observed, it must occur through the thermal quenching described above. Fourth, given reasonable estimations for the enthalpic (−132 kJ mol−1) and entropic (−188 J mol−1) changes associated with urethane formation,30 the equilibrium for this reaction should lie far to the reactants at the extreme temperatures (>800 K) known to be obtained under our photothermal conditions (K800 K ≈ 7 × 10−2 M−1).4 Thus, significant progress of the reaction can also only be ascribed to trapping of transiently formed products during thermal quenching – rather than simple biasing of the equilibrium at high temperatures. Finally, urethanes are widely employed in science and technology and the work described below provides the basis for on-demand thermal curing of urethane polymer films, which will extend the physical and chemical properties available for on-demand applications (e.g. 3D printing).
In addition to the kinetic, energetic, and practical considerations outlined above, the reaction also provides several excellent infrared (IR) markers that may be used to follow the progress of urethane formation. Fig. 1b shows the evolution of the IR spectrum during the reaction between HDI and BTEH (in equal parts alcohol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :isocyanate moieties) on an ATR crystal at room temperature. The most obvious change in the spectrum is the loss of intensity in the isocyanate stretching mode (2274 cm−1). However, several other changes are also observable. The intensity of the free OH stretch (3550 cm−1) also diminishes during the course of the reaction. Concomitantly, a band associated with C–O–C mode of the urethane (1242 cm−1) appears. The rate of disappearance of the isocyanate and alcohol modes are mirrored by the growth of the C–O–C mode (Fig. 1c) – confirming that these features of the IR spectra are reporting on the conversion of isocyanate and alcohols into urethane. Given the fact that the isocyanate mode presents the most isolated band, we choose this as our primary reporter on kinetics as we moved to study the efficacy of the photothermal effect for driving urethane formation.
:isocyanate moieties) on an ATR crystal at room temperature. The most obvious change in the spectrum is the loss of intensity in the isocyanate stretching mode (2274 cm−1). However, several other changes are also observable. The intensity of the free OH stretch (3550 cm−1) also diminishes during the course of the reaction. Concomitantly, a band associated with C–O–C mode of the urethane (1242 cm−1) appears. The rate of disappearance of the isocyanate and alcohol modes are mirrored by the growth of the C–O–C mode (Fig. 1c) – confirming that these features of the IR spectra are reporting on the conversion of isocyanate and alcohols into urethane. Given the fact that the isocyanate mode presents the most isolated band, we choose this as our primary reporter on kinetics as we moved to study the efficacy of the photothermal effect for driving urethane formation.
The heart of our study is a comparison between the impact of the photothermal effect upon the rate of catalysed and un-catalysed urethane formation. In order to isolate the effect of light upon these conditions, we investigated the curing rate of urethane under all permutations of adding AuNPs or DBTDL, or applying light (key, Fig. 2a). For this study, we worked with octanethiol-protected AuNPs with diameters of ∼2 nm. These particles are near the smallest that still support a SPR (see the ESI†), and thus have the desired photophysical properties that lead to the photothermal effect. Though larger particles would possess a stronger SPR (and associated photothermal effect), we chose to use small particles for their kinetics of thermal diffusion. Smaller particles display faster thermal quenching, making it more likely to trap transiently formed chemical bonds. For 2 nm AuNPs, the decay of the temperature has a lifetime of ∼10 ps,9 and can compete with the kinetics of bond formation/cleavage. Thus, for our first demonstration of the constructive use of the photothermal effect, we favored kinetic considerations over strength of the photothermal effect.
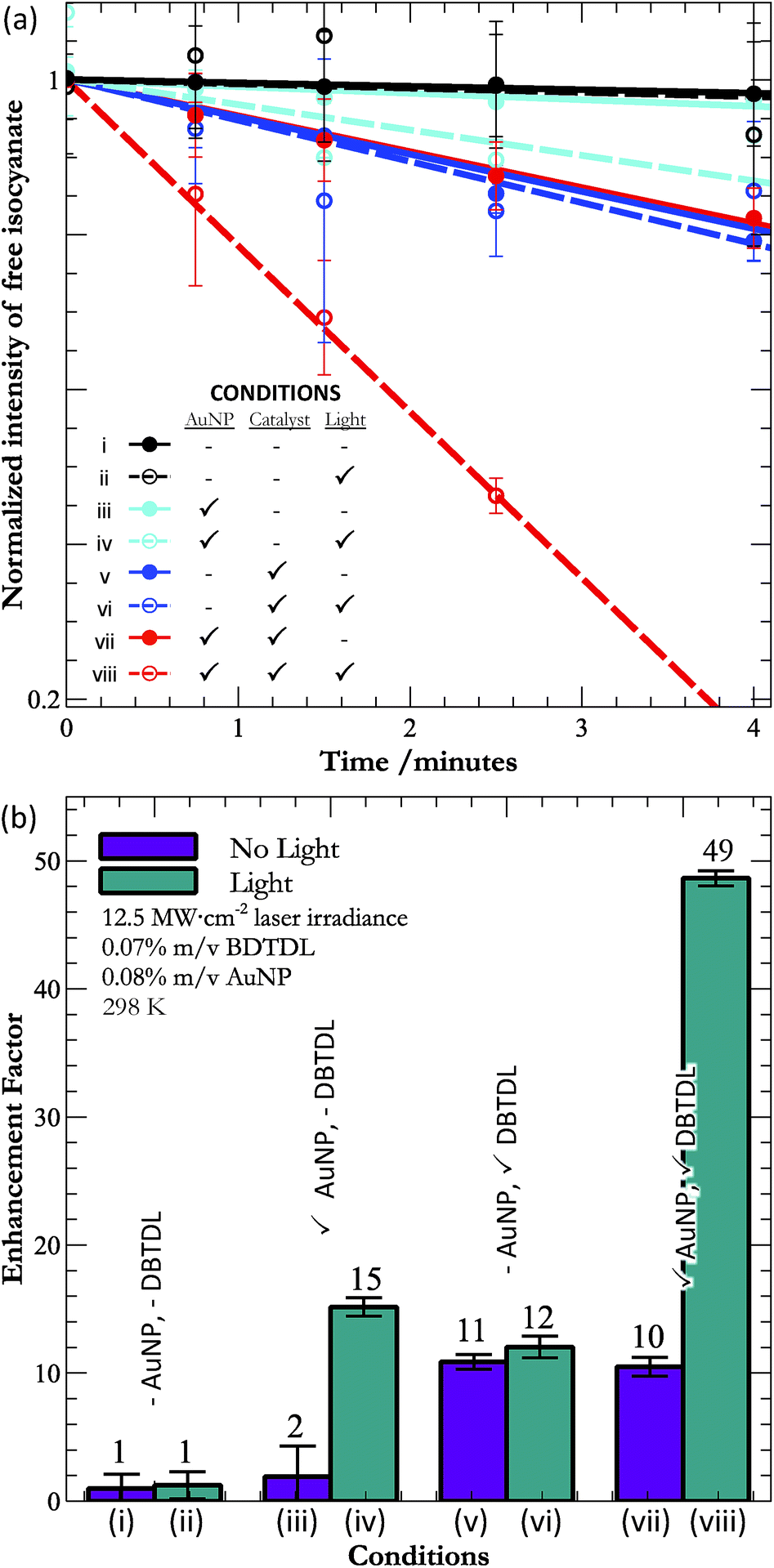 | ||
| Fig. 2 (a) Kinetic traces following the disappearance of the isocyanate band (2274 cm−1), and (b) the relative rates of reaction (with respect to pure polymer) under all eight conditions. | ||
A detailed description of our experimental procedures can be found in the ESI.† Briefly, the appropriate solutions were made by mixing HDI and BTEH in an approximately 1:1 ratio with either pure toluene, or toluene solutions containing either AuNPs or DBTDL, or both. For all solutions containing AuNPs or DBTDL, the final concentrations of these additives were 0.08% w/v and 0.07% w/v, respectively. These concentrations were chosen based upon preliminary data, such that the action of the photothermal effect would be comparable to the action of the catalyst. The final concentration of isocyanate was 13.7 M, which is similar to that used in industrial applications.
Once the solutions were prepared, the reaction between HDI and BTEH was allowed to proceed for four minutes, either in the presence or absence of light. For those exposed to light, we used 8 ns pulses (50 mJ per pulse) of 532 nm light from a QuantaRay 130 Nd:YAG laser operating at a repetition rate of 10 Hz. The peak irradiance for these pulses is 12.5 MW cm−2. The polymerisation of isocyanate and polyol to polyurethane was monitored following the disappearance of the free isocyanate band at 2274 cm−1.
Fig. 2a shows the kinetic traces obtained from the isocyanate band in the IR under all eight permutations. In all cases, we found that the early time kinetics (up to 4 minutes) were reasonably linear (χ2 better than 0.84) and so we also show the results of linear regressions to the data. At long times, the kinetics deviated strongly from linearity (Fig. 1c), as one might expect given the second order kinetics that underlie polyurethane curing.23,24 However, it is interesting that for the fastest observed reaction (condition (viii)), the reaction kinetics remain linear beyond 50% polymerisation. We did not anticipate this result, which demonstrated a linear response much closer to reaction completion than the kinetics observed for the pure polymer (Fig. 1c). This unusual behaviour under photothermal conditions may be an additional consequence of the localized nature of the heat, which allows the reaction to run close to completion near the surface of the nanoparticle. Diffusion of the particle between laser pulses would allow the particle to enter a region of unreacted isocyanate and alcohol, where the process would repeat. Though speculative, such behavior would provide a mechanism for the observed linearity beyond 50% completion under action of AuNPs, DBTDL, and light (condition (viii)).
Given that all conditions gave rise to linear early kinetics, we chose to focus on these kinetics, and our comparisons between the various conditions are in terms of the initial rates of reaction. From the IR, it is clear that substantial, though not complete, curing occurs during the 4 minutes that we followed. While it would be interesting to follow the enhancement of curing up to completion (>95% crosslinking), analysis of the rate enhancements during the later stages of curing would be difficult at present, as we do not yet know the reaction order or mechanism for the photothermal curing, though this is a clear topic for future research. For this reason, our focus is on the initial rates of reaction.
Because the exact mechanism of the photothermal curing is unknown, it is important to ensure we are forming a densely cross-linked polymer matrix. Though the IR results conclusively demonstrate urethane is being formed, it is also possible that the high temperatures experienced by the nanoparticles could lead to side reactions, such as chain scission, which would reduce the weight of the polymer. To the touch, there was no apparent physical difference between polymers cured with and without exposure to the photothermal effect. Nor did the IR show large differences between photothermally and non-photothermally cured films. Nevertheless, we also acquired mass spectra (MALDI – see ESI†) for conditions (v) and (vi), in order to determine any differences in the mass of polymer formed by photothermal curing. The only substantial difference we found between the mass spectra for these two conditions is a greater proportion of high mass components for the photothermally-cured films. Thus, it appears that the photothermal conditions result in greater cross-linking of the polyurethane. A possible explanation for this is that the high temperatures attained by the particles under photothermal conditions results in breaking apart of the HDI trimer, which would provide a greater density of free isocyanate sites to form cross-linking urethane moieties, when compared to the traditionally cured samples. Again, further work will be needed to confirm this hypothesis.
Returning to consideration of the rates of curing, we calculated the relative enhancement of bond formation for each condition (using the slopes from Fig. 2a) by dividing the rate for each condition by the rate of curing for pure alcohol and isocyanate components in the dark (condition (i)). The enhancement factors are shown in Fig. 2b and Table 1.
| Condition | Enhancement | ΔTobs | ΔTkinetics | ||
|---|---|---|---|---|---|
| REAL TIME | |||||
| HDI + BTEH | No light | i | 1 | 0 | 0 |
| Light | ii | 1 | 0 | 0 | |
| HDI + BTEH + AuNP | No light | iii | 2 | 0 | 0 |
| Light | iv | 15 | 12 | 65 | |
| HDI + BTEH + DBTDL | No light | v | 11 | 0 | 0 |
| Light | vi | 12 | 0 | 0 | |
| HDI + BTEH + AuNP + DBTDL | No Light | vii | 10 | 0 | 0 |
| Light | viii | 49 | 8 | 65 | |
|
|||||
| IRRADIATED TIME | |||||
| HDI + BTEH + AuNP | Light | iv | 1.55 × 109 | 12 | 305 |
| HDI + BTEH + AuNP + DBTDL | Light | viii | 4.84 × 109 | 8 | 322 |
There are a number of interesting results that are apparent from inspection of Fig. 2b and Table 1. To begin, only samples containing AuNPs experience rate enhancement upon exposure to light. These results imply that the AuNPs are the only significant source of photothermal heating – an important result given that DBTDL is a slightly coloured compound (see ESI†). It also implies that any increase in reaction rate upon exposure to light must stem from the actions of AuNPs. This is a point that will become more important below.
In addition, the photothermal enhancement for films with only AuNPs is comparable to the rate enhancement for films with only catalysts, which means that the photothermal effect of AuNPs competes on a weight-by-weight basis with the action of traditional catalysts. However, we can also consider the action on a per-number basis. The relative mass difference between the catalytic molecules (631.56 g mol−1) and the AuNPs (∼4.9 × 104 g mol−1) implies that, on a per-number basis, the photothermal effect of gold is approximately 90 times more efficient at accomplishing urethane formation than is the catalytic effect of DBTDL. Here it is important to realize that the molecular mass given for AuNPs is only a rough estimation (see ESI†) based upon the mean size of a polydisperse sample. The greater effectiveness per AuNP was an anticipated result, as the AuNP is able to create an area effect, while the catalyst interacts on a one-to-one basis with its substrate.
There is also a synergistic effect between the action of the DBTDL and the photothermal effect of the AuNPs (condition (viii)). That is, the enhancement of the rate is not the simple addition of the enhancements for DBTDL and AuNPs alone. Importantly, without light, the samples with DBTDL alone and DBTDL + AuNPs experience the same rate – meaning that the presence of AuNPs is not sufficient for this synergy; instead the SPR of the AuNP must be excited. In addition, irradiation of DBTDL produces no enhancement relative to the action of DBTDL alone. Thus, the large synergy must result from the excitation of the AuNPs' SPR in the presence of DBTDL. This conclusion implies that there is some interaction between DBTDL and the AuNPs, during irradiation, though the exact nature of this interaction is not clear at this time. Possible sources of synergy could be increased mobility of the liquid components, which would facilitate the diffusion-limited action of the catalyst. In addition, recalling that HDI exists (in part) as a trimer, the isocyanates bound in this moiety must be released before they are free to react with alcohols. Thus, if the photothermal effect breaks apart the trimer, this would make more free isocyanates available the DBTDL – providing another mechanism for synergistic rate enhancement. This hypothesis is consistent with the greater crosslinking of the urethane under photothermal conditions.
In order to ensure that the rate enhancements observed for the photothermal effect were not merely a result of bulk-scale temperature increases, we measured the temperature changes during the course of the reaction under all eight conditions reported in this manuscript. This was done using an IR thermal imager (Raytek ThermoView Ti30) to acquire temperature measurements before and after 4 minutes, for the same conditions used to generate Fig. 2. A summary of the observed temperature changes (ΔTobs) are given in Table 1. As can be seen, the only conditions that led to an observable temperature increase were those in which AuNPs were exposed to laser light. However, in these cases, the bulk-temperature jump was far too small (on the order of 10 K) to account for the observed rate increases. We confirmed this point by following the kinetics of polymer formation under several temperatures, attained by bulk heating in an oil bath (see ESI†). These results indicate that we would need bulk temperatures changes (ΔTkinetics) of ca. 65 K in order to observe the kinetic enhancement achieved by the photothermally driven reactions. Thus, the observed photothermal enhancement is not an effect of simple bulk-scale heating, but the result of transient and intense heat produced near the AuNPs' surface.
The above conclusion – that it is the localized and transient heat that gives rise to the rate enhancement – carries with it several additional implications. First, this implies that the reaction rate is only increased while the AuNPs are hot. Given the fast rise and decay of the temperature for these particles,9 we can approximate that the particles are only hot for the duration of the laser pulse (8 ns) – or a total of ca. 2 μs during the course of our 4 minute experiments. If we recalculate the rate of reaction using the total irradiation time (see ESI†), we arrive at an astonishing billion-fold enhancement of reaction rate during irradiation (Table 1). However, we also note that this is the lower bound for the enhancement – as it assumes enhanced reaction rates for the entire pulse and throughout the full sample. However, given the photophysics and size of the particles, it is more likely that the most extreme temperatures are only present for the first hundred picoseconds the laser pulse (before the onset of steady-state conditions), while the heating (enhancements) exists only near the particle surface. Thus, it is most likely that the rate enhancements near the particle surface are much greater then this billion-fold enhancement – though future work will be needed in order to quantify the actual enhancement.
The rate adjusted for irradiation time further implies a temperature of at least 600 K – though this would be for a uniform temperature and the actual temperature near the nanoparticle surface must be many times higher. Previous work from our laboratory suggests that the temperature near the nanoparticles should be well above 800 K under these conditions.4 Again, given the energetics of this reaction, the equilibrium should lie far to the side of the reactants at these temperatures and so the observed completion percentage must result from the trapping of transiently formed bonds during the thermal quenching of the particles. This conclusion highlights the unique ability to quickly drive bond formation at ‘effective’ temperatures that are far higher than those that would fail to give rise to appreciable reaction progress if applied in bulk.
These considerations further emphasize the benefits of photothermal heating over traditional catalysts, such as DBTDL. Unlike traditional catalysts, the efficiency of the AuNPs should be easily and dynamically tunable via alteration of the conditions. Indeed, increasing either the irradiance or the repetition rate of the laser should give rise to an increase in the efficacy of the photothermal effect. Simple consideration of the timescales associated with the photothermal effect suggests a further million-fold increase in repetition rate could be applied, while still realizing gains in efficacy. Thus, use of the photothermal effect promises the possibility of dynamic tuning of the reaction rate over 12 orders of magnitude.
Despite the clear advantages of photothermal effect for curing polyurethane, there are also a number of drawbacks to the approach as presented above. Gold is an expensive material, and complete recovery from the cured polymer will most likely prove difficult. In addition, the laser that we used to induce the photothermal effect is expensive, inefficient, and cannot illuminate large areas. Thus, it is unlikely that the use of gold and nanosecond pulsed lasers will be adopted on an industrial scale, though future work into the required properties of photothermal agents and light sources might mitigate these drawbacks. Nevertheless, the main conclusion of this paper is the remarkable fact that the extreme (but transient and localized) temperatures attained by nanoparticles under photothermal conditions can be used to drive an overall exothermic bond formation at astonishing rates. This result opens the door for examining the utility of the photothermal effect for accomplishing constructive chemical reactions.
Conclusions
In conclusion, we have used urethane formation to demonstrate the first application of photothermal heating for efficiently and effectively driving bond formation on the bulk scale. Though there have been other examples of driving bond formation using photothermal effects,31 the results were only observed on the nanoscale, making them impractical for large-scale transformations. Our bulk scale transformations are surprising given the fact that the heat produced is localised to the nanometre scale. Thus, this work shows that photothermal heating can provide new and powerful means by which to increase the rate of thermally activated reactions for large-scale transformations. Future work will focus on demonstrating the ultimate enhancements that can be realized, as well as the dependence upon the size of the nanoparticles, which controls the kinetics of thermal diffusion. Finally, we also seek to investigate other thermally activated reactions, in order to demonstrate the full generality of this approach.Acknowledgements
We thank Ray Schaak for many helpful discussions, and PPG Industries, Inc. for funding of this work.Notes and references
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115 CrossRef CAS PubMed.
- C. Fasciani, C. J. B. Alejo, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, Org. Lett., 2011, 13, 204 CrossRef CAS PubMed.
- I. H. El-Sayed, X. Huang and M. A. El-Sayed, Cancer Lett., 2006, 239, 129 CrossRef CAS PubMed.
- K. M. Haas and B. J. Lear, Nanoscale, 2013, 5, 5247 RSC.
- V. P. Zharov, K. E. Mercer, E. N. Galitovskaya and M. S. Smeltzer, Biophys. J., 2006, 90, 619 CrossRef CAS PubMed.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240 CrossRef CAS PubMed.
- L. Cheng, K. Yang, Y. Li, J. Chen, C. Wang, M. Shao, S.-T. Lee and Z. Liu, Angew. Chem., Int. Ed., 2011, 50, 7385 CrossRef CAS PubMed.
- H. H. Richardson, Z. N. Hickman, A. O. Govorov, A. C. Thomas, W. Zhang and M. E. Kordesch, Nano Lett., 2006, 6, 783 CrossRef CAS PubMed.
- G. V. Hartland, Chem. Rev., 2011, 111, 3858 CrossRef CAS PubMed.
- A. U. Zillohu, R. Abdelaziz, M. K. Hedayati, T. Emmler, S. Homaeigohar and M. Elbahri, J. Phys. Chem. C, 2012, 116, 17204 CAS.
- S. Maity, L. N. Downen, J. R. Bochinski and L. I. Clarke, Polymer, 2011, 52, 1674 CrossRef CAS PubMed.
- C. D. Jones and L. A. Lyon, J. Am. Chem. Soc., 2003, 125, 460 CrossRef CAS PubMed.
- A. O. Govorov, W. Zhang, T. Skeini, H. Richardson, J. Lee and N. A. Kotov, Nanoscale Res. Lett., 2006, 1, 84 CrossRef.
- Z. Fang, Y.-R. Zhen, O. Neumann, A. Polman, F. J. García De Abajo, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 1736 CAS.
- A. O. Govorov, H. Zhang, H. V. Demir and Y. K. Gun'ko, Nano Today, 2014,(1), 85–101 CrossRef CAS PubMed.
- L. Poon, W. Zandberg, D. Hsiao, Z. Erno, D. Sen, B. D. Gates and N. R. Branda, ACS Nano, 2010, 4, 6395 CrossRef CAS PubMed.
- A. AsadiRad, Z. Erno and N. R. Branda, Chem. Commun., 2013, 49, 5639 RSC.
- A. C. Estrada, A. Daniel-da-Silva and T. Trindade, RSC Adv., 2013, 3, 10828 RSC.
- D. T. Nguyen, R. Truong, R. Lee, S. A. Goetz and A. P. Esser-Kahn, Energy Environ. Sci., 2014, 7, 2603 CAS.
- D. A. Boyd, L. Greengard, M. Brongersma, M. Y. El-Naggar and D. G. Goodwin, Nano Lett., 2006, 6, 2592 CrossRef CAS PubMed.
- J. R. Adleman, D. A. Boyd, D. G. Goodwin and D. Psaltis, Nano Lett., 2009, 9, 4417 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869 CrossRef CAS PubMed.
- M. Sato, J. Org. Chem., 1961, 26, 770 CrossRef CAS.
- M. Sato, J. Org. Chem., 1962, 27, 819 CrossRef CAS.
- K. C. Frisch and L. P. Rumao, J. Macromol. Sci., Part C, 1970, 5, 103 CrossRef CAS PubMed.
- Q. Han and M. W. Urban, J. Appl. Polym. Sci., 2002, 86, 2322 CrossRef CAS PubMed.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 42, 1068 CrossRef PubMed.
- T. Hentshel and H. Munstedt, Polymer, 2001, 42, 3195 CrossRef.
- N. N. Nedyalkov, S. E. Imamova, P. A. Atanasov, R. A. Toshkova, E. G. Gardeva, L. S. Yossifova, M. T. Alexandrov and M. Obara, Appl. Surf. Sci., 2011, 257, 5456 CrossRef CAS PubMed.
- A. Y. Samuilov, L. A. Zenitova, I. N. Bakirova and Y. D. Samuilov, Russ. J. Appl. Chem., 2008, 81, 1419 CrossRef CAS.
- M. Fedoruk, M. Meixner, S. Carretero-Palacios, T. Lohmüller and J. Feldmann, ACS Nano, 2013, 7, 7648 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, experimental details, mass estimates for the AuNPs, temperature dependent rates, characterization of DBTDL, and TEM images. See DOI: 10.1039/c5sc02149a |
| This journal is © The Royal Society of Chemistry 2015 |