
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric direct aldol reaction of α-alkyl azlactones and aliphatic aldehydes†
Yang
Zheng
 and
Li
Deng
*
and
Li
Deng
*
Department of Chemistry, Brandeis University, Waltham, Massachusetts 02454-9110, USA. E-mail: deng@brandeis.edu
First published on 4th August 2015
Abstract
An unprecedented highly diastereoselective and enantioselective aldol reaction of α-alkyl azlactones and aliphatic aldehydes was achieved with cinchona alkaloid catalysts. To our knowledge, this reaction provides the first useful catalytic asymmetric access toward β-hydroxy-α-amino acids bearing alkyl substituents, which are structural motifs embedded in many natural products.
Optically active β-hydroxy-α-amino acids are an important class of amino acids as they are structural motifs in many biologically active natural products such as vancomycin,1 katanosins,2 cyclosporin,3 myriocin,4a,b mycestericins,4c,d sphingosine and threonine (Fig. 1). Furthermore, these amino acids are also useful chiral building blocks in organic synthesis as precursors to β-lactams,5 β-halo-α-amino acids,6 and aziridines.7 A variety of catalytic asymmetric approaches for the synthesis of β-hydroxy-α-amino acids has been reported.8–14 In a pioneering study,8a Ito, Hayashi and coworkers reported a gold-catalyzed highly diastereoselective and enantioselective aldol reaction for the generation of β-hydroxy-α-amino acids containing tertiary α-carbons. Since then other groups have also reported asymmetric direct aldol reactions with chiral transition-metal catalysts,8c–h organocatalysts9 and aldolases10 for the synthesis of β-hydroxy-α-amino acids and their derivatives. In addition, Sharpless asymmetric aminohydroxylation,11 transition-metal-catalyzed asymmetric hydrogenation,12 palladium-catalyzed allylic alkylation13 and chiral phosphoric acid-catalyzed addition to oxocarbenium ion14 have been utilized to achieve the same goal.
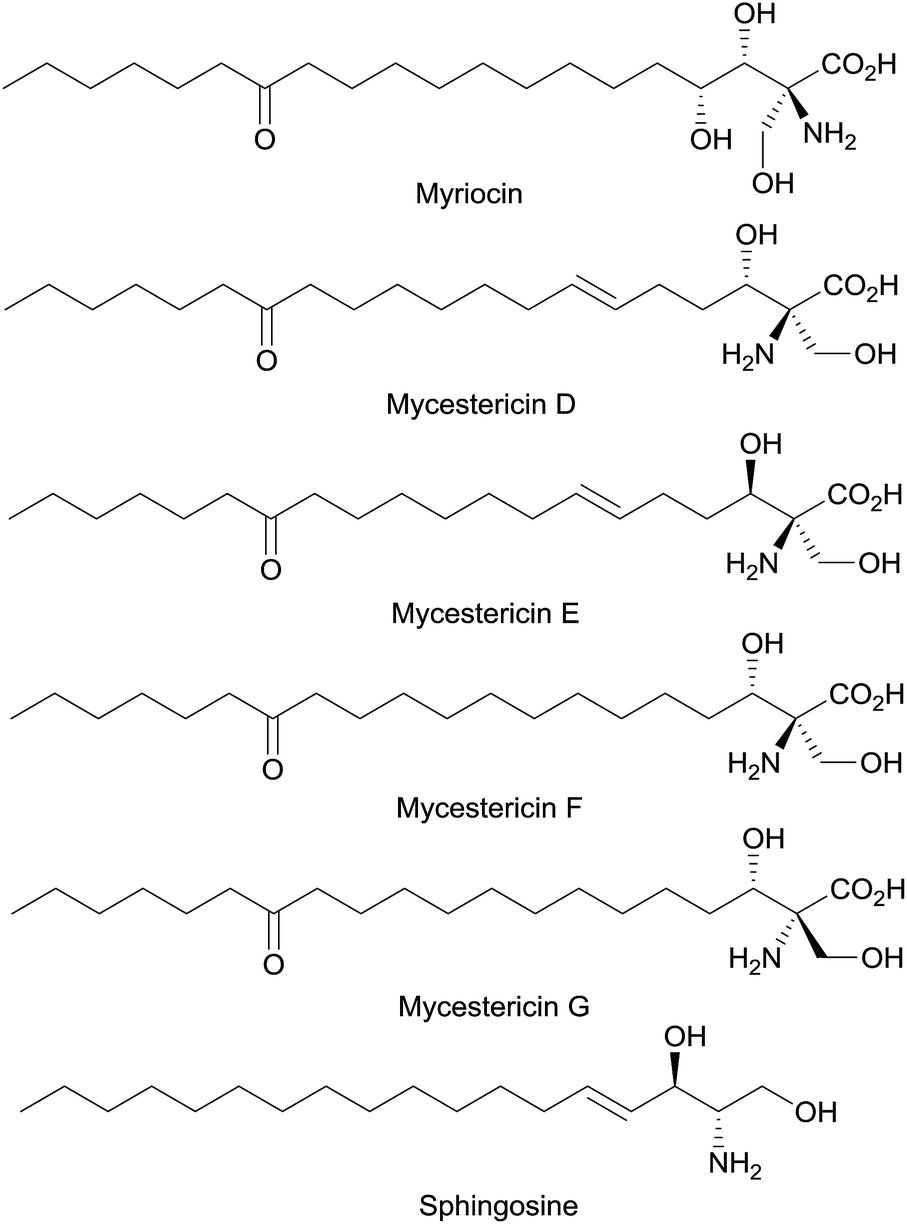 | ||
| Fig. 1 Mycestericins: potent immunosuppressant natural products | ||
We became interested in the development of catalytic asymmetric synthesis of β-hydroxy-α-amino acids because biologically interesting natural products such as mycestericins contain a chiral β-hydroxy-α-amino acid motif that could not be constructed from existing catalytic asymmetric aldol reactions. In particular this motif presents both a tertiary β-stereocenter and a quaternary α-stereocenter with alkyl substituents. In principle, an efficient catalytic asymmetric aldol reaction of α-alkyl enolates or the equivalents with aliphatic aldehydes could provide a direct access to this structural motif.15 However, to our knowledge, such an asymmetric transformation was not available. Herein, we report the first efficient catalytic asymmetric direct aldol reaction of α-alkyl azlactones 6 and aliphatic aldehydes 7 (Scheme 1), which provides, to our knowledge, the first useful asymmetric catalytic access toward β-hydroxy-α-amino acids bearing alkyl substituents at both the tertiary β-stereocenter and the quaternary α-stereocenter. The high anti-diastereoselectivity in combination with a broad substrate scope allows the reaction to complement existing methods to form a general strategy for the asymmetric synthesis of β-hydroxy-α-amino acids.
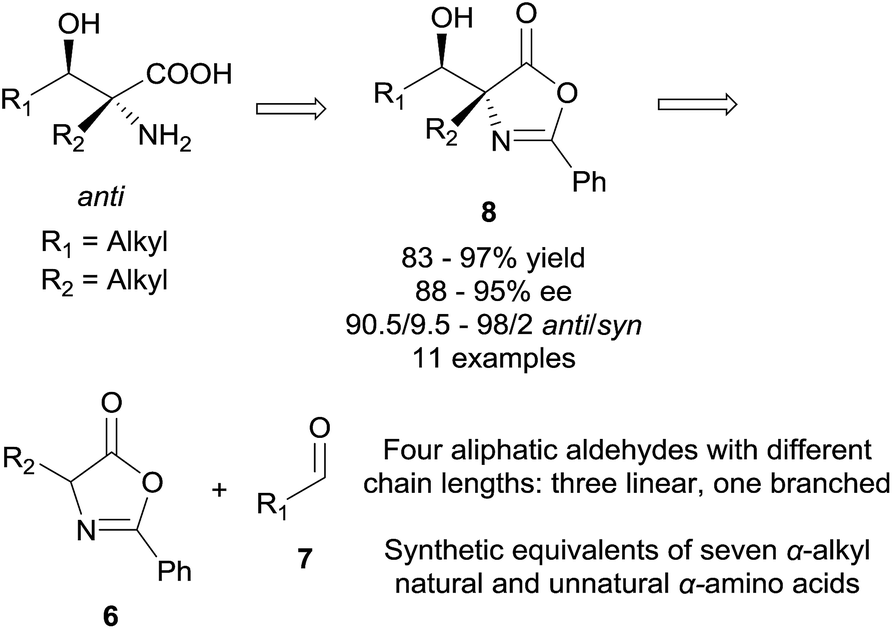 | ||
| Scheme 1 Reaction design | ||
We initiated our study by reacting azlactone 6a and aldehyde 7a in the presence of a stoichiometric amount of triethylamine. After considerable experiments, we found that a reaction could be reasonably fast and clean at −20 °C in chloroform. We next investigated the possibility of promoting an asymmetric variant of this reaction with cinchona alkaloid-derived catalysts (Fig. 2). Upon first screening of a series cinchona alkaloid derivatives, (entries 1–9, Table 1), we identified the 6′-OH cinchona alkaloid 3d as the most promising catalyst in terms of affording high diastereoselectivity and enantioselectivity (entry 6, Table 1). Catalyst 3e, the pseudo-enantiomer of 3d, gave comparable results with an expected reverse sense of asymmetric induction (entry 7, Table 1). Following these results, we carried out the 3d-promoted aldol reaction in a variety of solvents with azlactone 6a at a significantly decreased concentration of 0.5 M (entry 10–15). We found that the reaction at the reduced concentration proceeded in higher diastereo- and enantioselectivity (entry 10 vs. 6). Moreover, the reaction in dichloromethane occurred in a slightly higher diastereoselectivity than and the same enantioselectivity as the reaction in chloroform (entries 11–10, Table 1). Both the diastereoselectivity and enantioselectivity afforded by catalyst 3d could be improved significantly when the reaction was performed at significantly reduced temperature and concentration (entry 16 vs. 11), although a higher catalyst loading and an extended reaction time were required for the reaction to proceed to completion. Importantly, under these conditions, a highly diastereoselective and enantioselective aldol reaction was established to generate the desired aldol product 8aa in 92% isolated yield, 94% ee and 97.5/2.5 anti/syn ratio. It should be noted that no product resulted from the self-aldol reaction by aldehyde 7a was detected by NMR analysis.
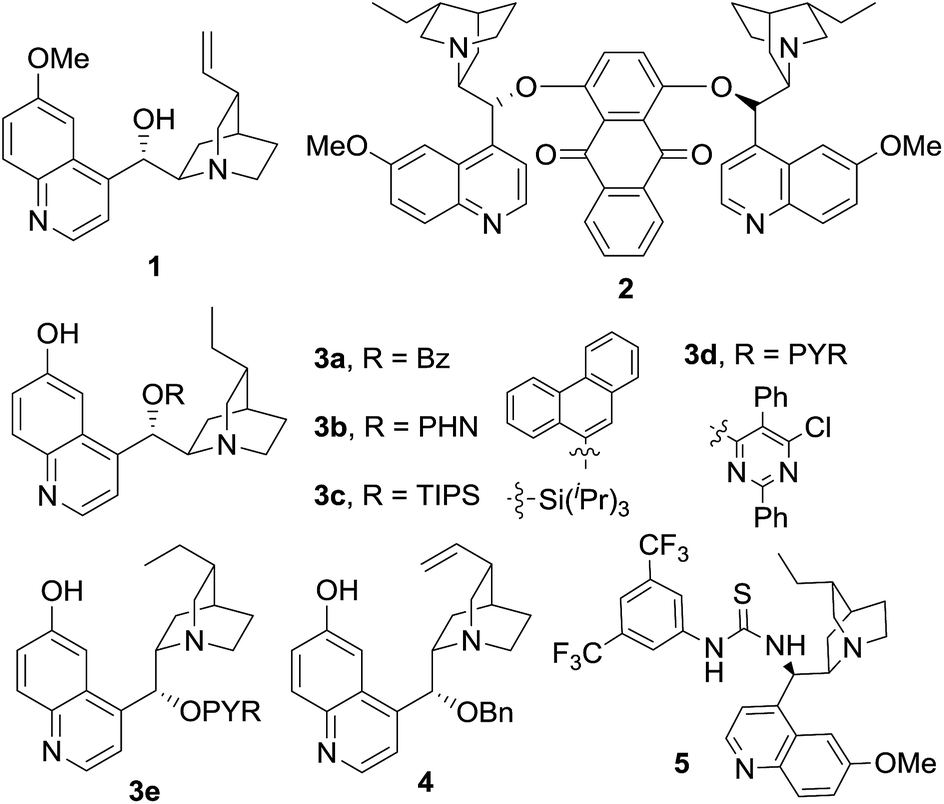 | ||
| Fig. 2 Cinchona alkaloid catalysts | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | Time | Conv.c (%) | eecd (%) | anti/sync |
| a Reactions were carried out with 0.1 mmol of 6a and 0.15 mmol of 7a. b Isolated yield. c Determined by chrial HPLC analysis. d ee (anti/syn). e 10 mg of 4 Å molecular sieves were added. f 15 mol% of 3d. | |||||||
| 1 | 1 | CHCl3 (2 M) | −20 | 15 h | >95 | 29/22 | 43.5/56.5 |
| 2 | 2 | CHCl3 (2 M) | −20 | 15 h | 93 | 43/12 | 40.5/59.5 |
| 3 | 3a | CHCl3 (2 M) | −20 | 15 h | >95 | 57/25 | 81/19 |
| 4 | 3b | CHCl3 (2 M) | −20 | 15 h | >95 | 51/19 | 73/27 |
| 5 | 3c | CHCl3 (2 M) | −20 | 15 h | 92 | −7/18 | 71/29 |
| 6 | 3d | CHCl3 (2 M) | −20 | 15 h | >95 | 75/13 | 88/12 |
| 7 | 3e | CHCl3 (2 M) | −20 | 15 h | >95 | −73/16 | 90/10 |
| 8 | 4 | CHCl3 (2 M) | −20 | 15 h | 91 | −64/−6 | 82/18 |
| 9 | 5 | CHCl3 (2 M) | −20 | 15 h | >95 | −71/22 | 66/34 |
| 10 | 3d | CHCl3 (0.5 M) | −20 | 34 h | 95 | 86/−11 | 91/9 |
| 11 | 3d | CH2Cl2 (0.5 M) | −20 | 34 h | 93 | 86/−39 | 93.5/6.5 |
| 12 | 3d | PhCH3 (0.5 M) | −20 | 34 h | 80 | 48/−28 | 80/20 |
| 13 | 3d | THF (0.5 M) | −20 | 34 h | >95 | 50/−28 | 79.5/20.5 |
| 14 | 3d | Et2O (0.5 M) | −20 | 34 h | >95 | 53/−28 | 83/17 |
| 15 | 3d | CH3CN (0.5 M) | −20 | 34 h | >95 | 72/−18 | 88/12 |
| 16e,f | 3d | CH2Cl2 (0.1 M) | −50 | 88 h | >95 (92)b | 94/ND | 97.5/2.5 |
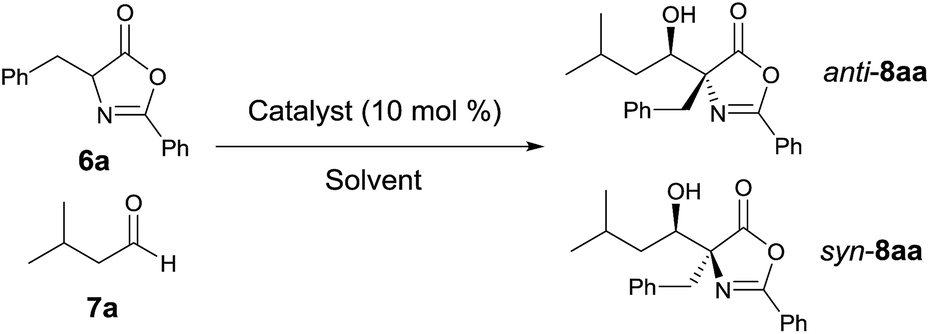
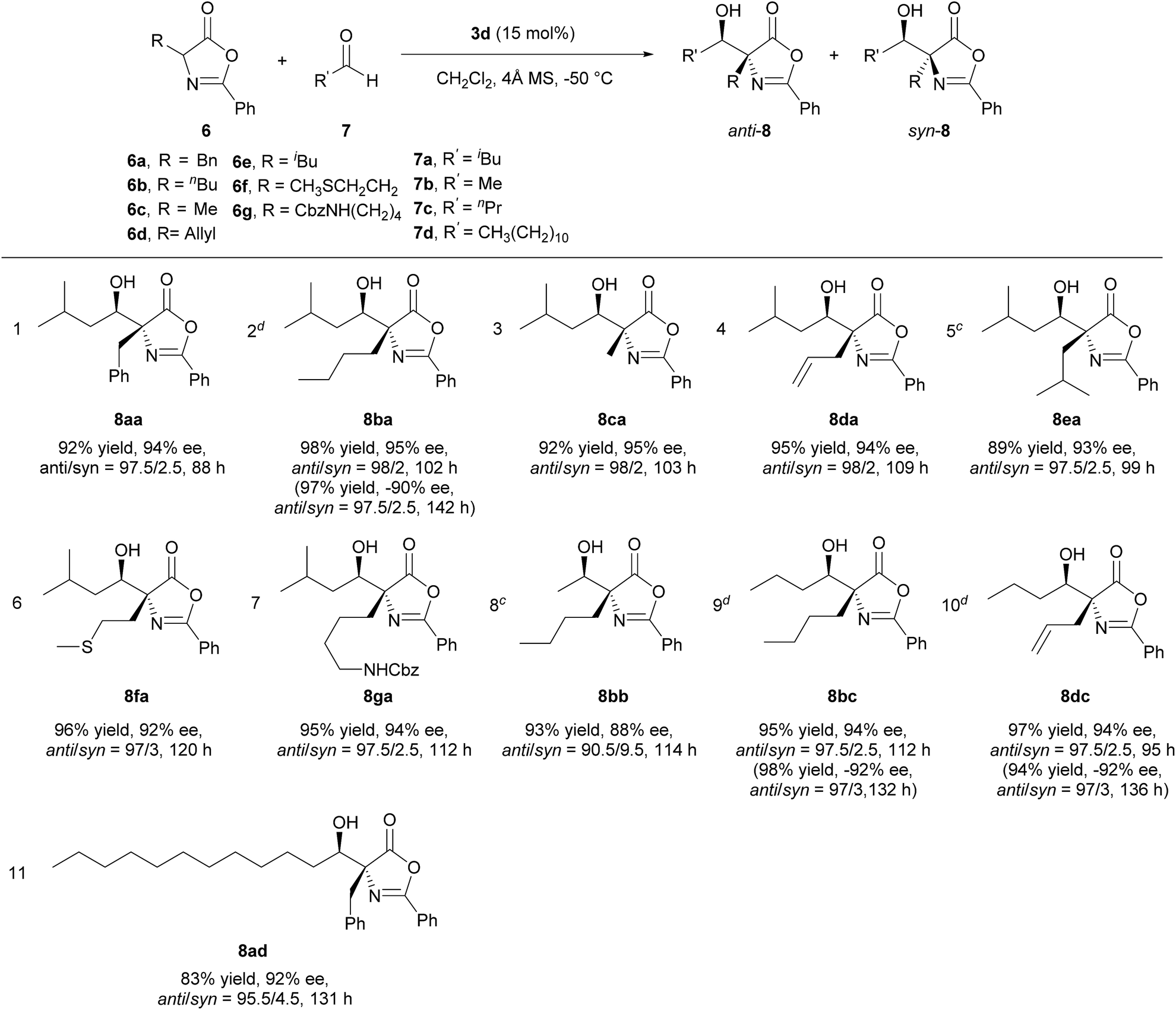
Applying the optimized reaction conditions for the model reaction, we investigated the substrate scope of this asymmetric aldol reaction (Table 2). The reactions of aldehyde 7a and azlactones 6a–g bearing different α-alkyl substituents gave consistently excellent yields, enantioselectivity and anti-selective diastereoselectivity (entries 1–7, Table 2). The catalyst could also accommodate variations in aliphatic aldehydes as shown by its high efficiency in the promotion of asymmetric aldol reactions involving a series of aliphatic aldehydes (entries 8–11, Table 2). The tolerance of aldehyde 7d, which bears a linear C12 alkyl chain, is noteworthy. With catalyst 3e, the reaction provide equally efficient access to the other enantiomer of the aldol product, as shown in the formation of aldol adduct ent-8ba, ent-8bc and ent-8dc (entries 2, 9, 10, Table 2). As detailed in the ESI,† the relative and absolute configurations of aldol products 8 were determined by 1D NOESY experiment and a modified Mosher's method, respectively.16
| a Unless noted, reactions were carried out with 0.1 mmol of 6, 0.15 mmol of 7, 0.015 mmol of 3d, 10 mg of 4 Å molecular sieves in 1 mL of dichloromethane. b ee value and anti/syn ratio determined by chiral HPLC analysis. c 0.2 mmol of 7b. d Results in parentheses obtained using 3e (15 mol%) as catalyst. e See ESI for determination of relative and absolute configurations. |
|---|
|
To demonstrate the potential synthetic utility of the chiral aldol adduct 8, ring opening transformations converting 8 into useful β-hydroxy-α-amino acid derivative 10 must be developed. We found that 8 were liable toward retro-aldol initiated decompositions under a variety of reaction conditions. After extensive experimental explorations, we were able to establish a high yield, three-step protocol to convert 8 into β-hydroxy-α-aminoester 10 (Scheme 2). Critical to the development of this useful conversion was the experimental discovery that the THP protected β-hydroxy-α-alkylazlactones 9, unlike 8, is inert toward retro-aldol decompositions.17 It should be noted that the four-step enantioselective preparations of β-hydroxy-α-aminoester 10 from azlactones 6 and aldehydes 7 require only a single purification for the isolation of 10, both intermediates 8 and 9 were used for the next step without subjecting to purifications. To establish enantioselective access to all four stereoisomers of β-hydroxy-α-amino acid derivative 10, we developed a one-pot conversion of anti-β-hydroxy-α-amino acid 10da into the corresponding syn-β-hydroxy-α-amino acid syn-12da involving the treatment of anti-10da with thionyl chloride followed by HCl in THF (Scheme 2).
 | ||
| Scheme 2 Transformation of aldol product 8. Reagents and conditions: (a) 3d (15 mol%), CH2Cl2, 4 Å MS, −50 °C; (b) PPTS, DHP, CH2Cl2, rt; then K2CO3, Na2SO4, MeOH, rt; (c) 2 N HCl, MeOH, rt; (d) HCl in MeOH (∼1.25 M), rt; (e) SOCl2, THF, rt; (f) 2 N HCl, THF, rt; (g) 3e (15 mol%), CH2Cl2, 4 Å MS, −50 °C. PPTS = pyridinum-p-toluenesulfonate; DHP = 3,4-dihydro-2-H-pyran. | ||
Conclusions
In summary, we have developed a highly enantioselective and diastereoselective direct aldol reaction of α-alkyl azlactones with aliphatic aldehydes catalyzed by cinchona alkaloid catalysts 3d and 3e. To our knowledge, this is the first efficient asymmetric direct aldol reaction of azlactones and aliphatic aldehydes. Providing an efficient catalytic asymmetric access to β-hydroxy-α-amino acids bearing alkyl substituents at both the tertiary β-stereocenter and the quaternary α-stereocenter, this new catalytic asymmetric aldol reaction should find applications in natural product synthesis and medicinal chemistry.18Acknowledgements
We are grateful for the generous financial support from National Institutes of General Medical Sciences (GM-61591).Notes and references
- (a) D. H. Williams, Acc. Chem. Res., 1984, 17, 364 CrossRef CAS; (b) C. M. Harris, H. Kopecka and T. M. Harris, J. Am. Chem. Soc., 1983, 105, 6915 CrossRef CAS; (c) R. Nagarajan, A. A. Schabel, J. L. Occolowitz, F. T. Counter and J. L. Ott, J. Antibiot., 1988, 41, 1431 CrossRef.
- (a) T. Kato, H. Hinoo, Y. Terui, J. Kikuchi and J. Shoji, J. Antibiot., 1988, 41, 719 CrossRef CAS; (b) J. Shoji, H. Hinoo, K. Matsumoto, T. Hattori, T. Yoshida, S. Matsuura and E. Kondo, J. Antibiot., 1988, 41, 713 CrossRef CAS; (c) S. A. Carr, E. Block and C. E. Costello, J. Org. Chem., 1985, 50, 2854 CrossRef CAS.
- (a) S. L. Schreiber, Science, 1991, 251, 283 CAS; (b) D. A. Evans and A. E. Weber, J. Am. Chem. Soc., 1986, 108, 6757 CrossRef CAS.
- (a) T. Fujita, K. Inoue, S. Yamamoto, T. Ikumoto, S. Sasaki, R. Toyama, K. Chiba, Y. Hoshino and T. Okumoto, J. Antibiot., 1994, 47, 208 CrossRef CAS; (b) T. Fujita, K. Inoue, S. Yamamoto, T. Ikumoto, S. Sasaki, R. Toyama, K. Chiba, Y. Hoshino and T. Okumoto, J. Antibiot., 1994, 47, 216 CrossRef CAS; (c) S. Sasaki, R. Hashimoto, M. Kiuchi, K. Inoue, T. Ikumoto, R. Hirose, K. Chiba, Y. Hoshino, T. Okumoto and T. Fujita, J. Antibiot., 1994, 47, 420 CrossRef CAS; (d) T. Fujita, N. Hamamichi, M. Kiuchi, T. Matsuzaki, Y. Kitao, K. Inoue, R. Hirose, M. Yoneta, S. Sasaki and K. Chiba, J. Antibiot., 1996, 49, 846 CrossRef CAS.
- (a) B. T. Lotz and J. Miller, J. Org. Chem., 1993, 58, 618 CrossRef CAS; (b) M. J. Miller, Acc. Chem. Res., 1986, 19, 49 CrossRef CAS.
- S. V. Pansare and J. C. Vederas, J. Org. Chem., 1987, 52, 4804 CrossRef CAS.
- D. Tanner, Angew. Chem., Int. Ed., 1994, 33, 599 CrossRef PubMed.
- For selected examples, see: (a) Y. Ito, M. Sawamura and T. Hayashi, J. Am. Chem. Soc., 1986, 108, 6405 CrossRef CAS; (b) Y. Ito, M. Sawamura, E. Shirakawa, K. Hayashizaki and T. Hayashi, Tetrahedron, 1988, 44, 5253 CrossRef CAS; (c) D. A. Evans, J. M. Janey, N. Magomedov and J. S. Tedrow, Angew. Chem., Int. Ed., 2001, 40, 1884 CrossRef CAS; (d) J. Kobayashi, M. Nakamura, Y. Mori, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 9192 CrossRef CAS PubMed; (e) M. C. Willis, G. A. Cutting, V. J.-D. Piccio, M. J. Durbin and M. P. John, Angew. Chem., Int. Ed., 2005, 44, 1543 CrossRef CAS PubMed; (f) F. Sladojevich, A. Trabocchi, A. Guarna and D. J. Dixon, J. Am. Chem. Soc., 2011, 133, 1710 CrossRef CAS PubMed; (g) T. Yoshino, H. Morimoto, G. Lu, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 17082 CrossRef CAS PubMed; (h) B. M. Trost and F. Miege, J. Am. Chem. Soc., 2014, 136, 3016 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Horikawa, J. Busch-Peterson and E. J. Corey, Tetrahedron Lett., 1999, 40, 3843 CrossRef CAS; (b) T. Ooi, M. Taniguchi, M. Kameda and K. Maruoka, Angew. Chem., Int. Ed., 2002, 41, 4542 CrossRef CAS; (c) T. Ooi, M. Kameda, M. Taniguchi and K. Maruoka, J. Am. Chem. Soc., 2004, 126, 9685 CrossRef CAS PubMed; (d) R. Thayumanavan, F. Tanaka and C. F. Barbas III, Org. Lett., 2004, 6, 3541 CrossRef CAS PubMed; (e) L. Li, K. G. Klauber and D. Seidel, J. Am. Chem. Soc., 2008, 130, 12248 CrossRef CAS PubMed; (f) W.-B. Chen, Z.-J. Wu, J. Hu, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2011, 13, 2472 CrossRef CAS PubMed.
- For selected examples, see: (a) V. P. Vassilev, T. Uchiyama, T. Kajimoto and C.-H. Wong, Tetrahedron Lett., 1995, 36, 4081 CrossRef CAS; (b) T. Kimura, V. P. Vassilev, G.-J. Shen and C.-H. Wong, J. Am. Chem. Soc., 1997, 119, 11734 CrossRef CAS; (c) M. L. Gutierrez, X. Garrabou, E. Agosta, S. Servi, T. Parella, J. Joglar and P. Clapés, Chem.–Eur. J., 2008, 14, 4647 CrossRef CAS PubMed; (d) K. Hernandez, I. Zelen, G. Petrillo, I. Usón, C. M. Wandtke, J. Bujons, J. Joglar, T. Parella and P. Clapés, Angew. Chem., Int. Ed., 2015, 54, 3013–3017 CrossRef CAS PubMed.
- For selected examples, see: (a) B. Tao, G. Schlingloff and K. B. Sharpless, Tetrahedron Lett., 1998, 39, 2507 CrossRef CAS; (b) A. Morgan, C. E. Masse and J. S. Panek, Org. Lett., 1999, 1, 1949 CrossRef CAS; (c) H. Park, B. Cao and M. M. Joullié, J. Org. Chem., 2001, 66, 7223 CrossRef CAS.
- For selected examples, see: (a) R. Noyori, T. Ikeda, T. Ohkuma, M. Widhalm, M. Kitamura, H. Takaya, S. Akutagawa, N. Sayo, T. Saito, T. Taketomi and H. Kumobayashi, J. Am. Chem. Soc., 1989, 111, 9134 CrossRef CAS; (b) R. Kuwano, S. Okuda and Y. Ito, J. Org. Chem., 1998, 63, 3499 CrossRef CAS; (c) C. Mordant, P. Dünkelmann, V. Ratovelomanana-Vidal and J. P. Genet, Eur. J. Org. Chem., 2004, 3017 CrossRef CAS PubMed; (d) C. Mordant, P. Dünkelmann, V. Ratovelomanana-Vidal and J. P. Genet, Chem. Commun., 2004, 1296 RSC; (e) K. Makino, T. Goto, Y. Hiroki and Y. Hamada, Angew. Chem., Int. Ed., 2004, 43, 882 CrossRef CAS PubMed; (f) K. Makino, Y. Hiroki and Y. Hamada, J. Am. Chem. Soc., 2005, 127, 5784 CrossRef CAS PubMed.
- (a) B. M. Trost and X. Ariza, Angew. Chem., Int. Ed., 1997, 36, 2635 CrossRef CAS PubMed; (b) B. M. Trost and C. B. Lee, J. Am. Chem. Soc., 1998, 120, 6818 CrossRef CAS; (c) B. M. Trost and C. B. Lee, J. Am. Chem. Soc., 2001, 123, 12191 CrossRef CAS PubMed.
- M. Terada, H. Tanaka and K. Sorimachi, J. Am. Chem. Soc., 2009, 131, 3430 CrossRef CAS PubMed.
- For total synthesis of Mycestericin D, E, F and G, see: (a) K. Shibata, K. Shingu, V. P. Vassilev, K. Nishide, T. Fujita, M. Node, T. Kajimoto and C.-H. Wong, Tetrahedron Lett., 1996, 37, 2791 CrossRef CAS; (b) T. Fujita, N. Hamamichi, T. Matsuzaki, Y. Kitao, M. Kiuchi, M. Node and R. Hirose, Tetrahedron Lett., 1995, 36, 8599 CrossRef CAS; (c) K. Nishide, K. Shibata, T. Fujita, T. Kajimoto, C.-H. Wong and M. Node, Heterocycles, 2000, 52, 1191 CrossRef CAS; (d) Y. Iwabuchi, M. Furukawa, T. Esumi and S. Hatakeyama, Chem. Commun., 2001, 2030 RSC; (e) L. Berhal, S. Takechi, N. Kumagai and M. Shibasaki, Chem.–Eur. J., 2011, 17, 1915 CrossRef PubMed; (f) N. W. G. Fairhurst, M. F. Mahon, R. H. Munday and D. R. Carbery, Org. Lett., 2012, 14, 756 CrossRef CAS PubMed.
- Please see ESI† for details.
- M. Miyashita, A. Yoshikoshi and P. A. Grieco, J. Org. Chem., 1977, 42, 3772 CrossRef CAS.
- C. R. Strader, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2011, 74, 900 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization for new compounds are provided. See DOI: 10.1039/c5sc02116b |
| This journal is © The Royal Society of Chemistry 2015 |