
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protonation state of the Cu4S2 CuZ site in nitrous oxide reductase: redox dependence and insight into reactivity†
Esther M.
Johnston
 a,
Simone
Dell'Acqua
b,
Sofia R.
Pauleta
c,
Isabel
Moura
c and
Edward I.
Solomon
*a
a,
Simone
Dell'Acqua
b,
Sofia R.
Pauleta
c,
Isabel
Moura
c and
Edward I.
Solomon
*a
aDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: edward.solomon@stanford.edu
bDipartimento di Chimica, Università di Pavia, Via Taramelli 12, 27100 Pavia, Italy
cUCIBIO, REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal
First published on 3rd July 2015
Abstract
Spectroscopic and computational methods have been used to determine the protonation state of the edge sulfur ligand in the Cu4S2 CuZ form of the active site of nitrous oxide reductase (N2OR) in its 3CuICuII (1-hole) and 2CuI2CuII (2-hole) redox states. The EPR, absorption, and MCD spectra of 1-hole CuZ indicate that the unpaired spin in this site is evenly delocalized over CuI, CuII, and CuIV. 1-hole CuZ is shown to have a μ2-thiolate edge ligand from the observation of S–H bending modes in the resonance Raman spectrum at 450 and 492 cm−1 that have significant deuterium isotope shifts (−137 cm−1) and are not perturbed up to pH 10. 2-hole CuZ is characterized with absorption and resonance Raman spectroscopies as having two Cu–S stretching vibrations that profile differently. DFT models of the 1-hole and 2-hole CuZ sites are correlated to these spectroscopic features to determine that 2-hole CuZ has a μ2-sulfide edge ligand at neutral pH. The slow two electron (+1 proton) reduction of N2O by 1-hole CuZ is discussed and the possibility of a reaction between 2-hole CuZ and O2 is considered.
1. Introduction
The main reductive part of the nitrogen cycle, known as bacterial denitrification, is performed by soil and marine bacteria as a means of anaerobic or microaerobic respiration. Denitrification involves the conversion of nitrate to dinitrogen via four successive reductive steps (NO3− → NO2− → NO → N2O → N2), each performed by a different metalloenzyme.1 The terminal product of denitrification can be either N2O or N2, depending on the regulatory control of the N2O reduction process and whether the bacterium involved contains the gene cluster for nitrous oxide reduction (the nos cluster; nosZ encodes the nitrous oxide reductase enzyme).2,3 The N2O reduction process and its regulation in vivo are of significant interest because N2O is a potent greenhouse gas, with a global warming potential 300× that of CO2,4,5 and depletes the ozone layer.6 Anthropogenic sources of environmental N2O, the majority of which is due to agricultural activity, is an increasing contribution to the global atmosphere.2 Soil studies have indicated that pH,7,8 temperature,9 acetylene,10 sulfide,11 and dioxygen12 all affect the production of N2O, but the molecular basis of these effects is still not known. A molecular understanding of nitrous oxide reduction and how this process is regulated could enable mitigation of N2O release from anthropogenic sources.5Nitrous oxide reductase contains two copper sites: a binuclear site known as CuA that functions as an electron transfer site, and an unusual tetranuclear copper sulfide cluster active site, where N2O binds and is reduced (Fig. 1). Two forms of this tetranuclear site have been structurally characterized. One, known as Cu*Z, has a μ4 sulfide ligand bridging all four coppers and a solvent derived ligand on an open edge (the CuI–CuIV edge) where N2O is proposed to bind (Fig. 1A).13 This edge ligand has previously been assigned as a bridging hydroxide ligand, due to the presence of a vibration in the resonance Raman spectrum of Cu*Z that shifts in H218O solvent at high pH and the absence of significant spectroscopic differences between Cu*Z at high and low pH.15 The other form of the cluster, known as CuZ, has an additional μ2 sulfur ligand bridging the CuI–CuIV edge (Fig. 1B).14 Whether the μ2 edge ligand in CuZ is a thiolate (SH−) or a sulfide (S2−) and how its protonation depends on the redox state of the cluster are not known. The Cu4S2 CuZ form of the cluster is dominantly isolated when N2OR is purified in the absence of oxygen16 or rapidly in the presence of oxygen,17 while the Cu4S Cu*Z form is isolated when the purification is performed aerobically or anaerobically from mutants in the accessory genes;16–18 however, all purifications typically yield enzyme with a mixture of the two sites.17,19 Which structural form of the cluster is responsible for N2O reduction in vivo is a matter of some debate.20–22 As isolated, neither N2OR containing a high percentage of CuZ nor N2OR containing a high percentage of Cu*Z shows high enough specific activity in steady-state assays to be consistent with N2OR activity in whole cells.17,23 N2OR containing CuZ can be activated by prolonged dialysis against base,23 while N2OR that contains Cu*Z can be reductively activated by preincubation with methyl viologen, which reduces Cu*Z to the active fully reduced (4CuI) redox state.24,25 After activation, both CuZ and Cu*Z show specific activities consistent with whole cell N2OR activity.20,23 However, it has recently been shown that the Cu*Z site in its fully reduced redox state is the form of the cluster that is responsible for the rapid N2O reduction in steady state assays with methyl viologen, based on its rapid single turnover reaction with N2O. Alternatively, in single turnover studies CuZ in its 1-hole redox state reduces N2O but at a rate too slow to be catalytically relevant (10−6 that of the fully reduced state of Cu*Z).26 Thus, the physiological role of the CuZ site in nitrous oxide reduction and whether it participates in N2O reduction in vivo is unknown.
 | ||
| Fig. 1 The two forms of the tetranuclear copper sulfide active site of nitrous oxide reductase. (A) Cu*Z in PdN2OR isolated aerobically (PDB ID 1FWX).13 (B) CuZ in PsN2OR isolated anaerobically (PDB ID 3SBP).14 | ||
The CuZ site in nitrous oxide reductase has been extensively studied in N2OR isolated from Pseudomonas stutzeri (PsN2OR)27,28 and Paracoccus pantotrophus (PpN2OR).19,29 In the latter enzyme, CuZ has been shown to access two redox states, the resting 2CuI2CuII (2-hole) redox state, and a 1 electron reduced 3CuICuII (1-hole) redox state (Eo = +60 mV).19 Both redox states of CuZ have previously been studied using EPR, absorption, MCD, and resonance Raman spectroscopies.19,28,30–33 However, these studies were performed before the elucidation of the presence of a second sulfur in the CuZ cluster, and so yielded limited direct insight into the cluster and the protonation state of the edge sulfur. Additionally, the previous studies were performed in the presence of background spectroscopic features from ∼30% Cu*Z, which complicates the analysis.19,29 These limitations lead to the conclusion that CuZ and Cu*Z were very similar and perhaps differed only in the second sphere.28 These results are now extended and correlated to the structural insight that CuZ contains an additional inorganic sulfur edge ligand.14 An understanding of the protonation state, electronic structure, and potential reactivity of the CuZ site is necessary to gain insight into its reactivity and role in vivo.
This study uses EPR, absorption, MCD and resonance Raman spectroscopies coupled with DFT calculations to determine the protonation state of the edge sulfur ligand in the 1-hole and 2-hole redox states of CuZ in Marinobacter hydrocarbonoclasticus N2OR (MhN2OR) and to define the electronic structures of these states. This leads to insight into the nature of the reactivity of the 1-hole and 2-hole states of CuZ and the origin of the spectroscopic similarity between 1-hole CuZ and 1-hole Cu*Z, despite significant differences in edge ligation in the two sites.
2. Methodology
2.1 Summary of experimental methodology
Full experimental methodology and computational details can be found in the ESI,† while a summary is presented here. Nitrous oxide reductase (N2OR) was isolated from Marinobacter hydrocarbonoclasticus 617 (formerly Pseudomonas nautica) grown under microaerobic conditions in the presence of nitrate after two aerobic chromatographic steps without added reductant, as described previously.17 These purification conditions were shown to maximize the amount of Cu4S2 CuZ content relative to Cu4S Cu*Z in the purified enzyme. Samples containing larger amounts of Cu*Z were purified in parallel with three chromatographic purification steps from a batch of cells grown under anaerobic conditions in the presence of nitrate, and that had been stored at −80 °C for a long period.17,26 Both MhN2OR samples showed copper quantitation results consistent with full occupancy of the CuA and CuZ/Cu*Z sites (6.4 ± 0.2 and 6.2 ± 0.7 respectively). The percentage of CuZversus Cu*Z in the samples used for this study was determined by EPR spin quantitation (Fig. S1†). Samples purified with high amounts of CuZ contained 60 ± 10% CuZ, while samples purified to obtain more Cu*Z contained 10 ± 10% CuZ. Spectroscopic samples of 1-hole and 2-hole CuZ were prepared in a glove box under N2 atmosphere. Samples of 1-hole CuZ were prepared from MhN2OR (60% CuZ and 40% Cu*Z) that had been incubated with 100 equivalents of reduced methyl viologen, with subsequent removal of the methyl viologen using a desalting column. Samples of 2-hole CuZ were prepared by reducing MhN2OR (60 ± 10% CuZ, 40 ± 10% Cu*Z) with 10 equivalents of sodium ascorbate, which reduces the CuA site rapidly and the 2-hole CuZ site very slowly, and spectra were collected within 1 hour so that minimal reduction of 2-hole CuZ was observed. In parallel, MhN2OR samples containing 90 ± 10% Cu*Z were reduced with 10 equivalents of sodium ascorbate to obtain the spectral features of 1-hole Cu*Z. For pH and deuteration studies, samples of 1-hole and 2-hole CuZ were buffer exchanged by centrifugation into different pH or pD buffers. Typical MhN2OR concentrations used for spectroscopic samples were 0.1–0.3 mM for absorption, MCD and EPR, and up to 0.5 mM for resonance Raman.2.2 Computational modeling
A computational model of CuZ was built from the atomic coordinates of the crystal structure of Pseudomonas stutzeri N2OR, the only known structure of the Cu4S2 cluster (PDB ID 3SBP, resolution 1.7 Å).14 The model included the Cu4S2 core and 7 ligating His residues, where the α carbon and distal nitrogen were constrained at their crystallographic positions. A computational model for Cu*Z with a hydroxide bridging ligand and identical α carbon and distal nitrogen constraints was constructed from the crystal structure of Paracoccus denitrificans N2OR (PdN2OR, PDB ID 1FWX).13 Calculations were performed using Gaussian 09 (version d01).34 Geometry optimizations were performed using the B3LYP functional, the TZVP basis set on all core atoms (Cu4S) and the ligating His nitrogens, and the SV basis set on all remaining atoms, and solvation was modeled with a PCM of 4.0. A larger basis set and different functionals were also explored, as described in the text. The optimized structures were then used for frequency, TD DFT, and single point calculations. To determine the relative energy of deprotonation (ΔΔE) of the edge SH− in the 2-hole versus 1-hole redox state, larger models were optimized that included two second sphere carboxylates, Asp127 and Asp240, which hydrogen bond to the His ligands of CuI and CuII. The energy of an internal proton transfer from the edge SH− to Asp127 was calculated for the 1-hole and 2-hole redox states and compared to obtain the ΔΔE.3 Results and analysis
3.1 Spectroscopy of 1-hole CuZ
Previous spectroscopic studies of CuZ, undertaken before identification of the presence of a second sulfur, were performed on samples of PpN2OR and PsN2OR that contained mixtures of the CuZ and Cu*Z sites (in a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio for PpN2OR) without a way to resolve the spectral features of the CuZ site from the mixture.19,28,29 Recently, it has been found that the two-sulfur CuZ site cannot be reduced by methyl viologen, which reduces both the CuA site and the Cu*Z form of the cluster.26 This provides an opportunity to cleanly resolve the spectral features of 1-hole CuZ by studying methyl viologen reduced samples after removal of the reductant. This approach allows correlation of the electronic structure of 1-hole CuZ, obtained from spectroscopy, with the recently determined Cu4S2 structure of the cluster, to determine the nature of the edge sulfur ligand in its 1-hole and resting 2-hole redox states.
:3 ratio for PpN2OR) without a way to resolve the spectral features of the CuZ site from the mixture.19,28,29 Recently, it has been found that the two-sulfur CuZ site cannot be reduced by methyl viologen, which reduces both the CuA site and the Cu*Z form of the cluster.26 This provides an opportunity to cleanly resolve the spectral features of 1-hole CuZ by studying methyl viologen reduced samples after removal of the reductant. This approach allows correlation of the electronic structure of 1-hole CuZ, obtained from spectroscopy, with the recently determined Cu4S2 structure of the cluster, to determine the nature of the edge sulfur ligand in its 1-hole and resting 2-hole redox states.
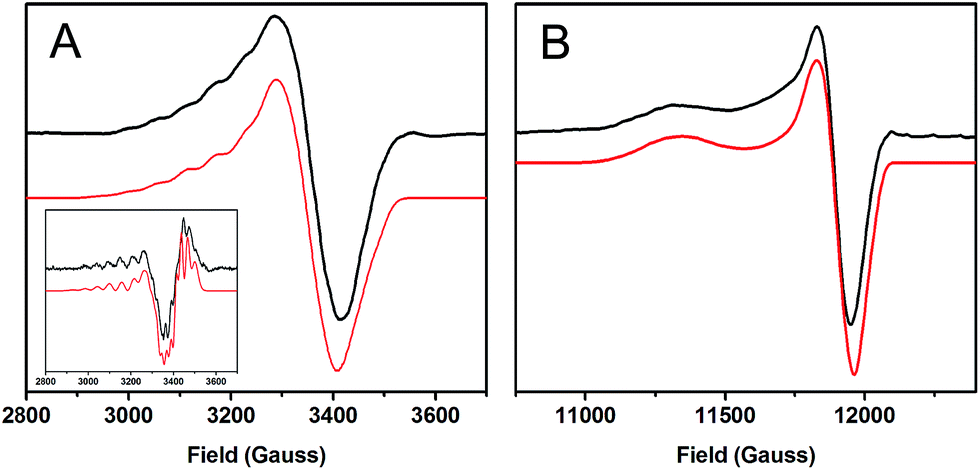 | ||
| Fig. 2 EPR spectra of 1-hole CuZ (black) with simulations (red). (A) X-band at 77 K, 9.6349 GHz. Inset: 2nd derivative of the X-band. (B) Q-band at 77 K, 34.082 GHz. | ||
| 1-hole CuZ | 1-hole Cu*Z | |
|---|---|---|
| g || | 2.152 | 2.160 |
| A || | 56 × 10−4 cm−1 | 61 × 10−4 cm−1 |
| 56 × 10−4 cm−1 | 23 × 10−4 cm−1 | |
| 56 × 10−4 cm−1 | ||
| g ┴ | 2.042 | 2.043 |
| A ┴ | 20 × 10−4 cm−1 | 25 × 10−4 cm−1 |
| 20 × 10−4 cm−1 | 20 × 10−4 cm−1 | |
| 20 × 10−4 cm−1 |
The A|| and A┴ values for 1-hole CuZ are similar in magnitude to those for Cu*Z, but fitting the hyperfine pattern requires three equivalent contributions rather than the ∼5:2 ratio of hyperfine values observed for Cu*Z (Table 1).36 This indicates that in the ground state of 1-hole CuZ the spin is distributed over three copper centers in dominantly dx2−y2 orbitals. The three coppers involved are likely CuI, CuII, and CuIV, since these copper centers are in the same plane as the two sulfur ligands and bonding with the strong donor μ4 sulfide and μ2 sulfur ligands should define a common x, y plane for these coppers, with the z axis of the local g tensor of each copper oriented perpendicular to the Cu3S2 plane. This is consistent with the axial nature of the g values and with the DFT calculations reported below.
600 cm−1 (ε ≈ 3000 M−1 cm−1), 1000 cm−1 lower than the absorption maximum of 1-hole Cu*Z (Fig. 3B). There are no additional low energy intense absorption features due to the edge sulfur. The low temperature absorption and MCD spectra can be simultaneously fit to yield a total of 11 transitions, which can be assigned by considering their energies and C0/D0 ratios, following ref. 15 (Table S1†). Comparison of the transition assignments and energies of the 1-hole CuZ and 1-hole Cu*Z sites reveals some key differences. While the absorption maximum of CuZ occurs at lower energy than that of Cu*Z, from MCD the three μ4S to Cu CT transitions, assigned in Cu*Z,35 occur at very similar energies in the two sites (bands 5, 6, and 7, numbering given in Fig. 3). The shift in the absorption maximum therefore arises from a different intensity pattern for these transitions, where in CuZ the lowest energy transition at 14600 cm−1 is the most intense (band 5) and the transition at 15600 cm−1 is weaker (band 6), but in Cu*Z this is reversed. The μ4S2− to Cu CTs in Cu*Z have previously been assigned as transitions from the three different 3p orbitals of the μ4S2− to the β LUMO of the cluster. From our current study of resting 1-hole Cu*Z and CuZ with different edge ligands,15,35 the β LUMO is delocalized in the plane that contains CuI, CuII, CuIV and the μ4S2−, with different amounts of spin distributed over CuI, CuII, and CuIV depending on the edge ligation. Two of the μ4S2− p orbitals are in the plane, oriented between CuI and CuIV (S px′) and between CuIV and CuII (S py′), while the third is perpendicular to the plane (S pz′). Scheme S1† reflects the orientation and simplified composition of these orbitals determined for 1-hole Cu*Z from DFT calculations. The CT intensities reflect the overlap of these three S p orbitals with the β LUMO. Since bands 5 and 6 show the highest intensity in the 1-hole forms of CuZ and Cu*Z, these must reflect charge transfer from the in-plane S px′ (band 6, dominant in Cu*Z due to higher overlap with CuI) and S py′ (band 5) orbitals. Bands 6 and 5 form a pseudo-A feature in the MCD spectrum (i.e. derivative-shaped) and thus must arise from two transitions with orthogonal transition moments that spin–orbit couple in a third, mutually perpendicular direction (i.e. Lz). Since band 6 arises from a transition to CuI (from its dominant intensity in Cu*Z), band 5 must reflect a transition to CuIV, since the CuI–S and CuIV–S bonds are close to perpendicular (96° from crystallography) while the CuI–S and CuII–S bonds are close to parallel (160°).13 The change in relative intensities of the μ4S2− to Cu CT transitions in CuZ relative to Cu*Z, where band 6 decreases in intensity while band 5 increases in intensity, thus indicates that there is less spin on CuI and more spin on CuIV in 1-hole CuZ relative to 1-hole Cu*Z. This is consistent with the EPR hyperfine values, which suggest that the spin in CuZ is delocalized 1:1:1 over CuI, CuII, and CuIV, while from ref. 35 in Cu*Z the spin is delocalized ∼5:2 over CuI and CuIV. Additionally, in Cu*Z, a band at 18000 cm−1 (band 8) was assigned as a high energy d–d transition due to its high C0/D0 ratio; this was assigned as a localized dxy → dx2−y2 transition on CuI, where most of the 1-hole is localized.35 No equivalent high energy d–d transition is observed in the MCD spectrum of CuZ. The lower energy of the d–d transitions in CuZ relative to Cu*Z is likely due to the decreased spin on CuI, the only four coordinate site, relative to CuII and CuIV, which are both 3 coordinate and have a weaker ligand field.
 | ||
| Fig. 3 Low temperature absorption and MCD spectra of (A) 1-hole CuZ, 10 K absorption, 5 K and 7 T MCD. (B) 1-hole Cu*Z, 5 K absorption, 5 K and 7 T MCD (adapted from ref. 15). | ||
 | ||
| Fig. 4 (A) Resonance Raman spectrum of 1-hole CuZ at 77 K, excitation energy 697 nm. (B) Excitation profile of the 203, 378, and 492 cm−1 vibrations. (C) H/D isotope shift of the vibrations of 1-hole CuZ, performed in pH or pD 7.8, 100 mM phosphate, excitation energy 676 nm. (D) Comparison of SH bending vibrations at pH/pD 7.8 (green) and pH/pD 10 (blue). | ||
3.2 Spectroscopy of 2-hole CuZ
300 cm−1 (ε ≈ 10000 M−1 cm−1) with a weaker low energy shoulder, consistent with absorption spectra previously reported for ascorbate reduced samples containing high amounts of 2-hole CuZ.19,32 The low temperature absorption spectrum of 2-hole CuZ (Fig. S5†) resolves this absorption maximum into three distinct absorption bands. Simulation of the room temperature absorption spectrum with transition energies derived from the low temperature spectrum distinguishes five transitions, all with absorption intensities higher than 1000 M−1 cm−1, sufficiently intense to be S to Cu CT transitions from the μ4S2− or the μ2S ligand (Fig. 5). The two most intense transitions (bands 2 and 3) are either from different ligands (μ2S and μ4S2−) or from the same ligand to two different acceptor orbitals (the α and β holes of the broken symmetry singlet ground state). Based on the correlation of resonance Raman excitation profiles of the vibrations of 2-hole CuZ to DFT calculations (vide infra), the assignment of the two transitions as CT transitions from the μ4S2− to two different holes is preferred.
 | ||
| Fig. 5 Absorption spectrum of 2-hole CuZ at room temperature, obtained after ascorbate reduction of CuA and subtraction of spectral contribution of 1-hole Cu*Z. | ||
300 cm−1 (Fig. 6A). Two vibrations are enhanced at 350 and 405 cm−1. The 34S isotope shifts of these vibrations have been previously reported to be −5.6 and −5.8 cm−1, respectively, indicating that they are Cu–S stretches.28 In contrast to 1-hole CuZ, no higher energy S–H bending vibration is observed (up to 800 cm−1). The excitation profile of the Cu–S stretching vibrations shows that they are enhanced differently in the most intense absorption bands 2 and 3 (Fig. 6B). The lower energy vibration at 350 cm−1 is enhanced in both transitions, while the higher energy vibration at 405 cm−1 is dominantly enhanced in the lower energy transition (band 2) and only weakly enhanced in band 3. This difference in profiling behavior is consistent with the Cu–S vibrations obtained computationally for a Cu4S2 cluster with a μ2S2− and μ4S2− and with the predicted enhancements of key vibrations in transitions from the μ4S2− to the α and β holes (see 3.3).
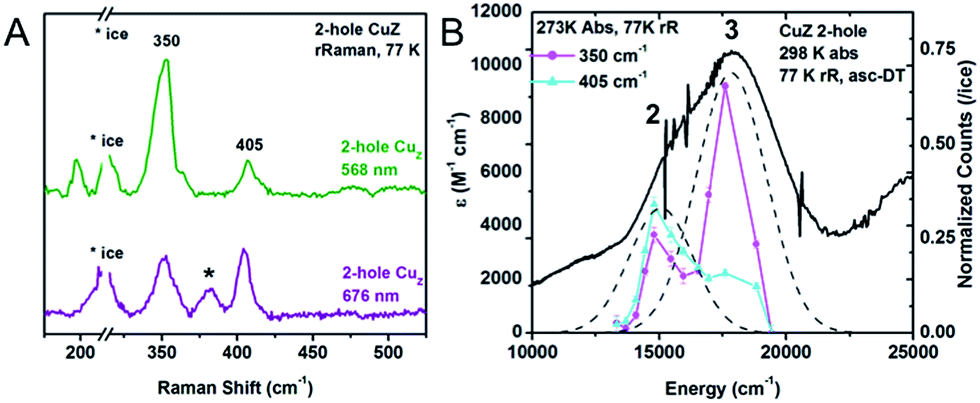 | ||
| Fig. 6 (A) Resonance Raman spectra of 2-hole CuZ at 77 K and two excitation energies, 568 nm (green) and 676 nm (purple). Starred vibration is due to 1-hole Cu*Z. (B) Excitation profiles of the 350 and 405 cm−1 vibrations overlaid with the room temperature absorption spectrum. | ||
The resonance Raman spectrum 2-hole CuZ shows no significant shift in the energies of the 350 and 405 cm−1 vibrations between pD 6 and pD 10 (Fig. S6†). This suggests that the edge ligand has a pKa either lower than 5.5 or higher than 10.5. A pKa higher than 10.5 in the 2-hole redox state is not consistent with observed pKa of 11–12 for 1-hole CuZ, as the increased charge of the 2-hole state will lead to a lower pKa relative to the 1-hole redox state. The possibility of a pKa less than 5.5 for 2-hole CuZ is evaluated computationally below.
3.3 Calculations
 | ||
| Fig. 7 Computational models of (A) 1-hole CuZ and (B) 1-hole Cu*Z (B3LYP, TZVP on Cu, S, and ligating N atoms, and SV on all remaining atoms, PCM of 4.0). | ||
The 1-hole model with an SH− edge ligand reproduces the key spectral features observed for the 1-hole CuZ site. The Mulliken atomic spin distribution of the cluster with an SH− edge ligand is delocalized over CuI, CuII, and CuIV in a 2:1:1 ratio. In going from Cu*Z to CuZ the calculated spin on CuI changes from 26% to 17% (Table 2), which is consistent with the decrease in intensity of band 6 observed in the absorption and MCD data for 1-hole CuZ and leads to a more equal distribution of spin over CuI, CuII, and CuIV, consistent with the EPR hyperfine values. The LUMO of the CuZ model contains dx2−y2 character on CuI, CuII, and CuIV, which are aligned, consistent with the ground state predicted from the EPR g values (Fig. S7†). It also contains significant antibonding μ4S2−and μ2SH− character, explaining why Cu–S stretching vibrations of both the μ4S2− and μ2SH− are enhanced in the charge transfer transitions to this acceptor orbital. Additionally, the computational model predicts the CuZ site to be more covalent than the Cu*Z site, with 10% less Cu character in the ground state wavefunction, reflecting delocalization of the spin from CuI onto the edge SH− ligand. The low g|| value for 1-hole CuZ is also predicted by the computational model (Table S7†). However, in contrast to experiment, the calculated g|| values for the CuZ and Cu*Z models differ, with a higher calculated g|| value for Cu*Z than that observed experimentally. This suggests that the calculated model of Cu*Z does not accurately predict the ligand field on CuI that leads to the higher energy dxy → dx2−y2 transition observed experimentally.
| Edge ligand | Mulliken atomic spin density | |||||
|---|---|---|---|---|---|---|
| CuI | CuII | CuIII | CuIV | μ4S2− | μ2L− | |
| SH− bridge | 0.17 | 0.11 | 0.06 | 0.10 | 0.34 | 0.16 |
| OH− bridge | 0.26 | 0.09 | 0.04 | 0.13 | 0.31 | 0.10 |
The Cu–S stretching vibrations and S–H bending vibrations for the 1-hole SH− model of CuZ are given in Table S8 and Fig. S8.† The model predicts two S–H bending modes at 426 and 461 cm−1 with H/D isotope shifts of −125 cm−1 and −123 cm−1, respectively, similar to the vibrations observed experimentally at 450 and 492 cm−1 (with a shift of −137 cm−1 for the 492 cm−1 vibration; the 450 cm−1 vibration cannot be observed after deuteration due to overlap with the ice scattering peak). Equivalent O–H bends are predicted for the OH bridged Cu*Z model at higher energies, but these are not experimentally observed. The CuZ model also predicts the presence of a low energy Cu–S stretching vibration of the μ2SH− (178 cm−1, observed at 203 cm−1 experimentally) and both models show similar energies for the Cu–μ4S stretching vibrations. The absolute energies of the Cu–S stretching vibrations for both the μ4 sulfide and μ2 thiolate are underestimated, as has been found for computational models of the Cu*Z site.15,35 The TD DFT calculated absorption spectrum for the CuZ model is also very similar to the calculated absorption spectrum for the Cu*Z model both with B3LYP and with the functional B98, which has been shown to predict the experimental absorption spectrum of a Cu3S2 model complex reasonably well.38 Interestingly, neither the experimental absorption spectrum nor the TD-DFT calculation predicts an intense low energy charge transfer transition from the μ2SH− ligand (Fig. S9†). While some weak transitions predicted computationally at lower energy than the μ4S2− to Cu CT transitions have μ2SH− to Cu CT character, they are predicted to lack intensity and are thus difficult to distinguish from the Cu d to d transitions that are also observed in this energy region.
Thus, a computational model of the tetranuclear copper cluster with an SH− edge ligand bridging CuI and CuIV provides a good structural model of 1-hole CuZ that reproduces its key spectral features. This spectroscopically calibrated model was then extended to the 2-hole redox state of the CuZ site, for which less experimental data are accessible.
| Mulliken spin density | |||||||
|---|---|---|---|---|---|---|---|
| μ2L | CuI | CuII | CuIII | CuIV | μ4S2− | ||
| 2-Hole SH−S = 0 | α LUMO | 0.16 | 0.37 | 0.06 | 0.06 | 0.04 | 0.20 |
| β LUMO | 0.17 | 0.03 | 0.17 | 0.09 | 0.16 | 0.30 | |
| 2-Hole S2−S = 0 | α LUMO | 0.33 | 0.22 | 0.06 | 0.04 | 0.07 | 0.23 |
| β LUMO | 0.37 | 0.05 | 0.06 | 0.05 | 0.14 | 0.27 | |
 | ||
| Fig. 8 Frontier molecular orbitals of 2-hole CuZ (two sulfide model). | ||
The energy of deprotonation calculated from the 2-hole models was compared to the calculated energy of deprotonation of the 1-hole SH− model, where the experimentally estimated pKa of the edge thiolate is 11–12 (vide supra). Examination of the energy required to deprotonate the 1-hole and 2-hole SH− models shows that deprotonation of the 1-hole is not energetically favored (ΔE = 26 kcal mol−1, relative to an energy of −268 kcal mol−1 for a solvated proton)39 while deprotonation of the 2-hole is favorable (ΔE = −9 kcal mol−1). However, the two models have different charges (+2/+1 and +3/+2 for protonated and deprotonated 1-hole and 2-hole models, respectively) and this will significantly affect the relative energies of deprotonation. To minimize the charge effect, the computational models were expanded to include two second sphere Asp residues near the CuZ site, such that the 1-hole CuZ model with an edge thiolate is neutral and the 2-hole CuZ Cu4S(SH) model has a +1 charge. The proton transfer was performed internally to one of the Asp residues, so the total charge of the model does not change upon deprotonation (Fig. S10†). The ΔΔE for deprotonation of the 2-hole edge thiolate relative to the 1-hole species is calculated to be −25 kcal mol−1 (with a dielectric of 4.0). This value is dependent on the dielectric (Fig. S11†) and, at high dielectric values, converges to a ΔΔE of −12 kcal mol−1. To estimate the difference in pKa between 1-hole and 2-hole CuZ, the ΔΔG was estimated from the ΔΔE using frequency calculations for structures with identical fixed atom constraints and thus the same number and magnitude of imaginary frequencies (these ΔG corrections vary by only 0.3 kcal mol−1 between the protonated and deprotonated 1-hole structures). This gives a ΔΔG of −12 kcal mol−1 for deprotonation of 2-hole versus 1-hole CuZ, which corresponds to a ΔpKa of −9. Given the experimental pKa value of 11–12 for the edge thiolate in 1-hole CuZ, these calculations predict a pKa for a thiolate in 2-hole CuZ of 3 or less, consistent with the absence of a pH effect in resonance Raman of 2-hole CuZ at pD 6. This strongly suggests that 2-hole CuZ is a two sulfide cluster at neutral pH.
The calculated spectral features for the Cu4S2 2-hole CuZ model can be compared with those determined experimentally. The TD DFT predicted absorption spectrum (using both B3LYP and B98) is qualitatively similar to the experimental absorption spectrum, showing two intense absorption maxima with a higher energy shoulder (Fig. S12†). The predicted vibrations of the 2-hole Cu4S2 model are given in Table S10 and Fig. S13.† All of the calculated vibrations are shifted up in energy in comparison to those of the 1-hole SH− model, with the most significant energy differences observed for the sulfur edge vibrations, due to the short and highly covalent Cu–μ2S2− bonds in the 2-hole Cu4S2 cluster. In particular, the μ2S2−–CuI stretch now occurs at a similar energy to and mixes with vibrations of the μ4S2−, leading to symmetric and antisymmetric combinations of the μ2S2−–CuI and μ4S2−–CuI stretches (predicted at 312 and 309 cm−1, respectively, see ESI†). The symmetric combination is allowed in resonance Raman and will be enhanced in all transitions due to the high amount of μ2S2− character in both the α and β holes. This is a good candidate for the 350 cm−1 vibration observed experimentally that profiles in both intense absorption bands (see Fig. 6B). The highest energy core vibration of the 2-hole μ2S2− cluster is a symmetric CuII–μ4S2−–CuIV stretch predicted at 344 cm−1 which will be selectively enhanced in a transition to the β LUMO localized on CuIV (Fig. 8). A symmetric CuII–μ4S2−–CuIV stretch is also computationally predicted in the 1-hole SH− model at 320 cm−1 and the calculated shift in energy of this mode between the 1-hole and 2-hole models (+24 cm−1) is similar to the energy increase of the highest energy Cu–S stretches observed experimentally in 1-hole and 2-hole CuZ (378 and 405 cm−1 respectively, Δν of +27 cm−1). Thus, the 2-hole Cu4S2 model qualitatively predicts a high energy Cu–S vibration that will be selectively enhanced only in a transition to the β hole and a lower energy Cu–S vibration that will be enhanced in both intense transitions. This is consistent with the enhancement profiles of the two vibrations observed experimentally in Fig. 6B. This establishes that a μ2S2− bridge is energetically favored and consistent with the spectral features of 2-hole CuZ.
4. Discussion
A combination of spectroscopic methods and DFT calculations has been used to define the protonation state of the μ2 sulfur ligand on the CuI–CuIV edge in 1-hole and 2-hole CuZ. This leads to insight into the spectroscopic similarities between 1-hole CuZ and 1-hole Cu*Z, the redox reactivity of 1-hole CuZ in the slow 2 electron reduction of N2O, and the interconversion between CuZ and Cu*Z, the reactive form of the cluster for N2O reduction in vitro.4.1 Protonation states of 1-hole and 2-hole CuZ
The protonation state of the edge ligand in 1-hole CuZ has been directly determined by resonance Raman spectroscopy. Two high energy vibrations are enhanced in the most intense μ4S2− to Cu CT transition of 1-hole CuZ, at 450 and 492 cm−1, and have large isotope shifts upon solvent deuteration (−137 cm−1 for the 492 cm−1 mode). This is consistent with S–H bending modes, indicating that the μ2S ligand is a thiolate. The energy and solvent isotope shift of these S–H bending modes are as predicted by DFT calculations for a model with a μ2SH− bridging the CuI–CuIV edge. The EPR spectrum of 1-hole CuZ indicates a ground state in which the spin is delocalized over 3 coppers in dominantly dx2−y2 orbitals. The absorption and MCD spectra show three μ4S2− to Cu charge transfer transitions that have very similar energies to those observed for 1-hole Cu*Z (which has a hydroxide bridged CuI–CuIV edge) but a different intensity pattern, consistent with a change in spin distribution in the cluster from dominantly on CuI in 1-hole Cu*Z to more evenly delocalized over CuI, CuII, and CuIV. This ground state spin distribution is consistent with that predicted from DFT calculations for the μ2SH−. Based on the absence of a pH effect in 1-hole CuZ up to a pH of 10, the pKa of the edge thiolate in 1-hole CuZ is estimated to be 11–12.The 2-hole state of CuZ was also spectroscopically defined, but no direct spectroscopic evidence for the protonation state of the edge ligand was obtained. DFT calculations of the deprotonation of a μ2SH− ligand in 2-hole CuZ relative to 1-hole CuZ were used to determine that 2-hole CuZ likely has a sulfide edge ligand. Deprotonation of a μ2SH− ligand in the 2-hole redox state is at least 12 kcal mol−1 more favorable than in the 1-hole redox state, after accounting for charge and dielectric effects. This yields a calculated pKa for a μ2SH− ligand in 2-hole CuZ of 3 or less, which strongly suggests that 2-hole CuZ has an edge sulfide ligand at physiological pH. The calculated spectroscopic properties of a model of 2-hole CuZ with a μ2S2− ligand are also consistent with those observed experimentally.
4.2 Similarities between 1-hole CuZ and 1-hole Cu*Z
It has previously been observed that the spectral features of 1-hole CuZ are rather similar to those of 1-hole Cu*Z, despite the change in the nature of the edge ligand from a thiolate to a hydroxide.19,28 The spectroscopic similarities between 1-hole CuZ and 1-hole Cu*Z reflect similar bonding interactions between the μ4S2− and the in plane coppers (CuI, CuII, and CuIV) which are not significantly perturbed by the nature of edge ligand. This results in similar transition energies in the absorption and MCD spectra, as the dominant transitions are due to μ4S2− to Cu charge transfer, and a similar intense core Cu–μ4S2− stretching mode in the resonance Raman spectrum, observed at 378 cm−1 in both sites. Small quantitative differences in the EPR hyperfine values and transition absorption and MCD intensities between the two sites arise from a perturbation of the spin density distribution of the cluster in 1-hole CuZ, where the more covalent μ2SH− leads to delocalization of the spin on CuI (dominant in Cu*Z) onto the edge SH−. Despite the higher covalency of the CuZ site, the g values in the EPR spectra are similar for CuZ and Cu*Z, as the localization of spin on the four coordinate CuI in 1-hole Cu*Z leads to higher energy d–d transitions, opposing the decreased covalency, leading to the net low g values also observed experimentally for CuZ. The difference in edge ligation in the two sites is observed primarily in the resonance Raman enhanced vibrations, where a low energy Cu–μ2SH− stretch at 203 cm−1 and higher energy S–H bending modes at 450 and 492 cm−1 are additionally enhanced in the dominant μ4S2− to Cu CT transition in CuZ but not Cu*Z, due to the more covalent interaction between the coppers and the edge SH−. Thus, the spectral similarities between 1-hole CuZ and 1-hole Cu*Z reflect similar bonding with the μ4S2− ligand and the distribution of spin over CuI, CuII, and CuIV. The differences in the vibrational spectra of the two sites reflect the μ2SH−versus μ2OH− edge ligation.4.3 Insights into reactivity of 1-hole and 2-hole CuZ
1-hole CuZ has been shown to perform a slow 2 electron reduction of N2O under single turnover conditions, with oxidation of both 1-hole CuZ and reduced CuA to generate resting 2-hole CuZ and 1 electron oxidized CuA.26 A structure of PsN2OR obtained from crystals pressurized with N2O shows a linear N2O molecule binding above the CuIV–CuII edge of the CuZ cluster (Fig. 9).14 The O of N2O is thought to be oriented towards a solvent filled cavity between CuZ and CuA, where there is a hydrogen bonding interaction with a localized solvent molecule, while the N end of the molecule is 2.8 Å from CuIV and 3.5 Å from the μ2SH− ligand. The spectroscopically and computationally defined protonation states for 1-hole and 2-hole CuZ indicate that the 1-hole CuZ site will donate both an electron and a proton upon oxidation, due to the significantly decreased pKa of the μ2SH− in the 2-hole redox state. The participation of a proton in the reduction of N2O by 1-hole CuZ avoids the thermodynamically unfavorable 1-electron reduction of N2O to N2O−, which is endergonic by 25.4 kcal mol−1, while the proton-coupled reduction of N2O to form N2 and a hydroxyl radical is exergonic by 7.4 kcal mol−1.40 However, a substantial barrier exists for this process due to the fact that N2O is not activated through direct interaction with CuZ (the rate of N2O reduction by 1-hole CuZ is 2 × 10−4 s−1).26 Thus the N2O may alternatively be oriented with the O atom pointed towards CuZ, where it can directly accept a proton and an electron from the μ2SH− to break the N–O bond and generate resting 2-hole CuZ, with transfer of the second electron from CuA. Since no intermediate is observed in the reduction of N2O by 1-hole CuZ,26 the hydroxide product that would be formed after N–O bond cleavage would likely be rapidly protonated and released into the nearby solvent-filled cavity, rather than coordinating to the CuZ cluster.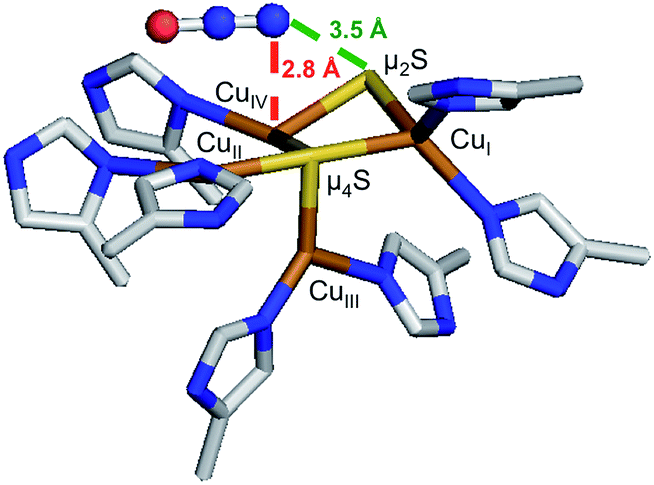
The 2-hole resting state of CuZ has been defined as having a highly covalent sulfide ligand bridging the CuI–CuIV edge. This resting species is potentially the starting point for the chemical conversion of CuZ to Cu*Z, the reactive form of the cluster for N2O reduction.26In vitro, the presence of O2 is thought to promote the conversion of CuZ to Cu*Z, as isolation of N2OR in the presence of O2 results in samples with a high proportion of resting 1-hole Cu*Z, while the resting 2-hole state of CuZ is obtained when the purification is performed in the absence of oxygen.16 DFT calculations on the μ2S2− model of 2-hole CuZ suggest that there are frontier molecular orbitals (FMOs) available to interact with O2. The α and β HOMOs of 2-hole CuZ are occupied μ2S2− orbitals with dominant S pz′ character (50% and 66% μ2S2− respectively, Fig. 8). This μ2S2− pz′ orbital is oriented perpendicular to the Cu3S2 plane, towards the solvent-filled cavity where N2O, and by analogy O2, would access the CuZ cluster. Based on these FMOs, reaction of the CuZ site with O2 would proceed via oxidation of the edge sulfide, rather than by a Cu-based oxidation. Since this is a four electron process, there will in principle also be electrons available from the sulfide for the reduction of the copper site, dependent on the nature of the oxidized sulfur product. However, it is unlikely that this is the mechanism involved in interconversion of CuZ and Cu*Zin vivo, since resting Cu*Z has been isolated under exclusion of oxygen conditions from anaerobically grown cells in bacterial strains with accessory genes knocked-out.41 Thus, the in vivo mechanism for interconversion of CuZ and Cu*Z, which is required to maintain N2OR in the reactive Cu*Z form, and the role of accessory proteins in this process, remain to be identified.
5. Conclusions
We have used a combination of spectroscopies and DFT calculations to determine the protonation states of the edge sulfur in the 1-hole and 2-hole redox states of CuZ. From resonance Raman spectroscopy, 1-hole CuZ has a μ2 thiolate ligand with a pKa of 11–12, due to the presence of S–H bending modes that are not perturbed up to pH 10. DFT calculations of a 1-hole cluster with a μ2SH− ligand reproduce the key spectral features of 1-hole CuZ. The computational modeling of the 2-hole CuZ site indicates that the edge ligand is a μ2S2− with a pKa of 3 or less, which is consistent with the absorption and resonance Raman features of 2-hole CuZ. The nature of this edge ligand has been used to obtain insight into the slow reduction of N2O by 1-hole CuZ and suggest how 2-hole CuZ might react with O2, a possible route for the conversion of CuZ to Cu*Zin vitro.Acknowledgements
The authors acknowledge support of this research from the National Institutes of Health (R01DK031450 from the National Institute of Diabetes and Digestive and Kidney Diseases to E.I.S.) and National funds by FCT (Fundação para a Ciência e a Tecnologia) under the project PTDC/QUI-BIQ/116481/2010 (I.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. No competing financial interests have been declared.Notes and references
- P. Tavares, A. S. Pereira, J. J. G. Moura and I. Moura, J. Inorg. Biochem., 2006, 100, 2087–2100 CrossRef CAS PubMed
.
- A. J. Thomson, G. Giannopoulos, J. Pretty, E. M. Baggs and D. J. Richardson, Philos. Trans. R. Soc., B, 2012, 367, 1157–1168 CrossRef CAS PubMed
- W. G. Zumft and P. M. H. Kroneck, Adv. Microb. Physiol., 2007, 52, 107–227 CrossRef CAS
- B. Bates, Z. W. Kundzewicz, S. Wu, N. Arnell, V. Burkett, P. Döll, D. Gwary, C. Hanson, B. Heij, B. Jiménez, G. Kaser, A. Kitoh, S. Kovats, P. Kumar, C. Magadza, D. Martino, L. J. Mata, M. Medany, K. Miller, T. Oki, B. Osman, J. Palutikof, T. Prowse, R. Pulwarty, J. Räisänen, J. Renwick, F. Tubiello, R. Wood, Z.-C. Zhao, J. Arblaster, R. Betts, A. Dai, C. Milly, L. Mortsch, L. Nurse, R. Payne, I. Pinskwar and T. Wilbanks, Technical Paper on Climate Change and Water, ed. I. Secretariat, 2008 Search PubMed.
- D. Richardson, H. Felgate, N. Watmough, A. Thomson and E. Baggs, Trends Biotechnol., 2009, 27, 388–397 CrossRef CAS PubMed
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed
- K. Butterbach-Bahl, E. M. Baggs, M. Dannenmann, R. Kiese and S. Zechmeister-Boltenstern, Philos. Trans. R. Soc., B, 2013, 368 Search PubMed
- L. Bergaust, Y. J. Mao, L. R. Bakken and A. Frostegard, Appl. Environ. Microbiol., 2010, 76, 8285–8285 CrossRef CAS PubMed
- L. R. Bakken, L. Bergaust, B. B. Liu and A. Frostegard, Philos. Trans. R. Soc., B, 2012, 367, 1226–1234 CrossRef CAS PubMed
- S. Saleh-Lakha, K. E. Shannon, S. L. Henderson, B. J. Zebarth, D. L. Burton, C. Goyer and J. T. Trevors, Appl. Environ. Microbiol., 2009, 75, 5082–5087 CrossRef CAS PubMed
- J. Sorensen, J. M. Tiedje and R. B. Firestone, Appl. Environ. Microbiol., 1980, 39, 105–108 CAS
- N. Morley and E. M. Baggs, Soil Biol. Biochem., 2010, 42, 1864–1871 CrossRef CAS PubMed
- K. Brown, K. Djinovic-Carugo, T. Haltia, I. Cabrito, M. Saraste, J. J. G. Moura, I. Moura, M. Tegoni and C. Cambillau, J. Biol. Chem., 2000, 275, 41133–41136 CrossRef CAS PubMed
- A. Pomowski, W. G. Zumft, P. M. H. Kroneck and O. Einsle, Nature, 2011, 477, 234–237 CrossRef CAS PubMed
- S. Ghosh, S. I. Gorelsky, S. D. George, J. M. Chan, I. Cabrito, D. M. Dooley, J. J. G. Moura, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 3955–3965 CrossRef CAS PubMed
- W. G. Zumft, C. L. Coyle and K. Frunzke, FEBS Lett., 1985, 183, 240–244 CrossRef CAS
- S. Dell'Acqua, S. R. Pauleta, J. J. G. Moura and I. Moura, Philos. Trans. R. Soc., B, 2012, 367, 1204–1212 CrossRef PubMed
- M. Prudencio, A. S. Pereira, P. Tavares, S. Besson, I. Cabrito, K. Brown, B. Samyn, B. Devreese, J. Van Beeumen, F. Rusnak, G. Fauque, J. J. G. Moura, M. Tegoni, C. Cambillau and I. Moura, Biochemistry, 2000, 39, 3899–3907 CrossRef CAS PubMed
- T. Rasmussen, B. C. Berks, J. N. Butt and A. J. Thomson, Biochem. J., 2002, 364, 807–815 CrossRef CAS PubMed
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed
- A. Wuest, L. Schneider, A. Pomowski, W. G. Zumft, P. M. H. Kroneck and O. Einsle, Biol. Chem., 2012, 393, 1067–1077 CAS
- S. R. Pauleta, S. Dell'Acqua and I. Moura, Coord. Chem. Rev., 2013, 257, 332–349 CrossRef CAS PubMed
- C. L. Coyle, W. G. Zumft, P. M. H. Kroneck, H. Korner and W. Jakob, Eur. J. Biochem., 1985, 153, 459–467 CrossRef CAS PubMed
- S. Ghosh, S. I. Gorelsky, P. Chen, I. Cabrito, J. J. G. Moura, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 15708–15709 CrossRef CAS PubMed
- J. M. Chan, J. A. Bollinger, C. L. Grewell and D. M. Dooley, J. Am. Chem. Soc., 2004, 126, 3030–3031 CrossRef CAS PubMed
- E. M. Johnston, S. Dell'Acqua, S. Ramos, S. R. Pauleta, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 614–617 CrossRef CAS PubMed
- T. Rasmussen, B. C. Berks, J. Sanders-Loehr, D. M. Dooley, W. G. Zumft and A. J. Thomson, Biochemistry, 2000, 39, 12753–12756 CrossRef CAS PubMed
- M. L. Alvarez, J. Y. Ai, W. Zumft, J. Sanders-Loehr and D. M. Dooley, J. Am. Chem. Soc., 2001, 123, 576–587 CrossRef CAS PubMed
- V. S. Oganesyan, T. Rasmussen, S. Fairhurst and A. J. Thomson, Dalton Trans., 2004, 996–1002 RSC
- T. Rasmussen, B. C. Berks and A. J. Thomson, J. Inorg. Biochem., 2001, 86, 393 Search PubMed
- J. A. Farrar, A. J. Thomson, M. R. Cheesman, D. M. Dooley and W. G. Zumft, FEBS Lett., 1991, 294, 11–15 CrossRef CAS
- J. A. Farrar, W. G. Zumft and A. J. Thomson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9891–9896 CrossRef CAS
- W. E. Antholine, P. M. H. Kroneck and W. G. Zumft, Mol. Phys., 1998, 95, 1247–1253 CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- P. Chen, I. Cabrito, J. J. G. Moura, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 10497–10507 CrossRef CAS PubMed
- P. Chen, S. D. George, I. Cabrito, W. E. Antholine, J. J. G. Moura, I. Moura, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 744–745 CrossRef CAS PubMed
- A. Pomowski, W. G. Zumft, P. M. H. Kroneck and O. Einsle, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 1541–1543 CrossRef CAS PubMed
- I. Bar-Nahum, A. K. Gupta, S. M. Huber, M. Z. Ertem, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2009, 131, 2812–2814 CrossRef CAS PubMed
- R. M. Noyes, J. Am. Chem. Soc., 1962, 84, 513–522 CrossRef CAS
- W. H. Koppenol, Free Radicals Biol. Med., 1991, 10, 85–87 CrossRef CAS
- P. Wunsch, H. Korner, F. Neese, R. J. M. van Spanning, P. M. H. Kroneck and W. G. Zumft, FEBS Lett., 2005, 579, 4605–4609 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and computational methodology, EPR quantification of the Cu*Z/CuZ ratio, 2nd derivative of the X band EPR of 1-hole CuZ, table of absorption band energies and assignments for 1-hole CuZ and 1-hole Cu*Z, resonance Raman spectrum and profile of 1-hole Cu*Z, pH dependence of the spectral features of 1-hole and 2-hole CuZ, low temperature absorption spectrum of 2-hole CuZ, and computational structures, vibrations, and TD-DFT absorption spectra for models of 1-hole and 2-hole CuZ (1-hole SH− and OH− and 2-hole SH− and S2− models). See DOI: 10.1039/c5sc02102b |
| This journal is © The Royal Society of Chemistry 2015 |