
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly enantioselective construction of tertiary thioethers and alcohols via phosphine-catalyzed asymmetric γ-addition reactions of 5H-thiazol-4-ones and 5H-oxazol-4-ones: scope and mechanistic understandings†
Tianli
Wang
a,
Zhaoyuan
Yu
b,
Ding Long
Hoon
a,
Kuo-Wei
Huang
c,
Yu
Lan
*b and
Yixin
Lu
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: chmlyx@nus.edu.sg
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400030, P. R. China. E-mail: lanyu@cqu.edu.cn
cDivision of Physical Sciences and Engineering, KAUST Catalysis Center, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
First published on 2nd June 2015
Abstract
Phosphine-catalyzed highly enantioselective γ-additions of 5H-thiazol-4-ones and 5H-oxazol-4-ones to allenoates have been developed for the first time. With the employment of amino-acid derived bifunctional phosphines, a wide range of substituted 5H-thiazol-4-one and 5H-oxazol-4-one derivatives bearing heteroatom (S or O)-containing tertiary chiral centers were constructed in high yields and excellent enantioselectivities. The reported method provides facile access to enantioenriched tertiary thioethers/alcohols. The mechanism of the γ-addition reaction was investigated by performing DFT calculations, and the hydrogen bonding interactions between the Brønsted acid moiety of the phosphine catalysts and the “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O” unit of the donor molecules were shown to be crucial in asymmetric induction.
O” unit of the donor molecules were shown to be crucial in asymmetric induction.
1. Introduction
Organosulfur compounds are important molecular architectures in synthetic organic chemistry1 and chemical biology.2 Therefore, it is not surprising that many methods have been devised in recent years to access optically active thiol derivatives. Common approaches include: conjugate addition of thiols,3 employment of sulfur-containing pronucleophiles,4 kinetic resolution of racemic thiols,5 and electrophilic sulfenylation reactions.6 In this context, the catalytic synthesis of chiral tertiary thiols is a challenging task and remains largely unexplored.7 Analogously, tertiary alcohol-containing structures are of great importance in the biological sciences and the pharmaceutical industry,8 and asymmetric synthesis of chiral tertiary alcohols is an intensively investigated area.9 A few selected biologically important tertiary thiols and alcohols are illustrated in Fig. 1.10 From the outset of this research, we aimed to devise a versatile catalytic approach that would allow us to access both tertiary thiols and alcohols in an enantioselective manner.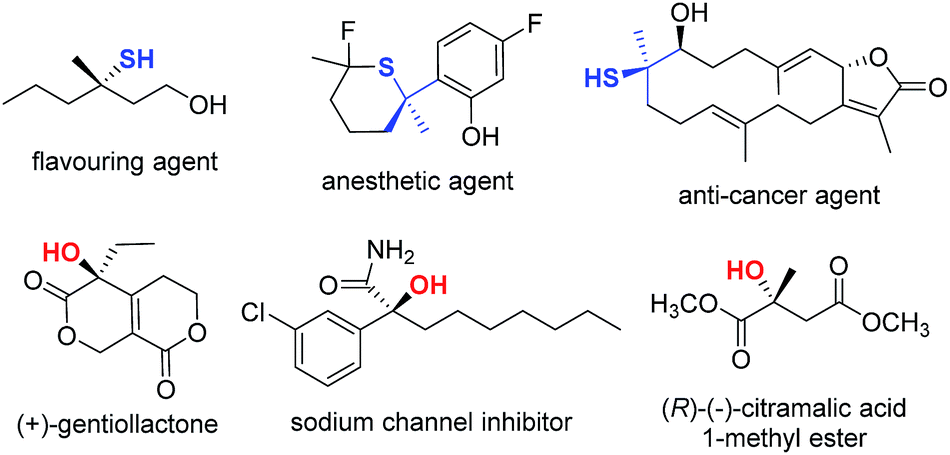 | ||
| Fig. 1 Representative bioactive tertiary thiol(ether)s and alcohols. | ||
To develop a method for the asymmetric synthesis of tertiary thiol molecules, it seems ideal to employ a readily accessible prochiral organosulfur compound. 5H-Thiazol-4-one and its derivatives, found to be useful in medicinal chemistry,11 are suitable donors; the acidic protons at the 5-position can be readily removed to facilitate their reactions with various electrophiles. Surprisingly, 5H-thiazol-4-ones have been rarely used in asymmetric synthesis, and there are only three examples to date describing their applications in asymmetric catalysis. Palomo and co-workers have reported the Brønsted base-catalyzed Michael additions of thiazolones to nitroalkenes12a and α′-silyloxy enone,12b for the synthesis of tertiary thiols. Very recently, Hartwig disclosed an Ir-catalyzed allylation of 5H-thiazol-4-ones to form enantioenriched tertiary thioethers.12c We envisioned that careful selection of the electrophilic reaction partners and catalytic systems, in combination with the utilization of pronucleophilic 5H-thiazol-4-ones, would lead to the discovery of novel synthetic methods for the asymmetric construction of tertiary thiols. Moreover, given the ready availability of the analogous 5H-oxazol-4-ones,13 we anticipated that the methodology developed for the thiol synthesis could be easily adapted to include α-oxygenated carboxylate surrogates, thus allowing facile preparation of chiral tertiary alcohols as well.
Our group has been actively investigating asymmetric phosphine catalysis14 in the past few years. We designed a series of amino acid-based bifunctional phosphine catalysts, and demonstrated their applications in a wide range of asymmetric transformations, including: (aza)-MBH reactions,15 [3 + 2], [4 + 2], and [4 + 1] annulations,16 allylic alkylations,17 and Michael additions.18 Very recently, we disclosed the utilization of 2,3-butadienoates in phosphine-catalyzed enantioselective γ-addition reactions.19,20 To further expand the range of phosphine-mediated asymmetric reactions, we envisaged that 5H-thiazol-4-ones and 5H-oxazol-4-ones could serve as valuable donors in phosphine-catalyzed γ-addition reactions to allenoates (Scheme 1). The chiral heteroatom-containing adducts formed have masked functionalities, and can be manipulated easily to give tertiary thiols/thioethers and alcohols.
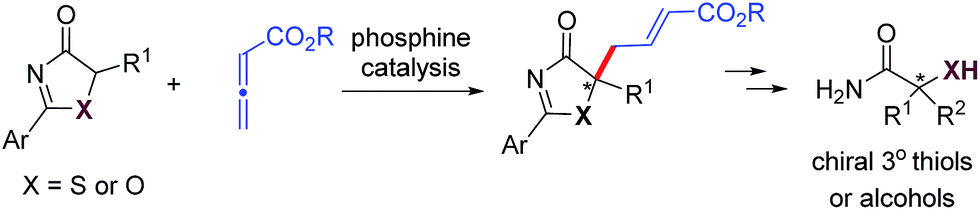 | ||
| Scheme 1 Construction of tertiary thiols/alcohols via phosphine-catalyzed γ-additions of 5H-thia(oxa)zol-4-ones. | ||
In this article, we disclose the first utilization of 5H-thiazol-4-ones and 5H-oxazol-4-ones in phosphine-catalyzed asymmetric γ-addition reactions, and the products can be readily converted to optically enriched tertiary thioethers and alcohols. In addition, we have also carried out DFT calculations to gain insights into the reaction mechanism and understand the origin of the stereochemical outcome of the reaction.
2. Results and discussion
2.1 Phosphine-catalyzed enantioselective γ-addition of 5H-thiazol-4-ones
In the past few years, amino acid-based bifunctional phosphines have been shown to be very powerful in phosphine catalysis. In this study, readily available L-valine and L-threonine were chosen as the starting chiral skeletons for the preparation of the phosphine catalysts. By installing different hydrogen bond donating groups and introducing various O-silyl protecting groups, we prepared a wide range of amino acid-derived bifunctional phosphines (Scheme 2), which were used for subsequent studies.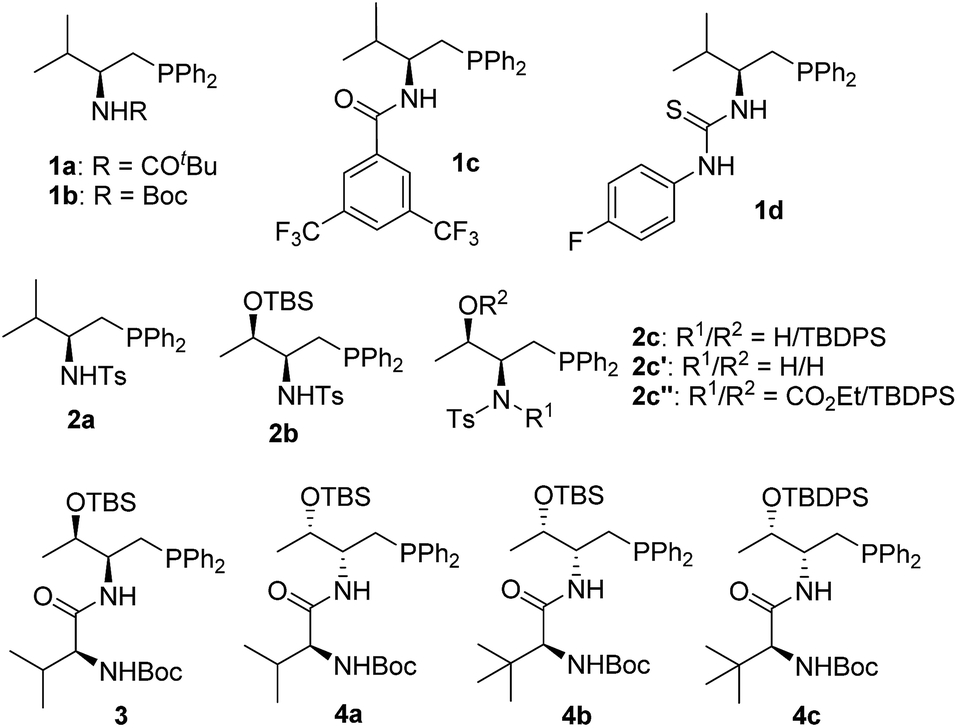 | ||
| Scheme 2 Phosphine catalysts investigated. | ||
We began our investigations by choosing 5-methyl-2-phenylthiazol-4(5H)-one 5a and allenoate 6c as substrates to evaluate the catalytic effects of the phosphine catalysts for the projected γ-addition (Table 1). To our delight, all the bifunctional phosphines could effectively promote the reaction. Among all the L-valine-derived phosphines, sulfonamide–phosphine 2a was found to be the most efficient (entries 1–5). L-Threonine-derived phosphine sulfonamide catalysts (2b & 2c) were then employed, and the enantioselectivity of the reaction could be improved to 89% ee (entries 6 and 7). The dipeptide phosphines were found to be less effective (entries 8–11).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Time (h) | Yieldb (%) | eec (%) |
| a Reactions were performed with 5a (0.1 mmol), 6c (0.12 mmol) and the catalyst (0.01 mmol) in toluene (1.0 mL) at room temperature. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. | ||||
| 1 | 1a | 12 | 86 | 10 |
| 2 | 1b | 12 | 90 | 62 |
| 3 | 1c | 12 | 86 | 34 |
| 4 | 1d | 12 | 92 | 34 |
| 5 | 2a | 12 | 91 | 66 |
| 6 | 2b | 12 | 90 | 78 |
| 7 | 2c | 12 | 95 | 89 |
| 8 | 3 | 12 | 88 | 47 |
| 9 | 4a | 12 | 92 | −57 |
| 10 | 4b | 12 | 88 | −65 |
| 11 | 4c | 12 | 92 | −68 |
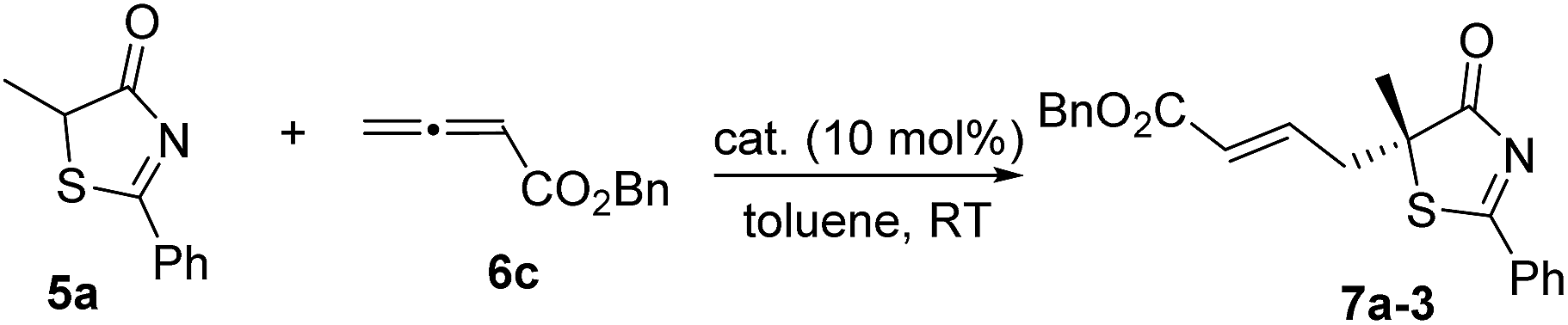
Subsequently, we further optimized the reaction conditions by varying the ester moiety in the allenoate structure (Table 2, entries 1–8). Among all the allenoates examined, the dibenzosuberyl ester proved to be the best, and the ee value of the reaction could be further improved to 91% (entry 6). Furthermore, solvent screening revealed that diethyl ether was the optimal solvent, and the desired product was obtained in 97% yield and with 95% ee under the optimized conditions (Table 2, entries 9–12). The use of different molecular sieves as additives did not result in a further improvement in enantioselectivity (entries 13–15). In addition, lowering the reaction temperature resulted in a significant decrease in reactivity coupled with reduced enantioselectivity (entry 16).
|
|
||||
|---|---|---|---|---|
| Entry | R/6 | Solvent | Yieldb (%) | eec (%) |
| a Reactions were performed with 5a (0.1 mmol), 6 (0.12 mmol) and 2c (0.01 mmol) in the solvent specified (1.0 mL) at room temperature for 12 h. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. d The reaction was stirred for 15 h. e 3 Å-MS were added. f 4 Å-MS were added. g 5 Å-MS were added. h The reaction was stirred at 0 °C for 36 h. | ||||
| 1 | Et/6a | Toluene | 95 | 87 |
| 2 | t Bu/6b | Toluene | 95 | 88 |
| 3 | Bn/6c | Toluene | 95 | 89 |
| 4 | CHPh2/6d | Toluene | 86 | 90 |
| 5 | 6e | Toluene | 88 | 70 |
| 6 | 6f | Toluene | 96 | 91 |
| 7 | 6g | Toluene | 93 | 87 |
| 8 | Ph/6h | Toluene | 91 | 57 |
| 9 | 6f | Xylene | 95 | 93 |
| 10 | 6f | Et2O | 97 | 95 |
| 11 | 6f | CHCl3 | 94 | 83 |
| 12d | 6f | CH2Cl2 | 87 | 92 |
| 13e | 6f | Et2O | 97 | 94 |
| 14f | 6f | Et2O | 97 | 94 |
| 15g | 6f | Et2O | 96 | 94 |
| 16h | 6f | Et2O | 86 | 90 |
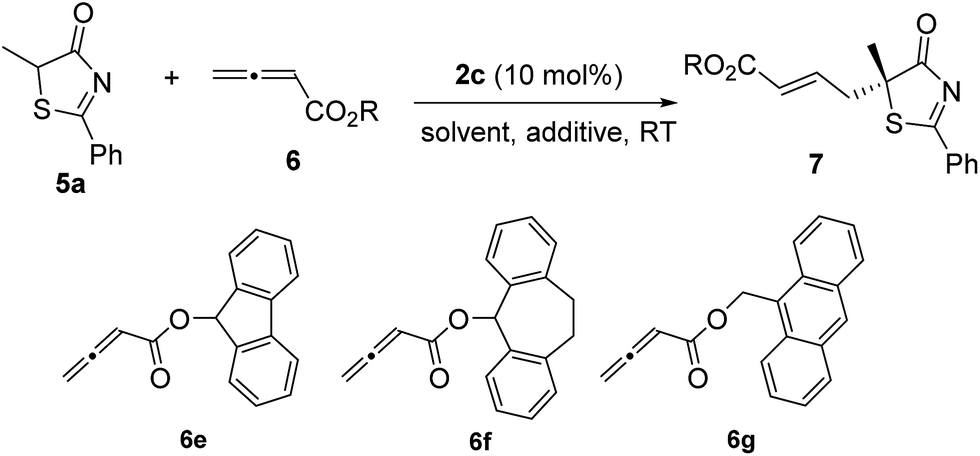
Having established the optimal reaction conditions, the substrate scope for the γ-addition of thiazolones to allenoates was then evaluated (Table 3). A wide range of 5-alkyl substituted 5H-thiazol-4-ones could be employed, the reaction was insensitive to the length of the alkyl chain, and both linear and branched alkyl groups were well tolerated (entries 1–10). In addition, benzyl and 2-(naphthalen-2-yl)-substituted thiazolones also proved to be suitable substrates (entries 11 and 12).
|
|
||||
|---|---|---|---|---|
| Entry | Ar/R1 | Product | Yieldb (%) | eec (%) |
| a Reactions were performed with 5 (0.1 mmol), 6f (0.12 mmol) and 2c (0.01 mmol) in Et2O (1.0 mL) at room temperature for 12–15 h. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. | ||||
| 1 | Ph/Me | 7a | 97 | 95 |
| 2 | Ph/Et | 7b | 95 | 94 |
| 3 | Ph/n-Pr | 7c | 97 | 94 |
| 4 | Ph/i-Pr | 7d | 92 | 92 |
| 5 | Ph/n-Bu | 7e | 94 | 94 |
| 6 | Ph/i-Bu | 7f | 89 | 88 |
| 7 | Ph/n-C6H13 | 7g | 96 | 94 |
| 8 | Ph/CH(CH2)5 | 7h | 95 | 93 |
| 9 | Ph/(CH2)2SCH3 | 7i | 93 | 90 |
| 10 | Ph/n-C10H21 | 7j | 86 | 93 |
| 11 | Ph/Bn | 7k | 90 | 92 |
| 12 | 2-Nap/Me | 7l | 96 | 89 |
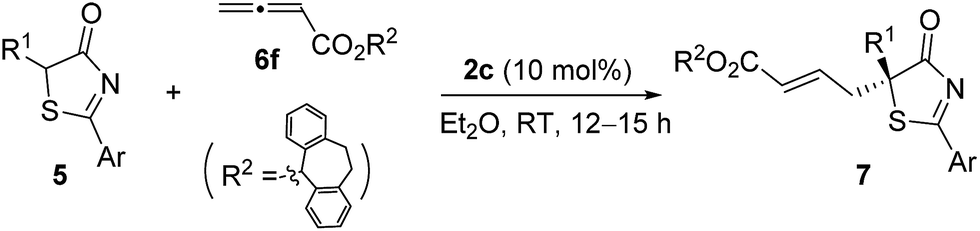
2.2. Enantioselective γ-addition of 5H-oxazol-4-ones
With the established protocol for the asymmetric γ-addition of 5H-thiazol-4-ones in hand, we next targeted access to the analogous α-oxygenated carboxylate surrogates by employing 5H-oxazol-4-ones. This task could be challenging as reports containing both sulfur- and oxygen-substituted substrates are rare.12c We hypothesized that the high tunability of our amino acid-based phosphine systems may provide a practical solution to this problem. The same set of phosphine catalysts were screened for the γ-addition of 5H-oxazol-4-ones to allenoate 6c, and the results are summarized in Table 4. The best catalyst for the previous addition of 5H-thiazol-4-ones, 2c, only afforded moderate enantioselectivity (entry 1). Switching to the dipeptide phosphines resulted in highly effective catalytic systems. While O-TBDPS-L-Thr-L-tert-Leu-based 3 led to the desired adduct with a slightly improved ee value, O-silyl-D-Thr-L-tert-Leu-derived phosphines offered excellent catalytic effects (entries 9–11). Finally, phosphine 4c was found to be the best catalyst, affording 9a in 95% yield and 76% ee (entry 11).|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Time (h) | Yieldb (%) | eec (%) |
| a Reactions were performed with 8a (0.1 mmol), 6c (0.12 mmol) and the catalyst (0.01 mmol) in toluene (1.0 mL) at room temperature for 12 h. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. | ||||
| 1 | 2c | 12 | 89 | 65 |
| 2 | 1a | 12 | 89 | 60 |
| 3 | 1b | 12 | 91 | 63 |
| 4 | 1c | 12 | 92 | 34 |
| 5 | 1d | 12 | 87 | 12 |
| 6 | 2a | 12 | 89 | 58 |
| 7 | 2b | 12 | 88 | 63 |
| 8 | 3 | 12 | 93 | 59 |
| 9 | 4a | 12 | 94 | −70 |
| 10 | 4b | 12 | 94 | −73 |
| 11 | 4c | 12 | 95 | −76 |
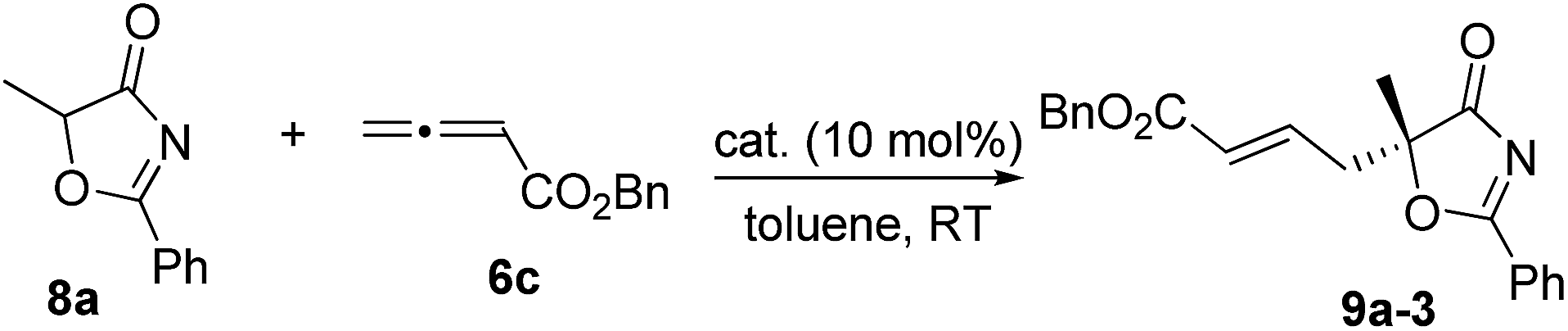
To further improve the enantioselectivity, we next optimized the ester moiety of the allenoate (Table 5). Among the different allenoate esters, the dibenzosuberyl ester was most ideal, affording the desired adduct in 96% yield and 86% ee (entry 6). Solvent screening identified diethyl ether as the most suitable solvent for the reaction. When the reaction was performed in the presence of 3 Å molecular sieves in diethyl ether, the γ-addition product was obtained in 97% yield with 92% ee (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | Allenoate | Solvent | Yieldb (%) | eec (%) |
| a Reactions were performed with 8a (0.1 mmol), 6 (0.12 mmol) and 4c (0.01 mmol) in the solvent specified (1.0 mL) at room temperature overnight. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. d 3 Å molecular sieves were added. e 4 Å molecular sieves were added. f 5 Å molecular sieves were added. g The reaction was stirred at 0 °C for 20 h. | ||||
| 1 | 6a | Toluene | 93 | 77 |
| 2 | 6b | Toluene | 95 | 82 |
| 3 | 6c | Toluene | 95 | 76 |
| 4 | 6d | Toluene | 96 | 84 |
| 5 | 6e | Toluene | 95 | 84 |
| 6 | 6f | Toluene | 96 | 86 |
| 7 | 6g | Toluene | 85 | 79 |
| 8 | 6h | Toluene | 89 | 67 |
| 9 | 6f | Xylene | 95 | 85 |
| 10 | 6f | Et2O | 97 | 88 |
| 11 | 6f | CHCl3 | 92 | 46 |
| 12 | 6f | CH2Cl2 | 90 | 50 |
| 13 | 6f | CH3CN | 82 | 67 |
| 14d | 6f | Et2O | 97 | 92 |
| 15e | 6f | Et2O | 96 | 91 |
| 16f | 6f | Et2O | 96 | 90 |
| 17d,g | 6f | Et2O | 86 | 91 |
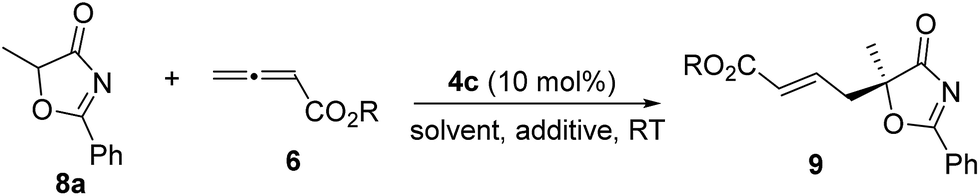
Under the optimal reaction conditions, the reaction was applicable to a wide variety of 5-alkyl substituted 5H-oxazol-4-ones. As shown in Table 6, the length of the alkyl chain can be varied, and both linear and branched alkyl groups can be employed, and high yields and excellent ee values were attainable in all cases (entries 1–10). When the 5-benzyl substrate was used, the enantioselectivity of the reaction dropped slightly (entry 11), which may be due to the unfavourable aromatic interactions induced by the Bn group. The absolute configuration of the γ-addition products was assigned by comparing the optical rotation of a derivative of 9b with the value reported in the literature.21
|
|
||||
|---|---|---|---|---|
| Entry | R1 | Time (h) | 9/Yieldb (%) | eec (%) |
| a Reactions were performed with 8 (0.1 mmol), 6f (0.12 mmol) and 4c (0.01 mmol) in the solvent specified (1.0 mL) at room temperature overnight. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. | ||||
| 1 | Me | 12 | 9a/97 | 92 |
| 2 | Et | 12 | 9b/93 | 93 |
| 3 | n-Pr | 12 | 9c/94 | 92 |
| 4 | i-Pr | 20 | 9d/93 | 93 |
| 5 | n-Bu | 12 | 9e/95 | 91 |
| 6 | i-Bu | 12 | 9f/96 | 93 |
| 7 | t-Bu | 36 | 9g/89 | 97 |
| 8 | n-C6H13 | 20 | 9h/94 | 93 |
| 9 | (CH2)2SCH3 | 12 | 9i/98 | 94 |
| 10 | n-C10H21 | 20 | 9j/91 | 92 |
| 11 | Bn | 20 | 9k/94 | 81 |
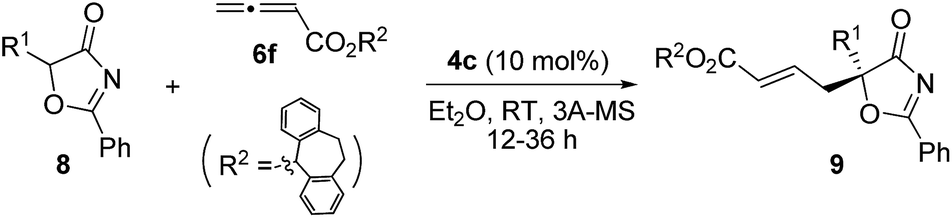
2.3. Scope of substrates and synthesis of tertiary thioethers and alcohols
Alkynes are common starting materials in organic synthesis, and the reaction here could be extended to alkyne substrates. Alkynoate (6′) could be employed, instead of allenoates, in the γ-addition reactions of both 5H-thiazol-4-ones and 5H-oxazol-4-ones. Although the reactions were slower, the chemical yields and enantioselectivities were the same [eqn (1) and (2)].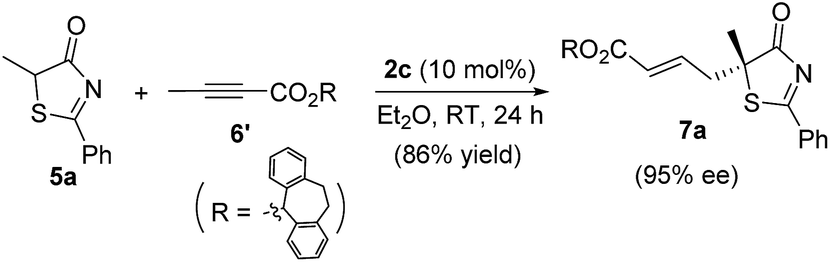 | (1) |
 | (2) |
For the thia(oxa)zolone substrates, the inclusion of 5-aryl-substituted thiazol-4-ones and oxazol-4-ones was unsuccessful. Thia(oxa)zolones are known to exist in tautomeric forms as thia(oxa)zoles,22 and the presence of a 5-aryl group makes the enol forms far more dominating. Indeed, the γ-addition products were not observed when 5-aryl substituted substrates were used. Instead, O-attack of the tautomeric thiazole/oxazole to allenoates took place, and the corresponding achiral adducts were obtained in high yields (Scheme 3).
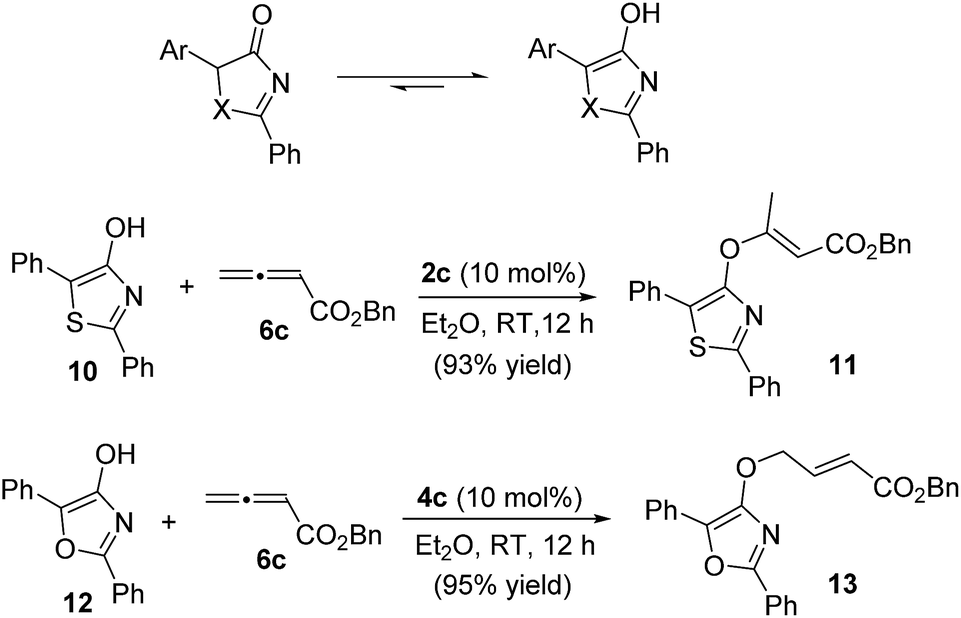 | ||
| Scheme 3 Reactions of 2,5-diphenyl-thiazol-4-ol 10 and 2,5-diphenyl-oxazol-4-ol 12 with allenoate 6c. | ||
The γ-addition products obtained possess a tertiary stereogenic center linked to a buried heteroatom, and they are valuable precursors for the convenient synthesis of enantiomerically enriched tertiary thiols/alcohols. As illustrated in Scheme 4, adduct 9b was converted to allyl oxazolone 15a, which was then treated with base to effect a ring opening, leading to the formation of a masked tertiary alcohol 15b in excellent yield. Reduction of the double bond and cleavage of the ester afforded a tertiary α-hydroxy acid derivative 17, which has both an ethyl and a propyl group present in the tertiary alcohol structure. Similarly, thiazolone 7a was transformed to allyl-substituted 18, which was readily converted to an enantiomerically enriched tertiary thioether 19via a base-catalyzed ring opening.23
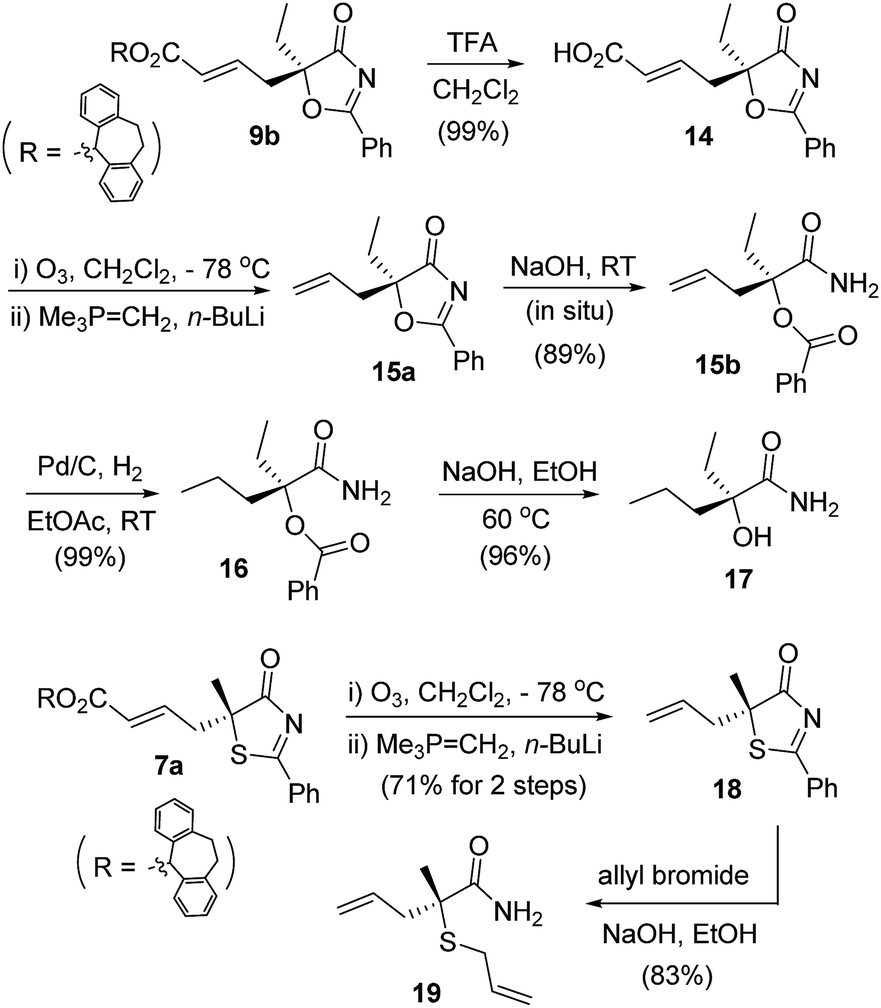 | ||
| Scheme 4 Elaboration of the γ-addition adducts into enantioenriched tertiary alcohols/thioethers. | ||
2.4. Mechanistic insights and DFT calculations
Despite the popularity of phosphine-catalyzed organic reactions, mechanistic investigations remain very limited; a few theoretical studies appearing in the literature were disclosed by the groups of Yu,24a–c Kwon,24d,e and others.24f–h We hypothesized that the hydrogen-bonding interactions between the Brønsted acid moiety of the phosphine catalyst and the donor molecules were essential for inducing enantioselectivity in our early reports on bifunctional phosphine catalyzed Michael and γ-addition reactions,18,20 and we believe such interactions are also crucial in our current reaction systems. The mechanism of the γ-addition of 5H-thiazol-4-one to allenoate is shown in Scheme 5, which follows the general mechanism described in the literature for γ-addition reactions.19,20 The reaction is initiated by the nucleophilic attack of the phosphorus atom on allenoate to form intermediate B, which is weakly basic. Deprotonation of the donor 5H-thiazol-4-one by B then affords the corresponding enolate, which subsequently attacks the γ-carbon of the allenoate to give intermediate E. Proton transfer takes place to afford 18,20 and this is followed by the elimination of the phosphine catalyst to furnish the final addition product. We propose that a hydrogen bonding interaction between the sulfonamide N–H and the thiazolone enolate dictates its addition to the C–C double bond, which is the key step for asymmetric induction.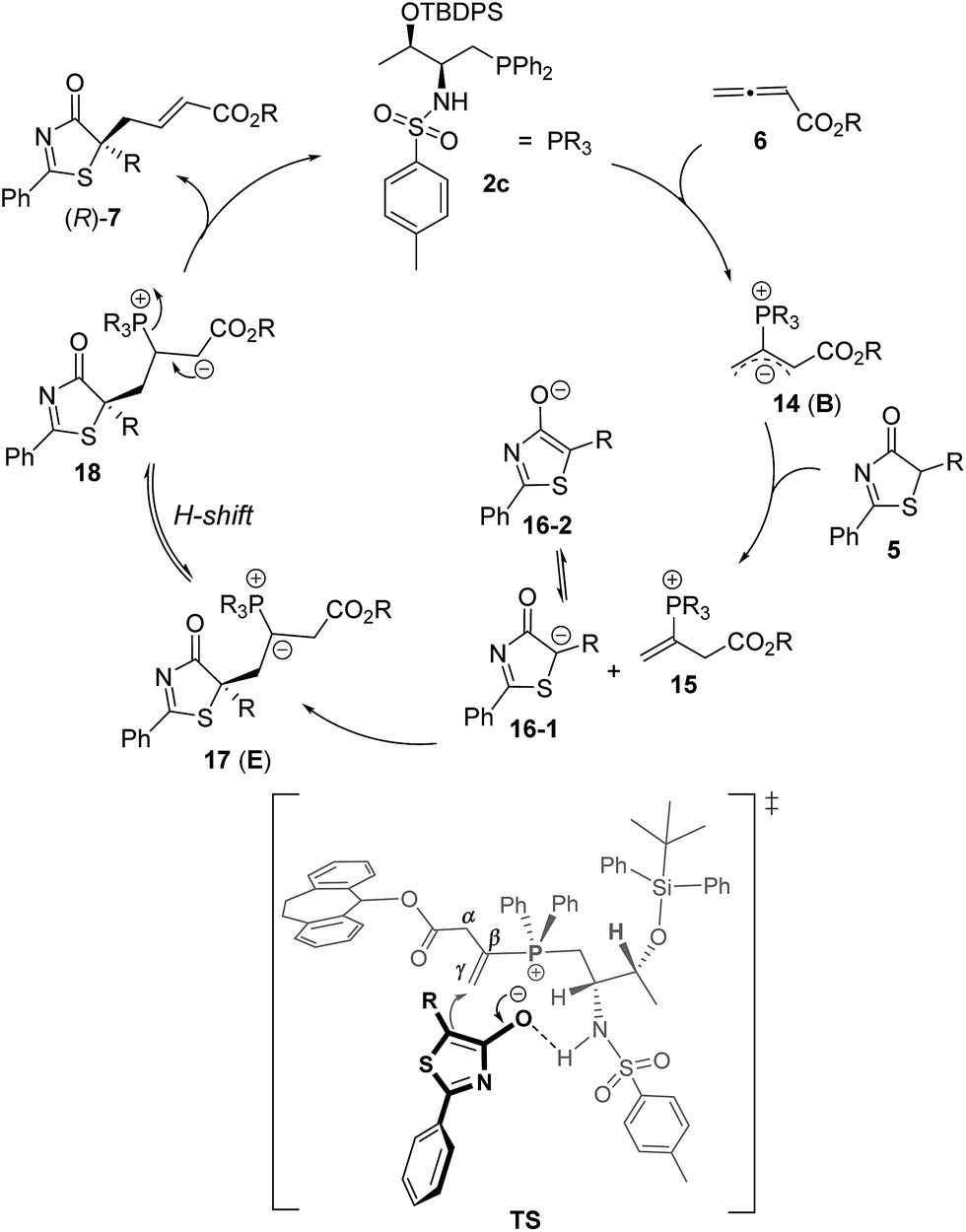 | ||
| Scheme 5 Proposed mechanism for the 2c-catalyzed γ-addition of 5H-thiazol-4-one to allenoate. | ||
In an effort to provide theoretical support for the proposed reaction mechanism and to rationalize the origin of enantioselectivity, detailed density functional theory (DFT) calculations were conducted. DFT methods, as implemented in the Gaussian 09 (ref. 25) program, have been employed to study the model reaction involving reactants 5H-thiazol-4-ones 5a, allenoate 6c and catalyst 2c. All the stationary points were optimized at the B3LYP26/6-31G (d) level27 of theory. The vibrational frequencies were computed at the same level of theory to determine whether the optimized structure was at an energy minimum or a transition state and to evaluate the corrections of enthalpy and Gibbs free energy. Solvent effects were computed by the IEFPCM28 solvation model at the M11 (ref. 29)/6-311+G (d)30 and B3LYP-D3 (ref. 31)/6-311+G (d) levels of theory using the gas phase optimized structures. The conclusions are similar for both methods, and the B3LYP-D3 calculated Gibbs free energies in toluene are discussed in the text.
The calculated Gibbs free energy profiles for the phosphine-catalyzed γ-addition of 5a to 6c are summarized in Fig. 2 (blue line). As proposed, the reaction is initiated by the nucleophilic attack of the phosphine catalyst 2c on allenoate 6cvia a transition state Ts1 with a barrier of 19.8 kcal mol−1. This process is facilitated by the NH⋯O hydrogen bond, which brings the phosphine and the allene groups into proximity. A zwitterionic intermediate B is first formed, endothermically and reversibly, with an overall barrier of 9.4 kcal mol−1. Subsequent proton transfer between intermediate B and reactant 5a takes place via transition state Ts2 with a barrier of 14.4 kcal mol−1 (an overall barrier of 23.8 kcal mol−1). The nucleophilic attack can then occur via two possible pathways: the Re-face attack occurs through transition state Ts4-Re with a barrier of 7.8 kcal mol−1 to give intermediate E with R-configuration ((R)-E); and the alternative Si-face attack proceeds via transition state Ts4-Si with a barrier of 9.7 kcal mol−1, 1.9 kcal mol−1 higher than that of Ts4-Re, leading to an intermediate with S-configuration ((S)-E). The corresponding addition product 7a can be generated by proton transfer and elimination of the catalyst, the pathway similar to those reported by Yu and co-workers.24a–c These observations suggest that the enantioselectivity is determined by the nucleophilic attack step and a value of 92% ee predicted by the B3LYP-D3 method based on the energy difference of Ts4-Re and Ts4-Si is in good agreement with the experimental result, where the R-product was formed preferentially (89% ee, entry 7 in Table 1). When sulfur is replaced by oxygen (5-methyl-2-phenyloxazol-4(5H)-one (8a)), the B3LYP-D3 calculations predict a value of 47% ee for the R-isomer (based on the energy difference of 0.6 kcal mol−1 between transition states Ts5-Re and Ts5-Si), consistent with the experimental observation (65% ee, entry 1 in Table 4).
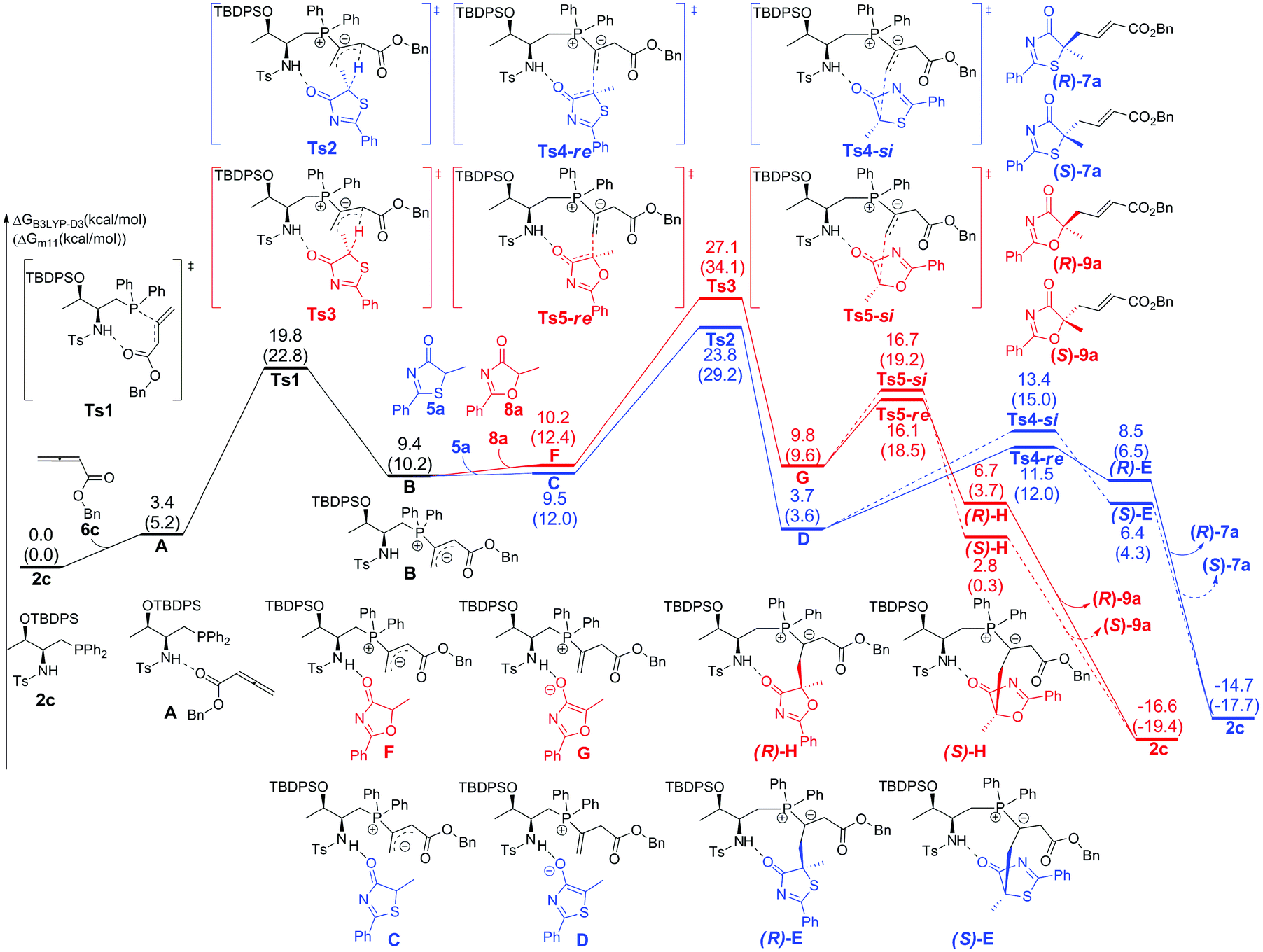 | ||
| Fig. 2 The DFT computed energy surfaces of the γ-addition reaction of 5a and 8a to allenoate 6c. The values given in kcal mol−1 are the B3LYP-D3 calculated relative free energies in toluene. The values in parentheses are the M11 calculated relative free energies in toluene. | ||
Upon evaluating transition states Ts4-Re and Ts4-Si (Fig. 3), it was found that the bond lengths for the forming C1–C2 bond are similar, in addition to the distances of the hydrogen bonds between H3 and O2 (about 1.8 Å). However, the short H2⋯C3 distance of 3.04 Å in Ts4-Si suggests a repulsion between the phenyl group of the reacting thiazolone and one of the phenyl groups in the phosphine catalyst, resulting in a higher transition state barrier. To better illustrate the steric repulsions in the nucleophilic addition step, a 2D contour map along the z-axis (defined as the forming C–C bond) of the van der Waals32 surface of Ts4-na is plotted (Fig. 4), representing the nucleophile moiety of transition state Ts4-Re without the thiazolone substrate. When the thiazolone group is deprotonated and bound to the catalyst via the N–H⋯O hydrogen bond during the formation of the C–C bond along the z-axis, the steric hindrance for the Re-face attack (labelled as R) is smaller than that for the Si-face attack (labelled as S). As such, Re-face attack becomes more favorable.
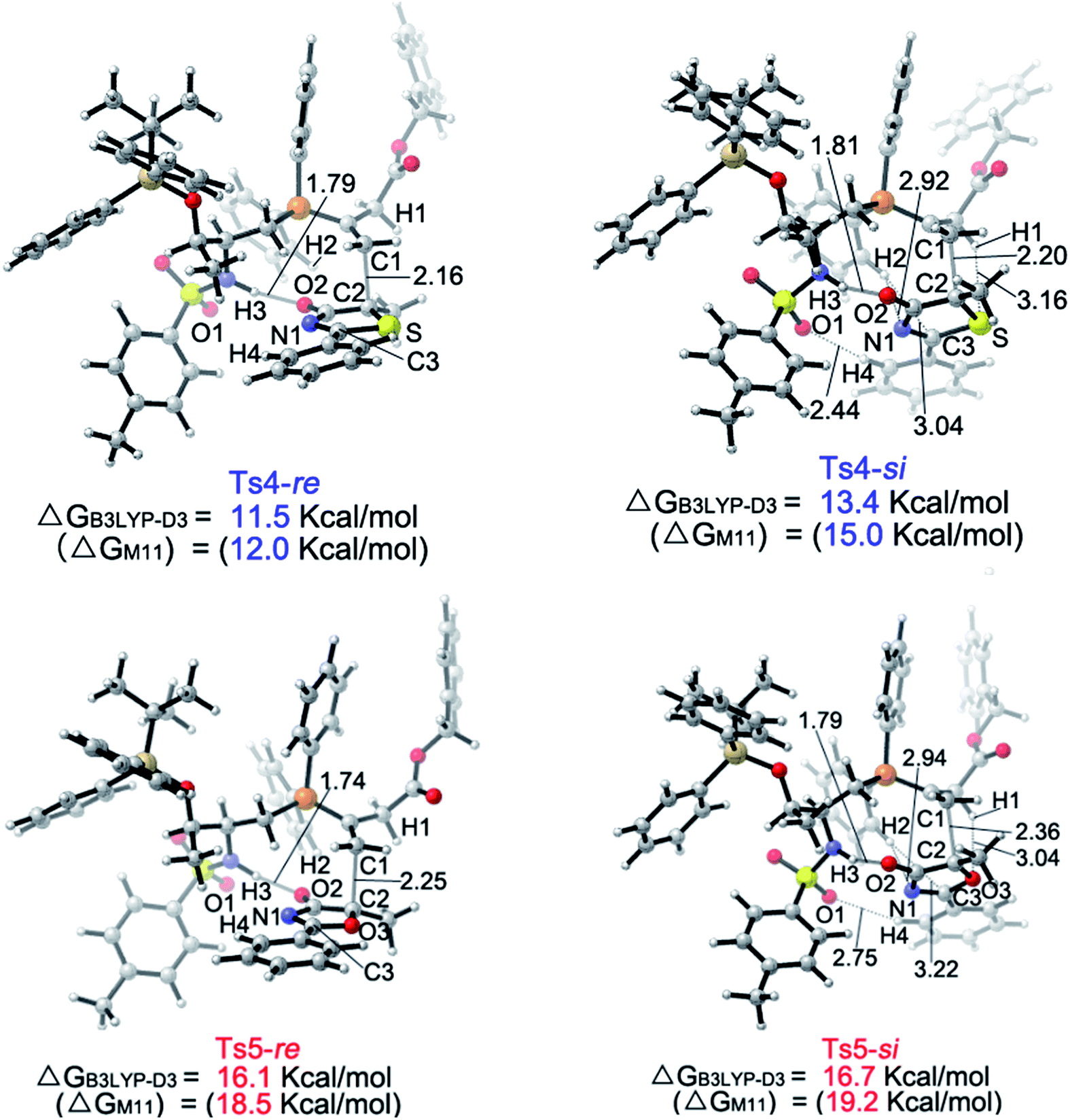 | ||
| Fig. 3 Geometries of the Ts4-Re, Ts4-Si, Ts5-Re and Ts5-Si transition states. The values for the bond lengths are given in angstroms. | ||
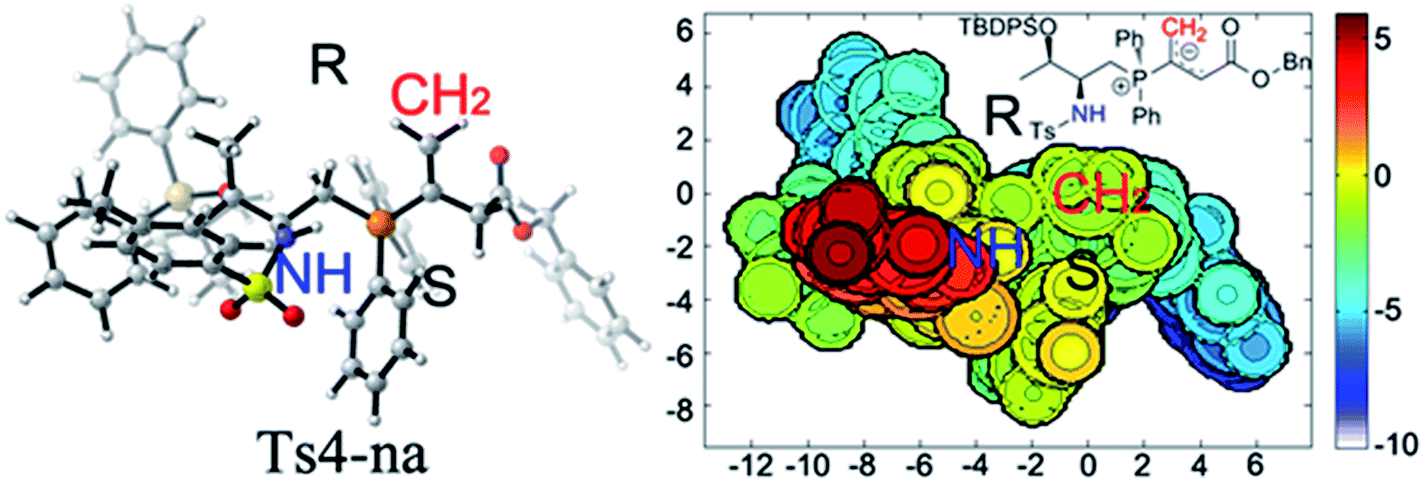 | ||
| Fig. 4 2D contour map of the van der Waals surface of catalyst 2c and allenoate 6c. Distances are reported in Å. The C atom of the CH2 group (labelled by red “CH2”) is located at the origin of the coordinate system in the contour map. A contour line of zero is defined as being in the same plane of the C atom. A negative distance (blue) indicates the atoms on the complex are farther away from substrate; a positive distance (red) indicates the atoms on complex are closer to substrate. | ||
2.5. Experimental confirmation
The importance of hydrogen bonding interactions for asymmetric induction has been clearly demonstrated in the above computational studies. Sulfonamide 2c and its close structural analogs 2c′ and 2c′′ were synthesized and applied to the γ-addition of 5H-thiazol-4-one 5a to allenoate 6f (Table 7). Blockage of the sulfonamide N–H led to a dramatic decrease in reactivity and enantioselectivity (entry 2). When the sterically hindered O-silyl group was replaced by a free OH, not only the enantioselectivity, but also the reactivity of the reaction decreased significantly (entry 3), suggesting that the bulky silyl group may be crucial for locking the transition state geometry and differentiating the Re- and Si-face attacks.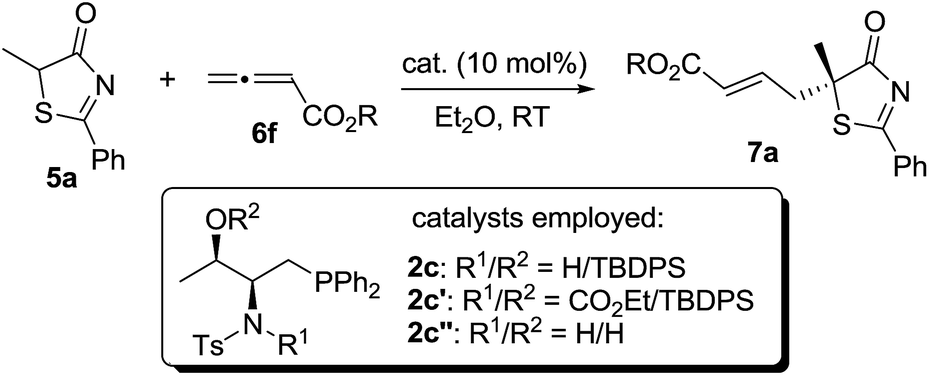
3. Conclusions
In summary, we have developed the first phosphine-catalyzed highly enantioselective γ-addition of 5H-thiazol-4-ones and 5H-oxazol-4-ones to 2,3-butadienoates. In the presence of amino acid-derived bifunctional phosphine catalysts, chiral thiazolones and oxazolones with a heteroatom (S or O)-containing tertiary chiral center were obtained in high yields and with excellent enantioselectivities. The optically enriched adducts are synthetically valuable, enabling the facile synthesis of optically enriched tertiary alcohols and thioethers. The method described in this report represents a method for rapid access to enantioenriched tertiary alcohol and thiol derivatives bearing an allylic chain, and may find wide applications in synthetic organic chemistry. DFT calculations for understanding the mechanism revealed that the observed enantioselectivity results from a combination of three factors: (1) the hydrogen-bonding interaction between the amino moiety of the phosphine catalyst and the “CO” unit of the thiazolone to activate the Michael donor, (2) the N–H⋯O interaction and the bulky O-silyl group to lock the conformation, and (3) the phenyl group of the thiazolone to differentiate the stereochemistry. It is noteworthy that this is the first complete theoretical study for phosphine-catalyzed γ-addition reactions. The theoretical results presented here are expected to offer new insight into the mechanisms of other phosphine-catalyzed asymmetric reactions, particularly those triggered by amino acid-derived phosphine catalysts.
Acknowledgements
Yixin Lu would like to thank the National University of Singapore (R-143-000-599-112), GSK–EDB (R-143-000-491-592), and the Ministry of Education (MOE) of Singapore (R-143-000-494-112) for generous financial support.Notes and references
- (a) A. Thuillier and P. Metzner, Sulfur Reagents in Organic Synthesis, ed. A. R. Katritzky, O. Meth-Cohn and C. S. Rees, Academic Press, 1995 Search PubMed; (b) Organosulfur Chemistry in Asymmetric Synthesis, ed. T. Toru and C. Bolm, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) C. Chatgilialoglu and K.-D. Asmus, Sulfur-Centered Reactive Intermediates in Chemistry and Biology, Springer, 1991 Search PubMed; (b) J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, Freeman, 5th edn, 2002 Search PubMed; (c) L. K. Moran, J. M. Gutteridge and G. J. Quinlan, Curr. Med. Chem., 2001, 8, 763 CrossRef CAS; (d) K. Pachamuthu and R. R. Schmidt, Chem. Rev., 2006, 106, 160 CrossRef CAS PubMed.
- For a review of sulfa-Michael reactions, see: (a) D. Enders, K. Lüttgen and A. A. Narine, Synthesis, 2007, 959 CrossRef CAS PubMed. For selected examples, see: (b) E. Emori, T. Arai, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1998, 120, 4043 CrossRef CAS; (c) C. Palomo, M. Oiarbide, F. Dias, R. López and A. Linden, Angew. Chem., Int. Ed., 2004, 43, 3307 CrossRef CAS PubMed; (d) C. Palomo, M. Oiarbide, R. López, P. B. González, E. Gómez-Bengoa, J. M. Saá and A. Linden, J. Am. Chem. Soc., 2006, 128, 15236 CrossRef CAS PubMed; (e) H. H. Lu, F. G. Zhang, X. G. Meng, S. W. Duan and W. J. Xiao, Org. Lett., 2009, 11, 3946 CrossRef CAS PubMed.
- (a) B. Strijtveen and R. M. Kellogg, J. Org. Chem., 1986, 51, 3664 CrossRef CAS; (b) J. M. McFadden, G.-L. Frehynot and C. A. Townsend, Org. Lett., 2002, 4, 3859 CrossRef CAS; (c) R. Alibés, P. Bayón, P. De March, M. Figueredo, J. Font and G. Marjanet, Org. Lett., 2006, 8, 1617 CrossRef PubMed; (d) A. Arpin, J. M. Manthorpe and J. L. Gleason, Org. Lett., 2006, 8, 1359 CrossRef CAS PubMed; (e) E. A. Tiong and J. L. Gleason, Org. Lett., 2009, 11, 1725 CrossRef CAS PubMed; (f) S. Takechi, N. Kumagai and M. Shibasaki, Org. Lett., 2013, 15, 2632 CrossRef CAS PubMed; (g) S. Takechi, S. Yasuda, N. Kunagai and M. Shibasaki, Angew. Chem., Int. Ed., 2012, 51, 4218 CrossRef CAS PubMed.
- (a) D. Bianchi and P. Cesti, J. Org. Chem., 1990, 55, 5657 CrossRef CAS; (b) N. Öhrner, C. Orrenius, A. Mattson, T. Norin and K. Hult, Enzyme Microb. Technol., 1996, 19, 328 CrossRef; (c) A. Peschiulli, B. Procuranti, C. J. O'Connor and S. J. Connon, Nat. Chem., 2010, 2, 380 CrossRef CAS PubMed.
- (a) M. Marigo, T. C. Wabnitz, D. Filenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794 CrossRef CAS PubMed; (b) S. Sobhani, D. Fielenbach, M. Marigo, T. C. Wabnitz and K. A. Jørgensen, Chem.–Eur. J., 2005, 11, 5689 CrossRef CAS PubMed; (c) Y. Cai, J. Li, W. Chen, M. Xie, X. Liu, L. Lin and X. Feng, Org. Lett., 2012, 14, 2726 CrossRef CAS PubMed; (d) X. Li, C. Liu, X.-S. Xua and J.-P. Cheng, Org. Lett., 2012, 14, 4374 CrossRef CAS PubMed; (e) Z. Hang, W. Chen, S. Dong, C. Yang, H. Liu, Y. Pan, L. Yan and Z. Jiang, Org. Lett., 2012, 14, 4670 CrossRef PubMed; (f) C. Wang, X. Yang, C. C. J. Loh, G. Raabe and D. Enders, Chem.–Eur. J., 2012, 18, 11531 CrossRef CAS PubMed; (g) S. E. Denmark, D. J. P. Kornfilt and T. Vogler, J. Am. Chem. Soc., 2011, 133, 15308 CrossRef CAS PubMed.
- (a) J. Clayden and P. MacLellan, Beilstein J. Org. Chem., 2011, 7, 582 CrossRef CAS PubMed and references cited therein; ; (b) C. Palomo, M. Oiarbide, F. Dias, A. Ortiz and A. Linden, J. Am. Chem. Soc., 2001, 123, 5602 CrossRef CAS; (c) C. Palomo, M. Oiarbide, F. Dias, R. Lopez and A. Linden, Angew. Chem., Int. Ed., 2004, 43, 3307 CrossRef CAS PubMed; (d) C. Palomo, M. Oiarbide, R. Lopez, P. Gonzalez, E. Gomez-Bengoa, J. M. Saa and A. Linden, J. Am. Chem. Soc., 2006, 128, 15236 CrossRef CAS PubMed.
- (a) Y. Hayashi, H. Yamaguchi, M. Toyoshima, K. Okado, T. Toyo and M. Shoji, Chem.–Eur. J., 2010, 16, 10150 CrossRef CAS PubMed; (b) D. A. Nicewicz, A. D. Satterfield, D. C. Schimitt and J. S. Johnson, J. Am. Chem. Soc., 2008, 130, 17281 CrossRef CAS PubMed; (c) A. N. Cuzzupe, R. D. Florio, J. M. White and M. A. Rizzacasa, Org. Biomol. Chem., 2003, 1, 3572 RSC; (d) K. Narasaka, T. Sakakura, T. Uchimaru and D. G. Vuong, J. Am. Chem. Soc., 1984, 106, 2954 CrossRef CAS.
- For selected examples, see: (a) R. Shintani, K. Takatsu and T. Hayashi, Org. Lett., 2008, 10, 1191 CrossRef CAS PubMed; (b) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS PubMed; (c) P. I. Dosa and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 445 CrossRef CAS; (d) K. Oisaki, D. Zhao, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 7164 CrossRef CAS PubMed; (e) M. Rueping, T. Theissmann, A. Kuenkel and R. M. Koenigs, Angew. Chem., Int. Ed., 2008, 47, 6798 CrossRef CAS PubMed; (f) H. Li, B. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 732 CrossRef CAS PubMed; (g) B. Török, M. Abid, G. London, J. Esquibel, M. Török, S. C. Mhadgut, P. Yan and G. K. S. Prakash, Angew. Chem., Int. Ed., 2005, 44, 3086 CrossRef PubMed; (h) S. Ogawa, N. Shibata, J. Inagaki, S. Nakamura, T. Toru and M. Shiro, Angew. Chem., Int. Ed., 2007, 46, 8666 CrossRef CAS PubMed.
- (a) A. Goeke, Sulfur Rep., 2002, 23, 243 CrossRef CAS PubMed; (b) J. J. Voss, Sulfur Chem., 2009, 30, 167 CrossRef CAS PubMed; (c) X.-F. Zhang, PCT Int. Appl. CN103833728 A, 2014; (d) J. M. Arif, S. S. Sawant, K. A. El Sayed, M. Kunhi, M. P. Subramanian, Y. M. Siddiqui, D. T. A. Youssef, K. Al-Hussain, M. N. Al-Ahdal and F. Al-Khodairy, J. Nat. Med, 2007, 61, 154 CrossRef CAS PubMed; (e) S. Sawant, D. Youssef, A. Mayer, P. Sylvester, V. Wali, M. Arant and K. E. Sayed, Chem. Pharm. Bull., 2006, 54, 1119 CrossRef CAS; (f) R. Kakuda, K. Machida, Y. Yaoita, M. Kikuchi and M. Kikuchi, Chem. Pharm. Bull., 2003, 51, 885 CrossRef CAS; (g) G. C. Davis, Y. Kong, M. Paige, Z. Li, E. C. Merrick, T. Hansen, S. Suy, K. Wang, S. Dakshanamurthy, A. Cordova, O. B. McManus, B. S. Williams, M. Chruszcz, W. Minor, M. K. Patel and M. L. Brown, Bioorg. Med. Chem., 2012, 20, 2180 CrossRef CAS PubMed; (h) M. L. Brown, S. Grindrod, T. H. Walls, T. Hansen, S. Sug and M. A. Paige, PTC Int. Appl. WO 2010027641 A2, 2010.
- (a) N. A. Khalil, E. M. Ahmed and H. B. El-Nassan, Med. Chem. Res., 2013, 22, 1021 CrossRef CAS; (b) U. V. Grummt, D. Weiss, E. Birckner and R. Beckert, J. Phys. Chem. A, 2007, 111, 1104 CrossRef CAS PubMed; (c) M. M. Véniant, C. Hale, R. W. Hungate, K. Gahm, M. G. Emery, J. Jona, S. Joseph, J. Adams, A. Hague and G. Moniz, et al. , J. Med. Chem., 2010, 53, 4481 CrossRef PubMed.
- (a) S. Diosdado, J. Etxabe, J. Izquierdo, A. Landa, A. Mielgo, I. Olaizola, R. López and C. Palomo, Angew. Chem., Int. Ed., 2013, 52, 11846 CrossRef CAS PubMed; (b) E. Badiola, B. Fiser, E. Gomez-Bengoa, A. Mielgo, I. Olaizola, I. Urruzuno, J. M. García, J. M. Odriozola, J. Razkin, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2014, 136, 17869 CrossRef CAS PubMed; (c) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 377 CrossRef CAS PubMed.
- For selected examples of recent asymmetric reactions of 5H-oxazol-4-ones. For metal-catalysis, see: (a) B. M. Trost, K. Dogra and M. Franzin, J. Am. Chem. Soc., 2004, 126, 1944 CrossRef CAS PubMed; (b) B. M. Trost and K. Hirano, Angew. Chem., Int. Ed., 2012, 51, 6480 CrossRef CAS PubMed; (c) D. Zhao, L. Wang, D. Yang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 7523 CrossRef CAS PubMed; (d) Z. Wang, Z. Chen, S. Bai, W. Li, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2012, 51, 2776 CrossRef CAS PubMed. For organocatalysis, see: (e) T. Misaki, G. Takimotoa and T. Sugimura, J. Am. Chem. Soc., 2010, 132, 6286 CrossRef CAS PubMed; (f) T. Misaki, K. Kawano and T. Sugimura, J. Am. Chem. Soc., 2011, 133, 5695 CrossRef CAS PubMed; (g) H. Huang, K. Zhu, W. Wu and J. Y. Jin, Chem. Commun., 2012, 48, 461 RSC; (h) Z. Han, W. Yang, C.-H. Tan and Z. Jiang, Adv. Synth. Catal., 2013, 355, 1505 CrossRef CAS PubMed; (i) B. Quiau, Y. An, Q. Liu, W. Yang, H. Liu, J. Shen, L. Yan and Z. Jiang, Org. Lett., 2013, 15, 2358 CrossRef PubMed; (j) P. Finkbeiner, N. M. Weckenmann and B. J. Nachtsheim, Org. Lett., 2014, 16, 1326 CrossRef CAS PubMed.
- For selected reviews, see: (a) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed; (b) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS PubMed; (c) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140 RSC; (d) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102 RSC; (e) A. Marinetti and A. Voituriez, Synlett, 2010, 174 CrossRef CAS PubMed; (f) S.-X. Wang, X. Han, F. Zhong and Y. Lu, Synlett, 2011, 2766 CAS; (g) Q.-Y. Zhao, Z. Lian, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 1724 RSC; (h) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588 RSC; (i) Z. Zhang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927 RSC; (j) Y. Xiao, Z. Sun, H. Guo and O. Kwon, Beilstein J. Org. Chem., 2014, 10, 2089 CrossRef PubMed; (k) Y. Wei and M. Shi, Chem.–Asian J., 2014, 9, 2720 CrossRef CAS PubMed.
- For a comprehensive review, see: (a) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS PubMed; (b) Y. Wei and M. Shi, Chem. Rev., 2013, 133, 6659 CrossRef PubMed. For selected examples, see: (c) F. Zhong, Y. Wang, X. Han, K.-W. Huang and Y. Lu, Org. Lett., 2011, 13, 1310 CrossRef CAS PubMed; (d) X. Han, Y. Wang, F. Zhong and Y. Lu, Org. Biomol. Chem., 2011, 9, 6734 RSC.
- For selected examples of phosphine catalyzed annulation reactions, see: (a) C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CrossRef CAS; (b) G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Am. Chem. Soc., 1997, 119, 3836 CrossRef CAS; (c) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426 CrossRef CAS PubMed; (d) B. J. Cowen and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 10988 CrossRef CAS PubMed; (e) Y. Q. Fang and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 5660 CrossRef CAS PubMed; (f) A. Voituriez, A. Panossian, N. Fleury-Brégeot, P. Retailleau and A. Marinetti, J. Am. Chem. Soc., 2008, 130, 14030 CrossRef CAS PubMed; (g) M. Sampath and T.-P. Loh, Chem. Sci., 2010, 1, 739 RSC; (h) H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467 CrossRef CAS PubMed; (i) Y. Fujiwara and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 12293 CrossRef CAS PubMed; (j) H. Guo, Q. Xu and O. Kwon, J. Am. Chem. Soc., 2009, 131, 6318 CrossRef CAS PubMed; (k) R. Na, C. Jing, Q. Xu, H. Jiang, X. Wu, J. Shi, J. Zhong, M. Wang, D. Benitez, E. Tkatchouk, W. A. Goddard III, H. Guo and O. Kwon, J. Am. Chem. Soc., 2011, 133, 13337 CrossRef CAS PubMed; (l) Q. Zhang, L. Yang and X. Tong, J. Am. Chem. Soc., 2010, 132, 2550 CrossRef CAS PubMed; (m) C. E. Henry, Q. Xu, Y. C. Fan, T. J. Martin, L. Belding, T. Dudding and O. Kwon, J. Am. Chem. Soc., 2014, 136, 11890 CrossRef CAS PubMed; (n) D. T. Ziegler, L. Riesgo, T. Ikeda, Y. Fujiwara and G. C. Fu, Angew. Chem., Int. Ed., 2014, 53, 13183 CrossRef CAS PubMed; (o) S. Kramer and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 3803 CrossRef CAS PubMed. For selected examples reported by us, see: (p) X. Han, Y. Wang, F. Zhong and Y. Lu, J. Am. Chem. Soc., 2011, 133, 1726 CrossRef CAS PubMed; (q) X. Han, F. Zhong, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 767 CrossRef CAS PubMed; (r) F. Zhong, X. Han, Y. Wang and Y. Lu, Chem. Sci., 2012, 3, 1231 RSC; (s) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837 CrossRef CAS PubMed; (t) F. Zhong, G.-Y. Chen, X. Han, W. Yao and Y. Lu, Org. Lett., 2012, 14, 3764 CrossRef CAS PubMed; (u) X. Han, W. Yao, T. Wang, Y. R. Tan, Z. Yan, J. Kwiatkowski and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 5643 CrossRef CAS PubMed; (v) X. Han, S.-X. Wang, F. Zhong and Y. Lu, Synthesis, 2011, 1859 CAS; (w) W. Yao, X. Dou and Y. Lu, J. Am. Chem. Soc., 2015, 137, 55 Search PubMed.
- F. Zhong, J. Luo, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2012, 134, 10222 CrossRef CAS PubMed.
- F. Zhong, X. Dou, X. Han, W. Yao, Q. Zhu, Y. Meng and Y. Lu, Angew. Chem., Int. Ed., 2013, 52, 943 CrossRef CAS PubMed.
- For pioneering studies on phosphine-mediated γ-additions, see: (a) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 10819 CrossRef CAS; (b) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 3167 CrossRef CAS; (c) B. M. Trost and G. R. Dake, J. Org. Chem., 1997, 62, 5670 CrossRef CAS; (d) C. Zhang and X. Lu, Synlett, 1995, 645 CrossRef CAS PubMed; (e) Z. Chen, G. Zhu, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Org. Chem., 1998, 63, 5631 CrossRef CAS; (f) C. Zhang and X. Lu, Synlett, 1995, 645 CrossRef CAS PubMed. For selected asymmetric γ-addition, see: (g) Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2009, 48, 2225 CrossRef CAS PubMed; (h) S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2009, 131, 14231 CrossRef CAS PubMed; (i) R. Sinisi, J. Sun and G. C. Fu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20652 CrossRef CAS PubMed; (j) J. Sun and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 4568 CrossRef CAS PubMed; (k) Y. Fujiwara, J. Sun and G. C. Fu, Chem. Sci., 2011, 2, 2196 RSC; (l) R. J. Lundgren, A. Wilsily, N. Marion, C. Ma, Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 2525 CrossRef CAS PubMed; (m) Y.-Q. Fang, P. M. Tadross and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 17966 CrossRef CAS PubMed; (n) J. Chen, Y. Cai and G. Zhao, Adv. Synth. Catal., 2014, 356, 359 CrossRef CAS PubMed.
- T. Wang, W. Yao, F. Zhong, G. H. Pang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 2964 CrossRef CAS PubMed.
- J. L. G. Ruano, A. M. M. Castro and J. H. Rodríguez, J. Org. Chem., 1992, 57, 7235 CrossRef.
- (a) F. A. J. Kerdesky, J. H. Holms, J. L. Moore, R. L. Bell, R. D. Dyer, G. W. Carter and D. W. Brooks, J. Med. Chem., 1991, 34, 2158 CrossRef CAS; (b) E. Täuscher, D. Weiß, R. Beckert, J. Fabian, A. Assumpção and H. Görls, Tetrahedron Lett., 2011, 52, 2292 CrossRef PubMed; (c) N. W. Jacobsen and A. Philippides, Aust. J. Chem., 1985, 38, 1335 CrossRef CAS.
- Allyl thioether was prepared since the free thiol readily formed the corresponding disulfide compound.
- (a) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS PubMed; (b) Y. Liang, S. Liu, Y. Xia, Y. Li and Z.-X. Yu, Chem.–Eur. J., 2008, 14, 4361 CrossRef CAS PubMed; (c) Y. Liang, S. Liu and Z.-X. Yu, Synlett, 2009, 905 CAS; (d) T. Dudding, O. Kwon and E. Mercier, Org. Lett., 2006, 8, 3643 CrossRef CAS PubMed; (e) E. Mercier, B. Fonovic, C. Henry, O. Kwon and T. Dudding, Tetrahedron Lett., 2007, 48, 3617 CrossRef CAS PubMed; (f) P. Xie, W. Lai, Z. Geng, Y. Huang and R. Chen, Chem.–Asian J., 2012, 7, 1533 CrossRef CAS PubMed; (g) L. Zhao, M. Wen and Z.-X. Wang, Eur. J. Org. Chem., 2012, 3587 CrossRef CAS PubMed; (h) Y. Qiao and K.-L. Han, Org. Biomol. Chem., 2012, 10, 7689 RSC.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed; (b) J. D. Dill and J. A. Pople, J. Chem. Phys., 1975, 62, 2921 CrossRef CAS PubMed; (c) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS PubMed.
- (a) E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef PubMed; (b) E. Cancès and B. Mennucci, J. Math. Chem., 1998, 23, 309 CrossRef; (c) B. Mennucci, E. Cancès and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506 CrossRef CAS; (d) G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 2, 2810 CrossRef CAS.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS PubMed; (b) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS; (b) P. Liu, J. Montgomery and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 6956 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and characterization data for the prepared compounds. See DOI: 10.1039/c5sc01614b |
| This journal is © The Royal Society of Chemistry 2015 |