
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Remote stereoselective deconjugation of α,β-unsaturated esters by simple amidation reactions†
Mahesh
Vishe
a,
Radim
Hrdina
a,
Amalia I.
Poblador-Bahamonde
a,
Céline
Besnard
b,
Laure
Guénée
b,
Thomas
Bürgi
c and
Jérôme
Lacour
 *a
*a
aDepartment of Organic Chemistry, University of Geneva, Quai Ernest Ansermet 30, CH-1211 Geneva 4, Switzerland. E-mail: jerome.lacour@unige.ch; Web: http://www.unige.ch/sciences/chiorg/lacour/ Fax: +41 22-379-3215
bLaboratory of Crystallography, University of Geneva, Quai Ernest Ansermet 24, CH-1211 Geneva 4, Switzerland
cDepartment of Physical Chemistry, University of Geneva, Quai Ernest Ansermet 30, CH-1211 Geneva 4, Switzerland
First published on 25th May 2015
Abstract
The thermodynamically disfavored isomerization of α,β-unsaturated esters to deconjugated β,γ-unsaturated analogues occurs readily when coupled to an amidation. Within the framework of macrocyclic derivatives, it is shown that 15, 16, and 18 membered macrocycles react with tBuOK and anilines to generate, in one-pot, β,γ-unsaturated amides (yields up to 88%). Importantly, single (chiral) diastereomers are isolated (d.r. > 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1H NMR) irrespective of the size and nature of the rings, showing an effective transmission of remote stereochemistry during the isomerization process. CSP-chromatographic resolution and absolute configuration determination by VCD are achieved.
:1, 1H NMR) irrespective of the size and nature of the rings, showing an effective transmission of remote stereochemistry during the isomerization process. CSP-chromatographic resolution and absolute configuration determination by VCD are achieved.
Introduction
The isomerization of α,β-unsaturated esters to their deconjugated β,γ-unsaturated analogues is a valued reaction as it allows, in a single step, alkenes and esters to be decoupled leading to benefits from the individualized reactivity and properties of the separated functional groups. However, due to the thermodynamic stability associated with conjugation, achieving such an isomerization is intrinsically difficult. To solve this problem, photochemical1 or strongly basic conditions2 are used to generate dienol or dienolate intermediates, which are then protonated selectively in the α position to the ester, under kinetic control (Scheme 1, top).3 Such reactions have been used in both academic and industrial laboratories.2b,4 Herein, we report that the isomerization becomes facile when coupled to an amidation reaction. Using unsaturated macrocycles as substrates for practical reasons,5 we report that the 15 and 18-membered derivatives 1 and 2 react with tBuOK and anilines to generate, in one-pot, the β,γ-unsaturated amides 3 and 4 respectively (Scheme 1, bottom, yields up to 88%). In the case of 4, even if the stereogenic centers are separated by more than 7 atoms, single diastereomers are observed (d.r. > 49:1, 1H NMR).6,7 This highly stereoselective isomerization can be extended to other ring compositions and sizes showing the effectiveness of the remote (intramolecular) transmission of stereochemistry. Calculations at the DFT/B3PW91 level validate the preferred formation of the chiral diastereomeric series over the meso. CSP-chromatographic separation of the enantiomers was also achieved and the absolute configuration determined by VCD.
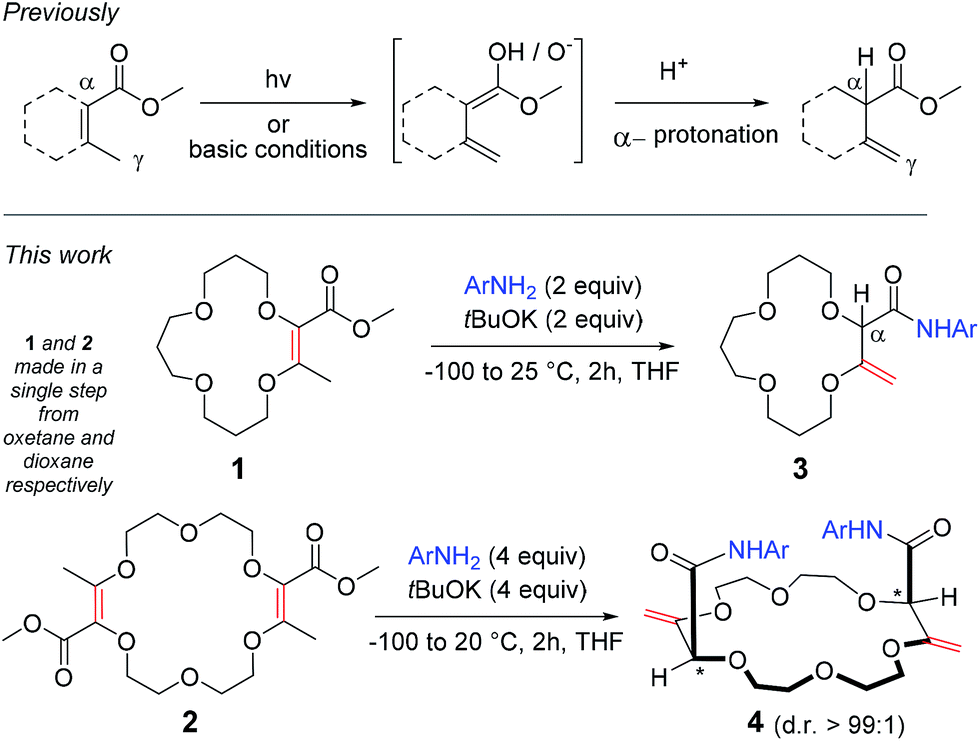 | ||
| Scheme 1 Deconjugation of 2,3-dioxo-methyl butenoate moieties. | ||
Results and discussion
Recently, we an others5,8 have reported the synthesis of functionalized polyether macrocycles through metal-catalyzed decompositions of α-diazo β-ketoesters in the presence of cyclic ethers such as oxetane and 1,4-dioxane. In these one pot reactions that occur at high concentration (typically 1 M), four building blocks are condensed. Compounds 1 and 2, which possess respectively one and two dioxobutanoate motifs, are afforded in generally good yields (up to 84%).5 The presence of both α,β-unsaturated ester moieties and β-methyl groups led us to consider the possibility of base or photochemically-induced alkene isomerizations. Unfortunately, attempts following established protocols2b led either to unreacted starting materials or to the complete decomposition of the substrates.However, treatments of 1 and 2 with tBuOK at 20 °C in the presence of 3,5-bis(trifluoromethyl) aniline (a, two and four equivalents respectively),9 led to products 3a and 4a after 2 hours of reaction (70 and 60% yield respectively). In both cases, 1H NMR spectroscopic analysis indicated the presence of amide groups and, importantly, displayed signals typical for enol ether protons (δ 4.33–4.35 ppm and 4.33–4.51 ppm respectively) and also singlets which could be assigned to α-hydrogen atoms (δ 4.27 and 4.40 ppm for 3 and 4 respectively). Interestingly, in the case of 4a, only one set of signals was obtained, indicative of the presence of a single diastereomeric species despite a separation of the stereogenic centers by more than 7 atoms (d.r. > 49:1, 1H NMR)10 – the nature and the relative (chiral) configuration of which was established unambiguously by X-ray diffraction analysis (ESI† and Fig. 1).11
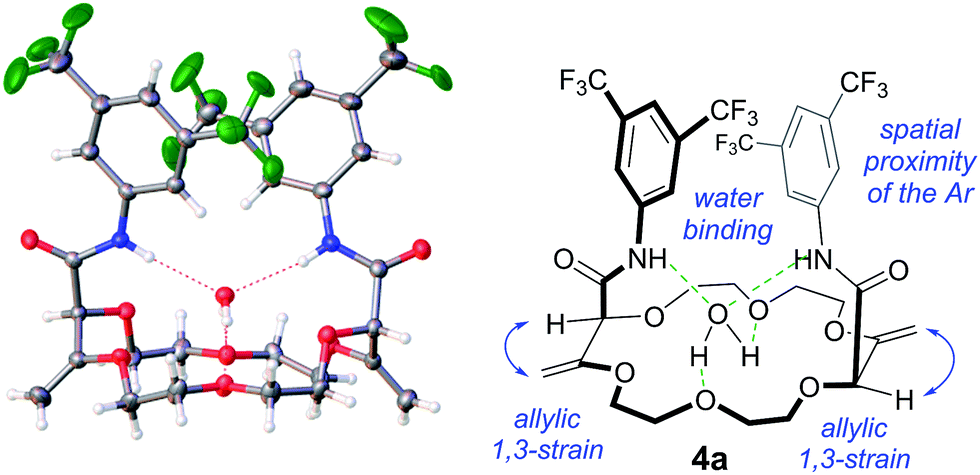 | ||
| Fig. 1 “Diaxial”-like conformation of the isomerized macrocycles, e.g.4a. Displacement ellipsoid plot of the crystal structure of 4a. Thermal ellipsoids are drawn at 50% probability. Solvent molecules and the disorder of the CF3 groups are omitted for clarity. | ||
With this result in hand, an optimization study was conducted using 3,5-bis(trifluoromethyl) aniline (a) for the amidation (Table 1). A 1:1 ratio between the base and the aniline was strictly maintained throughout the study. With both substrates 1 and 2, the addition of the base at 20 °C leads to an exothermic reaction (entries 1 and 4). Care was then taken to add tBuOK at a low temperature (−100 °C) and then allow the reaction to warm up to 20 °C on its own. Satisfactorily, yields of products increased in both cases (entries 2 and 5).
| Entry | Substrate | Base | Equiv. | Temp (°C) | Yieldb | d.r. |
|---|---|---|---|---|---|---|
| a Addition of base performed at −100 °C (using an ethanol liquid nitrogen bath) and, after 2 minutes, the reaction was allowed to warm to 20 °C. b Isolated yields. Average of at least two runs [%]. | ||||||
| 1 | 1 | tBuOK | 2 | 20 | 70 | — |
| 2a | 1 | tBuOK | 2 | −100 | 80 | — |
| 3a | 1 | tBuOK | 4 | −100 | 95 | — |
| 4 | 2 | tBuOK | 4 | 20 | 60 | >49 |
| 5a | 2 | tBuOK | 4 | −100 | 85 | >49 |
| 6a | 2 | tBuOK | 8 | −100 | 85 | >49 |
| 7a | 2 | tBuONa | 4 | −100 | 83 | >49 |
| 8a | 2 | tBuOLi | 4 | −100 | — | — |
| 9a | 2 | KHMDS | 4 | −100 | 60 | >49 |
| 10a | 2 | NaHMDS | 4 | −100 | 35 | >49 |
| 11a | 2 | LiHMDS | 4 | −100 | — | — |
| 12a | 2 | BuLi | 4 | −100 | — | — |
| 13a | 2 | LDA | 4 | −100 | 35 | >49 |
Addition to 1 and 2 of a larger amount of base and aniline (four and eight equivalents respectively) was beneficial only in the case of monoester 1 (entry 3). A stoichiometry of two equivalents of base and aniline for each unsaturated ester moiety was thus chosen for the remainder of the study.
Care was also taken to investigate the influence of the base and its counterion as, with crown-ether like compounds, an influence of the cationic atom was expected. While tBuONa afforded 4a in a similar yield (83 vs. 85%), a total lack of product was noticed with tBuOLi (entry 8). With hexamethyldisilazane salts (entries 9 to 11), lower yields were globally obtained and a clear difference was noticed between the potassium, sodium and lithium derivatives. Again, no product was formed with the Li+ salt; a result confirmed with BuLi (entry 12). Only in the case of LDA (entry 13) was a modest yield of 4a obtained. In view of these results, tBuOK was selected and the reaction was generalized with a series of anilines as reactants (Fig. 2).
 | ||
| Fig. 2 Substrate scope using macrocycles 1 and 2. | ||
First, 1 was reacted with regular aniline b and pyridine-derived k to afford the corresponding 15C4 products 3b and 3k in moderate yields (50% and 66% respectively). Using 2 as the substrate, 18C6 products 4a–4o were obtained in low to excellent yields (18–85%). Electron-donating and withdrawing substituents were equally introduced at the ortho, meta and para positions of the anilines (products 4c–4i). Amino pyridines and pyrazines (j–n) were well tolerated as products 4j, 4k, 4l, 4m and 4n were isolated in 30 to 60% yields. Finally, when 1,8-diaminonaphthalene was used with 2 as the substrate, a double sequence of three consecutive reactions occurred to afford product 4o in 64% yield. In this case, the resulting amides react spontaneously with the free amino naphthalene groups to form cyclic perimidines under the reaction conditions.
Moreover, it was shown that macrocycles 5, 6, 7 (Fig. 3), derived in one-step from THP (tetrahydropyran), THF (tetrahydrofuran) and benzofuran,5a,c also reacted well to afford compounds of type 8, 9 and 10 respectively. A selection of products is detailed in Fig. 4 using some of the anilines or pyridines already introduced on 3 and 4. Similar yields were globally obtained for these 18C4 and 16C4 macrocycles demonstrating the generality of the process.
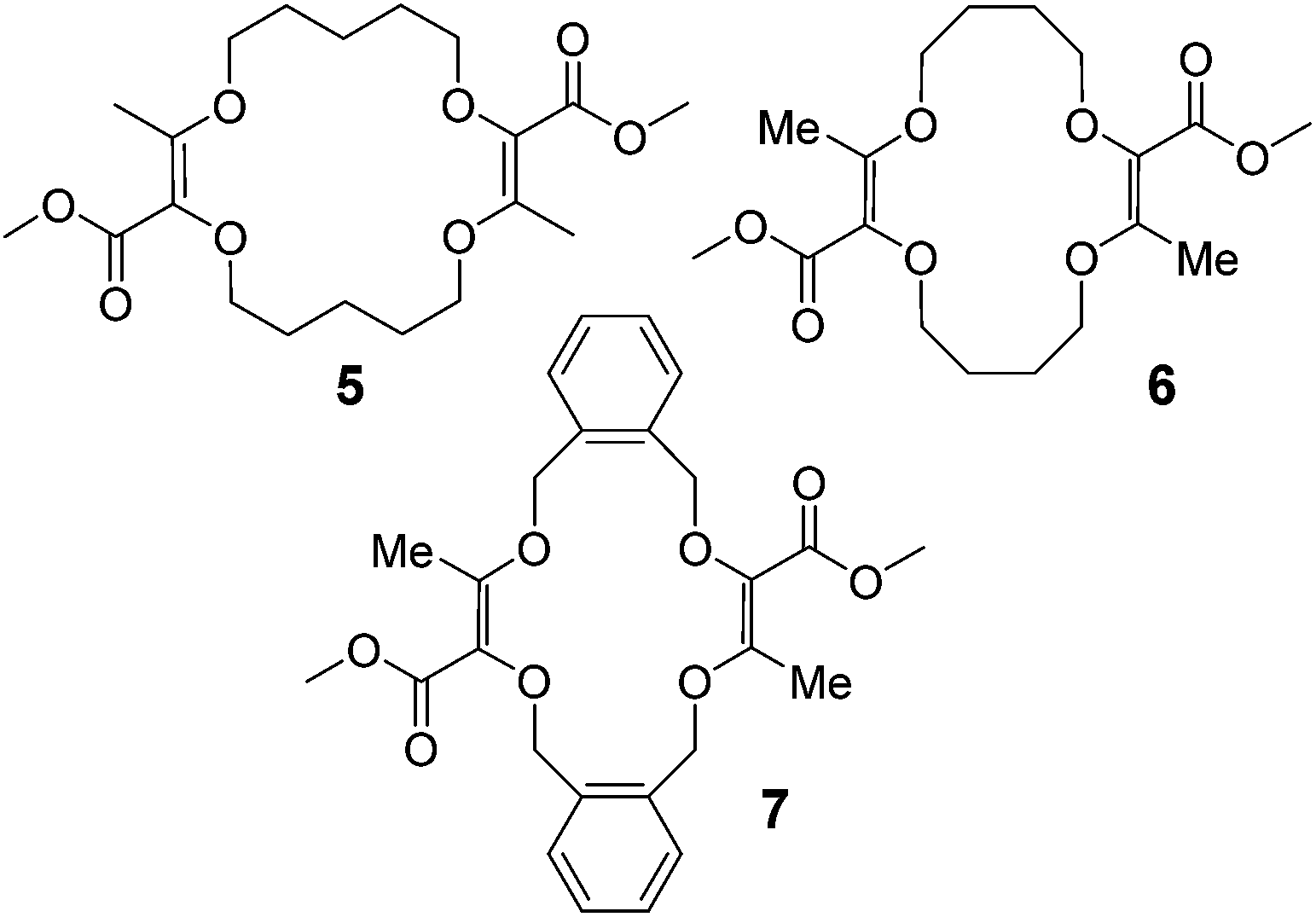 | ||
| Fig. 3 THP, THF and benzofuran-derived macrocycles 5, 6 and 7. | ||
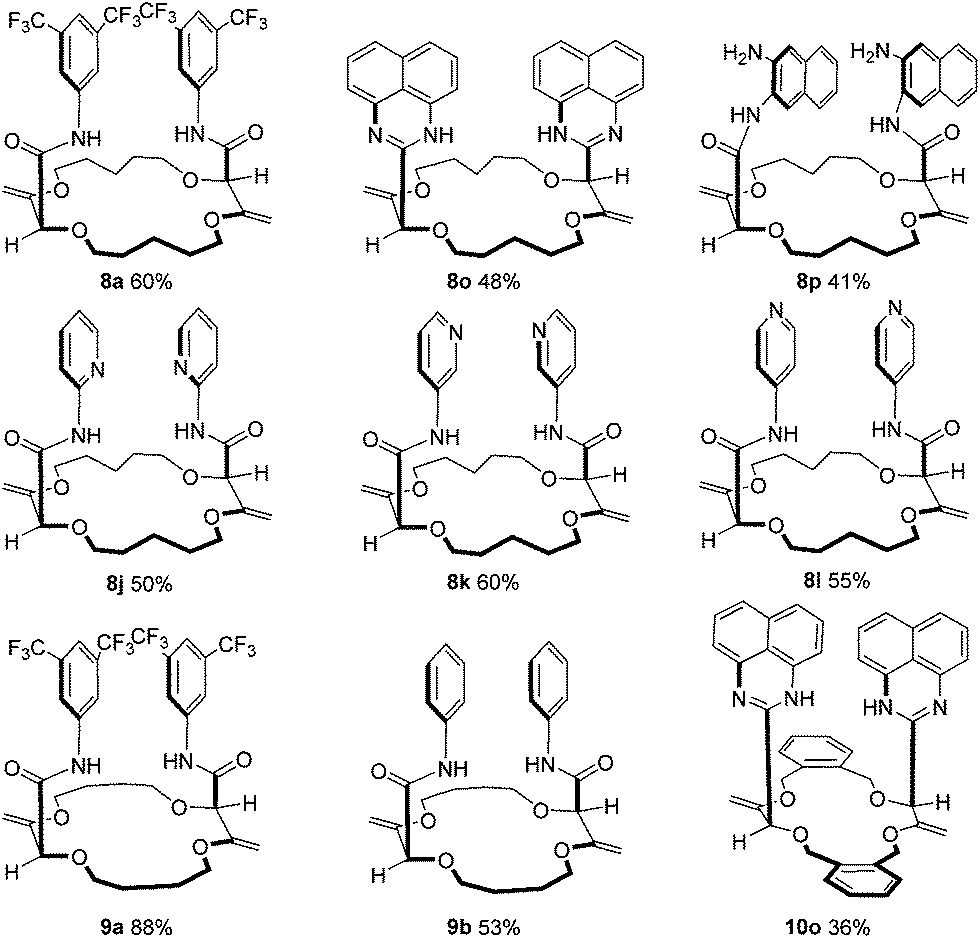 | ||
| Fig. 4 Substrate scope using macrocycles 5, 6 and 7. | ||
Several of these macrocycles were analyzed successfully by X-ray diffraction (see Fig. 1 (4a) and the ESI† for 4l, 8a, and 10o). Of importance, strong evidence was found for allylic 1,3-strain interactions between the exocyclic double bonds and the adjacent stereogenic centers.12 In fact, in all macrocycles, irrespective of their size, the nature of the ring and of the substituents, the α-C–H bonds are periplanar to the exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 bonds.13 As a consequence, the amide moieties are disposed in an essentially perpendicular orientation to the mean plane of the macrocycles. This geometry then favors (i) close interactions between the aromatic moieties and (ii) hydrogen bonding interactions between the amides N–H and neighboring oxygen atoms or, in the case of the 18C6 macrocycles of type 4, with guest water molecules.14
CH2 bonds.13 As a consequence, the amide moieties are disposed in an essentially perpendicular orientation to the mean plane of the macrocycles. This geometry then favors (i) close interactions between the aromatic moieties and (ii) hydrogen bonding interactions between the amides N–H and neighboring oxygen atoms or, in the case of the 18C6 macrocycles of type 4, with guest water molecules.14
Mechanistic rationale
To gain some insight on the transformation, two isotope labeling experiments were performed. First, compound 1 was treated with aniline-d7 and tBuOK to yield deuterated products 3b-d with 80% and 45% incorporations of deuterium at the α and γ positions respectively (see Scheme 2). This result indicates that the aniline moiety acts both as a nucleophile and proton source in the transformation. The resulting 3b-d were then resubmitted to the reaction conditions but with regular aniline PhNH2 in the medium. After two hours, the amount of deuteration was found to be unchanged, indicating a lack of D/H exchange in compound 3b-d. With this experimental information, a mechanistic rationale can be proposed (Scheme 2).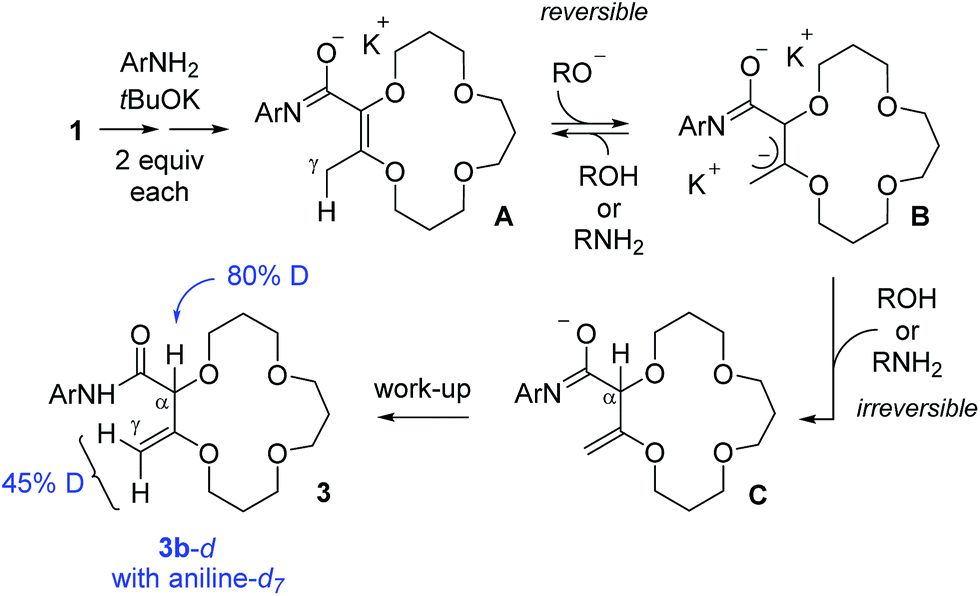 | ||
| Scheme 2 Condensed mechanistic rationale for the tandem amidation–isomerization. Results of deuteration experiments are indicated in blue. | ||
First, a reaction between the anilines and tBuOK occurs to generate anilides that react both as a nucleophile and base to yield, in a few steps, anionic intermediates of type A. γ-Deprotonation then occurs to form allylic B; this step being reversible to account for the 45% of deuteration at the γ position of 3b-d in the presence of aniline-d7. However, α-protonation of B also occurs. This step is proposed to be irreversible to explain the lack of D/H exchange when 3b-d is resubmitted to the reaction conditions and this explains the predominant formation of the deconjugated products.15,16 Imidate intermediates C are then quenched during the work-up, yielding products 3.
With substrates, 2, 5, 6 and 7 that contain two butenoate motifs, this isomerization sequence occurs twice to generate compounds 4, 8, 9 and 10 with a very high stereoselectivity (d.r. > 49:1, 1H NMR) even if the stereogenic centers are separated by several atoms (seven or more).6 A rationale for this general asymmetric induction is detailed in Scheme 3 and Fig. 5. In intermediates D, which possess an already transposed double bond and an anionic allylic imidate functional group, the presence of the first stereogenic center controls remotely the stereoselectivity of the protonation – and hence the configuration of the second center. It is proposed that a potassium (or sodium) cation serves as a bridge (relay) between the two imidate anions and, under these chelating conditions, the pathway leading the racemic product is highly favored.
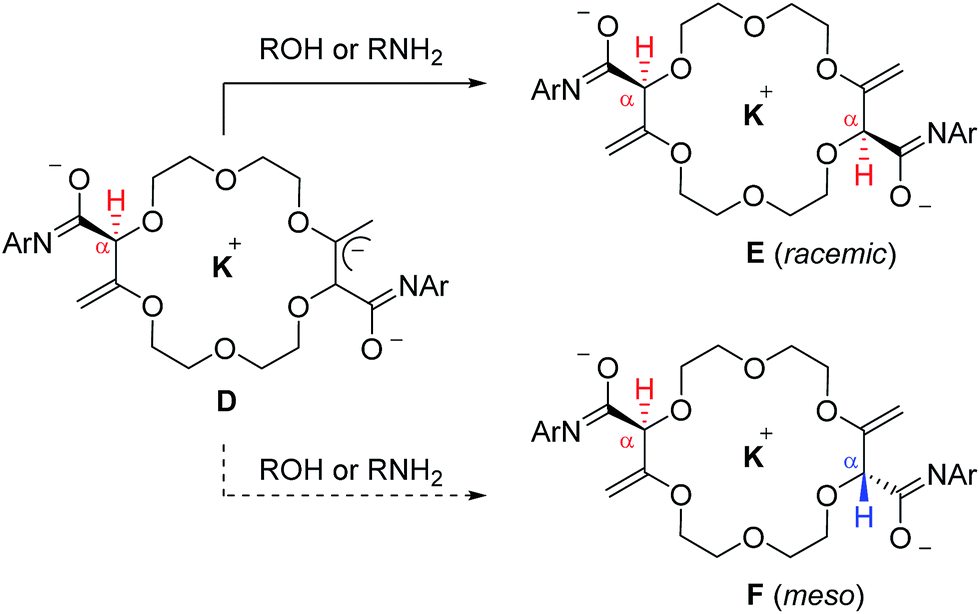 | ||
| Scheme 3 Stereoselective protonation in favor of the racemic over meso diastereomers. | ||
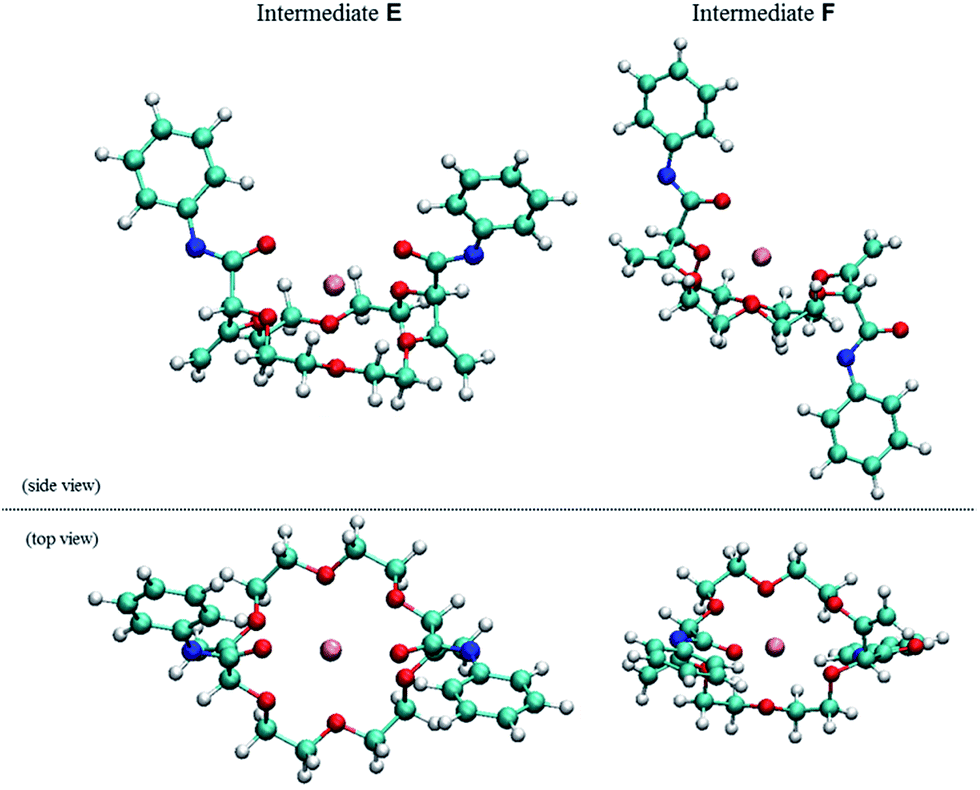 | ||
| Fig. 5 Side and top view of the calculated geometries (DFT/B3PW91) of racemic E (right) and meso F (left, +5.4 kcal mol−1) intermediates. | ||
Full optimization of structures E and F were conducted at the DFT/B3PW91 level of theory to assess the coordination mode of the potassium cation and the relative energy of both diastereomers. Care was taken to account for the presence of the two imidate moieties that allow a coordination of the potassium cation with either oxygen or nitrogen atoms (see ESI†). Geometry optimizations for all possible conformers of E and F were thus modeled. Only intermediate E favors the chelation of the cation with both oxygens of the imidate moieties.17 The comparison of the relative energies of the most stable conformers for intermediates E (racemic, Ar = Ph) and F (meso, Ar = Ph) indicates that the chiral diastereomer is more stable that its achiral analogue by 5.4 kcal mol−1 (Fig. 5).18 This energy difference between E and F is sufficiently large to consider with confidence that there is also a large energy gap between the transition states which leads to these intermediates; this ΔΔG‡ gap is decisive for the stereoselectivity as the α-protonation is irreversible (see above).19
Finally, the resolution of 4a was tackled.20 The enantiomeric separation was performed by CSP-chromatography on a semi-preparative scale using a Chiralpak IC column and a mixture of n-hexane:i-PrOH 85:15 as the eluent. From 50 mg of rac-4a, after several runs, two major separated fractions were afforded, 20 mg (ee > 99%) of (−)-4a and 18 mg (ee > 99%) of (+)-4a (see the ESI† for details). The electronic circular dichroism (ECD) spectra display totally symmetric curves in the 250 nm to 300 nm domain. The spectra are reported in Fig. S4.† With the separated enantiomers in hand, care was taken to determine the absolute configuration with certainty.21 This was established by vibrational circular dichroism (VCD) in view of the rigidity of compounds 2.22 IR absorption and VCD spectra were measured for solutions (CD2Cl2) of both (+) and (−)-4a and compared to the averaged spectrum calculated for (R,R)-4a and its water complex (R,R)-4a·H2O (Fig. 6). Overall, considering that the macrocycle is present in solution as a mixture of the hydrated and non-hydrated forms, a good agreement between the experimental and theoretical spectra was observed. Some discrepancies are observed in the region of the amide II vibrations (N–H deformation) around 1560 cm−1. This spectral region is strongly affected by the interaction with water, which is not well described by the calculations. Despite this difficulty the VCD measurements allow the assignment of (−)-4a as the (R,R) enantiomer.
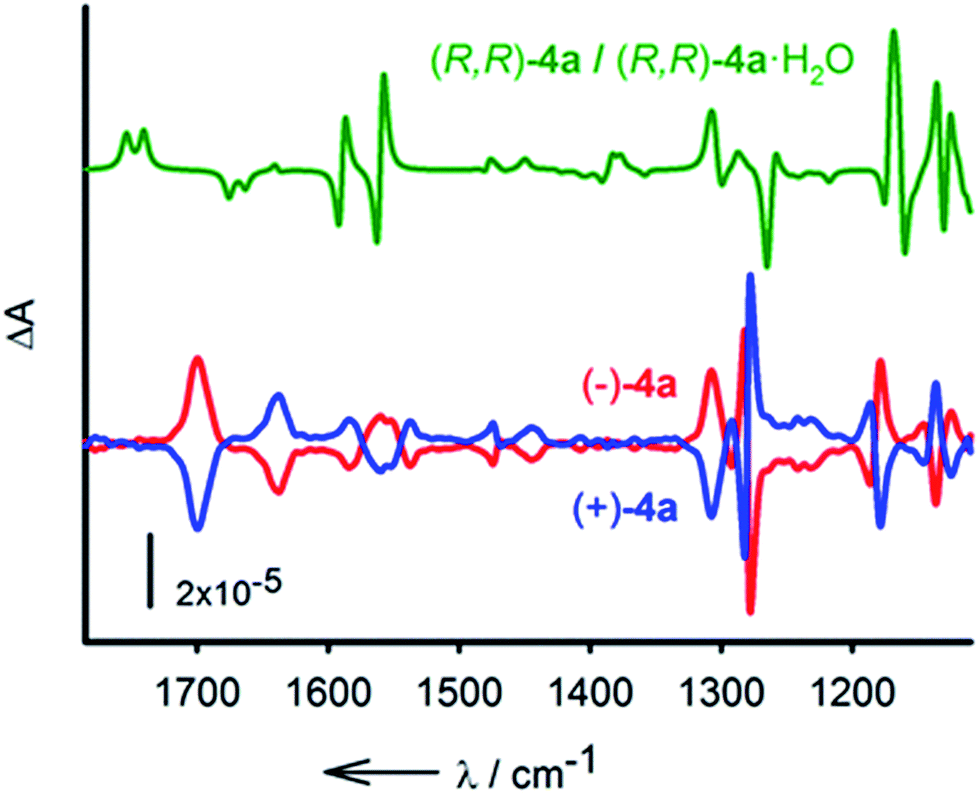 | ||
| Fig. 6 Calculated (1:1 average) spectrum of (R,R)-4a and (R,R)-4a·H2O (green). Experimental VCD spectra (CD2Cl2, 298 K) of (−)-4a (red) and (+)-4a (blue). | ||
Conclusions
In conclusion, a new deconjugation reactivity of α,β-unsaturated esters is reported owing to an amidation–isomerization sequence. This transformation is favored by the presence of anilines that act not only as nucleophiles but also, importantly, as effective proton sources in the isomerization step. Applied to macrocycles, the process is highly stereoselective (d.r. > 49:1) despite a separation of the stereogenic centers by more than 7 atoms. Calculations at the DFT/B3PW91 level validate the preference for the chiral diastereomers via a cation acting as a relay for the stereochemical transmission. CSP-chromatographic separation of the enantiomers was also achieved and the absolute configuration determined by VCD. As such, well-defined conformationally-constrained chiral macrocycles are obtained – and this in only two steps from simple and commercial cyclic ether precursors. Applications in enantioselective catalysis are looked for.23
Acknowledgements
We thank the University of Geneva and the Swiss National Science Foundation, the COST Action CM0802 and the State Secretariat for Education and Science for financial support. We also acknowledge the contributions of the Sciences Mass Spectrometry (SMS) platform at the Faculty of Sciences, University of Geneva.Notes and references
-
(a) M. J. Jorgenson and N. C. Yang, J. Am. Chem. Soc., 1963, 85, 1698–1699 CrossRef CAS
; (b) M. J. Jorgenson and T. Leung, J. Am. Chem. Soc., 1968, 90, 3769–3774 CrossRef CAS
-
(a) A. C. Weedon, Can. J. Chem., 1984, 62, 1933–1939 CrossRef CAS
-
(a) L. Duhamel and J. C. Plaquevent, J. Am. Chem. Soc., 1978, 100, 7415–7416 CrossRef CAS
-
(a) C. Fehr and J. Galindo, J. Am. Chem. Soc., 1988, 110, 6909–6911 CrossRef CAS
-
(a) W. Zeghida, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2010, 49, 7253–7256 CrossRef CAS PubMed
- For a review on remote stereocontrol, see: J. Clayden, Chem. Soc. Rev., 2009, 38, 817–829 RSC
-
(a) W. C. Still and I. Galynker, Tetrahedron, 1981, 37, 3981–3996 CrossRef CAS
-
(a) W. Kirmse and R. Lelgemann, Chem. Ber., 1991, 124, 1865–1866 CrossRef CAS PubMed
- B. R. Kim, H.-G. Lee, S.-B. Kang, G. H. Sung, J.-J. Kim, J. K. Park, S.-G. Lee and Y.-J. Yoon, Synthesis, 2012, 44, 42–50 CrossRef CAS PubMed
- LC analysis of the crude confirms the excellent stereoselectivity with a ratio higher than 200:1.
- In fact, upon formation of compounds 4, two stereogenic centers are created at the α positions possibly leading to meso or racemic stereoisomers. In this study, all solid-state and solution studies with NMR chiral solvating agents point towards the formation of racemic adducts only.
- R. W. Hoffmann, Chem. Rev., 1989, 89, 1841–1860 CrossRef CAS
- In the reported X-ray structures, the mean average for the H–Cα–CCH2 dihedral angle is in fact 6.5° ± 4.
- Proton signals of guest water molecules in compounds 4 are shifted up to 4.20 ppm at high concentration (CDCl3, 1H NMR).
- As discussed later, the conformation induced by the allylic 1,3-strain interaction imposes a periplanar alignment of the Cα–H bond with the exocyclic olefin. This orientation strongly disfavors the reformation of the allylic system by deprotonation as the Cα–H bond and π electrons are orthogonally disposed. A similar analysis can be done for the adjacent amide group which also tends to a periplanar arrangement with the Cα–H bond.
- An alternative to explain the result would be to consider a strong kinetic isotope effect that would disfavor the Cα–D cleavage and hence the reformation of the allylic enolate. In this case, the protonation could then be reversible.
- The preference of the oxygen over the nitrogen atoms is probably due to a steric effect.
- Analogous calculations were performed with two potassium ions. The difference in energy between the racemic and meso diastereomers is much larger (33.5 kcal mol−1) in favor of the chiral geometry. In the most stable species, one cation is imbedded within the crown ether and the other is trapped between two adjacent aryl moieties; the presence of these cation–π interactions strongly favor the chiral structure.
- The energy difference between the chiral and meso diastereomers is even larger for the potassium complexes of the final bisamide products: a difference of 14.5 kcal mol−1 being calculated in this case. See ESI.†.
- The chiral nature of compounds 4 can also be assessed in 1H NMR spectroscopy using chiral solvating reagents and Eu(hfc)3 in particular. See ESI.†.
- A. Aamouche, F. J. Devlin and P. J. Stephens, J. Am. Chem. Soc., 2000, 122, 2346–2354 CrossRef CAS
-
(a) G. Holzwarth, E. C. Hsu, H. S. Mosher, T. R. Faulkner and A. Moscowitz, J. Am. Chem. Soc., 1974, 96, 251–252 CrossRef CAS
-
(a) D. J. Cram and G. D. Y. Sogah, J. Chem. Soc., Chem. Commun., 1981, 625–628 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1034974, 1034976–1034978. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01118c |
| This journal is © The Royal Society of Chemistry 2015 |