
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of (diarylmethyl)amines using Ni-catalyzed arylation of C(sp3)–H bonds†
José A.
Fernández-Salas
a,
Enrico
Marelli
a and
Steven P.
Nolan
*ab
aEaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK. E-mail: stevenpnolan@gmail.com
bChemistry Department, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
First published on 12th June 2015
Abstract
The first nickel catalyzed deprotonative cross coupling between C(sp3)–H bonds and aryl chlorides is reported, allowing the challenging arylation of benzylimines in the absence of directing group or stoichiometric metal activation. This methodology represents a convenient access to the (diarylmethyl)amine moiety, which is widespread in pharmaceutically relevant compounds.
Cross-coupling catalysis holds a preferred position in the synthetic chemist's arsenal as it provides a myriad of options for the efficient and user-friendly access to organic motifs that are otherwise difficult or impossible to obtain.1 In this context efforts have been devoted to extend the use of cross-coupling to the functionalization of C–H bonds, as this highly attractive strategy leads to an atom-economical formation of new bonds while generating minimal waste.2 The use of directing groups and/or activated C–H bonds in this chemistry has been thoroughly studied.3–5 In contrast, the use of less acidic C(sp3)–H pro-nucleophiles in the absence of any directing group6,7 has proven more challenging, and such examples remain scarce.8
 | ||
| Scheme 1 Context of the present work. | ||
The arylation of benzylamines-derived imines belongs to this reaction class. It was initially reported by Yorimitsu and Oshima9 in 2008 and more recently alternative protocols have been described.10 As most of the deprotonative cross couplings reported to date, these protocols are based on palladium catalysis (see Scheme 1). In recent years, attention has focused on the use of less expensive and more earth-abundant first-row transition metals as catalysts.11 Amongst these, nickel has long been used in a number of industrial applications,12 and its utility as a powerful catalyst has been revisited in several areas of homogenous catalysis, ranging from coupling reactions13 to C–H bond functionalization14 and small molecules activation.15 However, deprotonative functionalization of benzylic C–H bond under nickel catalysis is, to date, unprecedented. As the (diarylmethyl)amine moiety is a well-known pharmacophore motif found in pharmaceuticals,16 the development of more sustainable synthetic methodologies towards its synthesis is of significant interest.
We therefore envisioned the use of a Ni–NHC (NHC: N-heterocyclic carbene) system, known to be highly active in cross-coupling chemistry,13d,h,nin lieu of Pd-based catalysts in this challenging transformation. Our initial hypothesis relied on the existence, for nickel, of a mechanistically closely related process to palladium (see Scheme 2).
 | ||
| Scheme 2 The mechanism of the deprotonative cross coupling of benzylimines. | ||
Our study began with the examination of a model reaction involving 1a and chlorotoluene (Table 1). The role of the base was examined early on and full conversion and good NMR yields of the product were obtained using a [Ni(COD)2](1)/IPr catalytic system in toluene (see Fig. 1) when potassium hexamethyldisilylamide (KHMDS) was used as base. All other bases tested gave no conversion to the desired product (for full solvent-base system optimization, see the ESI†).17
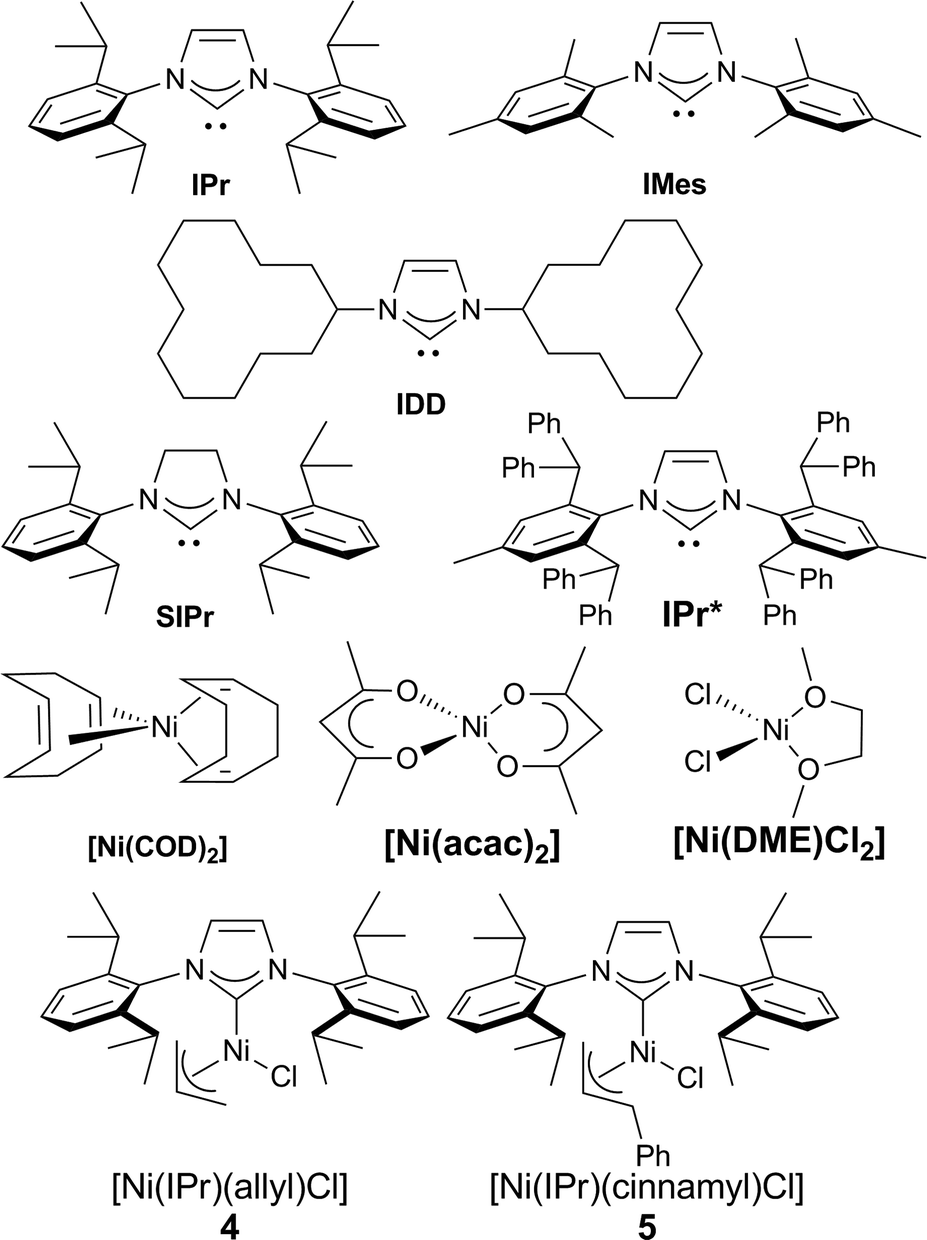 | ||
| Fig. 1 Ligands and complexes tested. | ||
We proceeded to examine the influence of the ligand: the use of smaller NHCs resulted in poor or no conversion (entries 2–3), while SIPr (entry 4) gave only a moderate yield. The use of the very bulky IPr* ligand, which usually provides the best outcome when employed in cross coupling chemistry,18 resulted in a lower yield (entry 5). As we identified IPr as the optimal ligand, in our initial reaction, the IPr-bearing well-defined catalysts 4 and 5 (entries 6–7) were tested. In contrast to previous examples of Ni–NHC catalyzed reactions,13n,15d both pre-catalysts gave poorer results compared to the in situ prepared [Ni(COD)2]/IPr system. We suspect the lower efficiency shown by the preformed pre-catalysts is due to the inability of the 2-azaallyl anion to effectively activate the Ni(II) center. This is an issue we are currently addressing in the design and synthesis of novel nickel-based pre-catalysts. To complete the optimization, temperature effects were examined and yields decreased with higher temperature (entry 8). The concentration could be increased to 0.17 M (entry 9), but further increase led to dramatic decrease in yield (entry 10). The optimal metal/ligand ratio was found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (entry 11). The use of a representative phosphine ligand, PCy3, resulted in no conversion. Similar results were obtained when other Ni sources, such as [Ni(acac)2] and [Ni(DME)Cl2] were employed. Interestingly, further increasing the amount of ligand completely suppressed the reaction. This result suggests that a monoligated Ni species is possibly the catalytically active species, and large excess of ligand moves the equilibrium towards the more stable but inactive bis-ligated species. Gratifyingly, the relatively mild operating temperature does not lead to the formation of isomeric mixtures. It is important to underline that, contrarily to previous reports,10a,b slow addition of the base is unnecessary, thus making our protocol operationally simple.
:2 (entry 11). The use of a representative phosphine ligand, PCy3, resulted in no conversion. Similar results were obtained when other Ni sources, such as [Ni(acac)2] and [Ni(DME)Cl2] were employed. Interestingly, further increasing the amount of ligand completely suppressed the reaction. This result suggests that a monoligated Ni species is possibly the catalytically active species, and large excess of ligand moves the equilibrium towards the more stable but inactive bis-ligated species. Gratifyingly, the relatively mild operating temperature does not lead to the formation of isomeric mixtures. It is important to underline that, contrarily to previous reports,10a,b slow addition of the base is unnecessary, thus making our protocol operationally simple.
|
|
||||
|---|---|---|---|---|
| Entry | [Ni] (5 mol%) | L (mol%) | Conc. (mol L−1) | Conv.b (yield)c |
| a Conditions: 4-chlorotoluene (0.25 mmol), imine 1a (2.0 equiv.), KHMDS (2.0 equiv.), toluene (1.0–2.5 mL), Ni source (2.5–5 mol%), ligand (3–10 mol%), 45 °C, 16 hours. b Calculated by G.C. analysis. c Yield calculated by NMR analysis using dimethyl malonate as an internal standard. d Reaction performed at 60 °C. | ||||
| 1 | [Ni(COD)2] | IPr (6) | 0.10 | >95 (81) |
| 2 | [Ni(COD)2] | IMes (6) | 0.10 | 24 |
| 3 | [Ni(COD)2] | IDD (6) | 0.10 | — |
| 4 | [Ni(COD)2] | SIPr (6) | 0.10 | 94 (65) |
| 5 | [Ni(COD)2] | IPr* (6) | 0.10 | >95 (60) |
| 6 | 4 | — | 0.10 | 70 (45) |
| 7 | 5 | — | 0.10 | >95 (70) |
| 8d | [Ni(COD)2] | IPr (6) | 0.10 | >95 (72) |
| 9 | [Ni(COD)2] | IPr (6) | 0.17 | >95 (85) |
| 10 | [Ni(COD)2] | IPr (6) | 0.25 | >95 (53) |
| 11 | [Ni(COD)2] | IPr (10) | 0.17 | >95 (93) |
| 12 | [Ni(COD)2] | PCy3 (10) | 0.17 | — |
| 13 | [Ni(acac)2] | IPr (10) | 0.17 | Traces |
| 14 | [Ni(DME)Cl2] | IPr (10) | 0.17 | — |
| 15 | [Ni(COD)2] | IPr (15) | 0.17 | — |

Once the optimal reaction conditions were established, we sought to explore the generality of the new protocol by varying the nature of the aryl chloride coupled with 1a (see Scheme 3). We were pleased to find that both the electron-rich 4-chloroanisole 2b and the electron-poor chlorides 2c and 2d led to high yields of the desired products. The compatibility of functionalized aryl chlorides, bearing functional groups such as amines (3e), benzodioxole (3g) and relatively sensitive ketone and nitrile derivatives (3g and 3h) was then examined. In all cases, good to very good yields were obtained. The use of heterocyclic (3i) and hindered aryl-chlorides (3j and 3k) was also possible; in these cases, complete conversion required a catalyst loading of 7.5 mol%. Compound 3k was isolated after hydrolysis, as the reaction mixture contained a small impurity that was not possible to remove by column chromatography (see ESI†). The results obtained in the coupling of bulky aryl chlorides clearly improve on previous Pd-based reports. Imines 1b and 1c, bearing respectively a 4-methoxyphenyl and a 4-fluorophenyl moiety on the benzylamine starting material afforded the coupling products with chlorobenzene in good yields (see entries 3b-2 and 3c-2). To highlight some of the limitations of the method, heterocyclic substrates 6–10 proved unsuitable in this transformation.
 | ||
| Scheme 3 Reaction scope of the Ni-catalyzed arylation of C(sp3)–H bonds. Reaction conditions: aryl chloride (2) (0.25 mmol), 1 (2.0 equiv.), KHMDS (2.0 equiv.), toluene (1.5 mL), [Ni(COD)2] (5 mol%), IPr (10 mol%), 45 °C, 16 h. [a] 4 (7.5 mol%), IPr (15 mol%). [b] Isolated yield of the corresponding ammonium chloride salt. | ||
Encouraged by these results and to further increase the scope and demonstrate the versatility of this catalytic system, the methodology was tested on the commercially available imine 1d. As the deprotonation of 1a and 1d converge to the same intermediate Int-1, a unique final product was expected (see Scheme 2). Under the optimized reaction conditions, the desired coupling products were indeed obtained and in very good yields (see Scheme 4).
 | ||
| Scheme 4 Reaction scope of the Ni-catalyzed arylation of C(sp3)–H bonds. Reaction conditions: aryl chloride (2) (0.25 mmol), 1 (2.0 equiv.), KHMDS (2.0 equiv.), toluene (1.5 mL), [Ni(COD)2] (5 mol%), IPr (10 mol%), 45 °C, 16 h. | ||
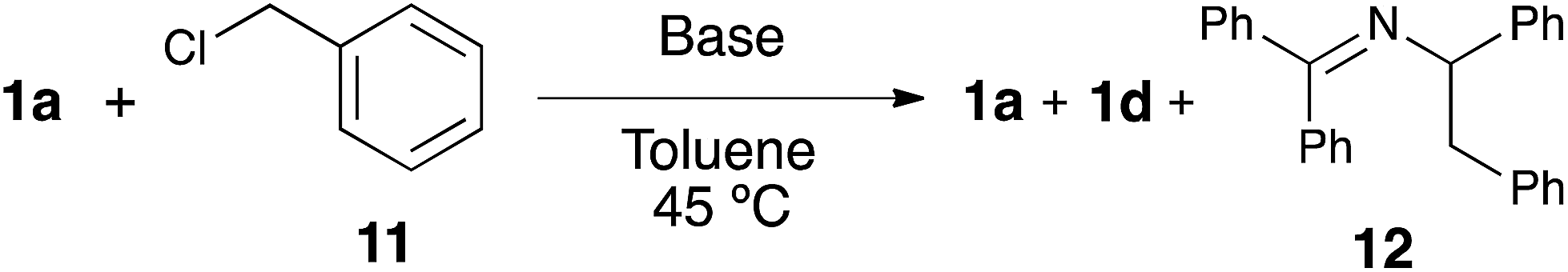
In order to shed light on the exact role of the base in this reaction, we performed the alkylation of 1a using benzyl chloride under the catalytic arylation reaction conditions (toluene, 45 °C) in the absence of the catalyst, using three different bases: KOtBu, NaHMDS and the optimal KHMDS (Table 2). We found that in the presence of a base weaker than the azaallyl anion, such as a t-butoxide, lower amounts of alkylated product were observed (entry 3), and the crude NMR analysis showed the formation of significant amounts of side-products, which were absent in the reactions using HMDS containing-bases (entries 1 and 2). This observation led us to test NaHMDS and KHMDS in the absence of any electrophile, finding that while the latter leads only to the formation the expected starting material and the isomerized form 1d (entry 4), the use of NaHMDS caused side-products to arise (entry 5). No side-products were observed using KHMDS even when the catalytic Ni/IPr system was present in the reaction medium (entry 6). Although further studies are needed to elucidate the mechanism of this reaction, the fact that KHMDS is the only base which cleanly affords the azaallyl indicates that this could be a reasonable explanation for the lack of reactivity of other bases in the catalytic arylation reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | 11 (equiv.) | 12 (%) | Notes |
| a Conditions: benzyl chloride (0.24 mmol, 1.2 equiv., or none), imine 1a (0.2 mmol, 1.0 equiv.), base (0.4 mmol or 0.1 mmol, 2.0 equiv. or 0.5 equiv.), toluene (0.6 mmol), 45 °C, 3 hours. b Calculated by NMR analysis using dimethyl malonate as an internal standard. c Reaction performed in the presence of 5% [Ni(COD)2]/10% IPr. | ||||
| 1 | KHMDS (2.0) | 2.0 | 81 | — |
| 2 | NaHMDS (2.0) | 0.10 | 80 | — |
| 3 | KOtBu (2.0) | 0.10 | 60 | Side-products observed |
| 4 | KHMDS (0.5) | — | — | Only 1a and 1d observed |
| 5 | NaHMDS (0.5) | — | 24 | Side-products observed |
| 6c | KHMDS (0.5) | — | — | Only 1a and 1d observed |
Conclusions
In summary, we have developed a synthetic methodology to access the (diarylmethyl)amine motif via a high yielding Ni-catalyzed coupling between C(sp3)–H bonds of benzylimine pro-nucleophiles and aryl chlorides. This work discloses the use of a commercially available Ni-based catalytic system under mild and operationally simple conditions. We hope to soon report on related Ni-catalyzed processes, as well as on the details of the reaction mechanism.Acknowledgements
We thank the ERC (Advanced Researcher award-FUNCAT), the EPSRC (grant no EP/J011053/1) and King Saud University for funding. We thank the EPSRC NMSSC in Swansea for mass spectrometric analyses. SPN is a Royal Society Wolfson Research Merit Award holder. We thank Dr Marcel Brill for useful discussions during the assembly of this paper.Notes and references
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (d) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644–4680 CrossRef CAS PubMed; (e) J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed; (f) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed; (g) J. G. De Vries, Top. Organomet. Chem., 2012, 42, 1–34 CAS; (h) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- (a) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS PubMed; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (c) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (d) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (e) F. Bellina and R. Rossi, Chem. Rev., 2009, 110, 1082–1146 CrossRef PubMed; (f) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (g) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191–206 RSC; (h) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (i) R. Ferraccioli, Synthesis, 2013, 45, 581–591 CrossRef CAS PubMed.
- (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (b) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed; (c) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS PubMed.
- (a) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed; (b) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676–707 CrossRef CAS PubMed; (c) T. Ankner, C. C. Cosner and P. Helquist, Chem.–Eur. J., 2013, 19, 1858–1871 CrossRef CAS PubMed; (d) S. T. Sivanandan, A. Shaji, I. Ibnusaud, C. C. C. J. Seechurn and T. J. Colacot, Eur. J. Org. Chem., 2015, 2015, 38–49 CrossRef CAS PubMed; for recent reports, see: (e) E. Marelli, M. Corpet, S. R. Davies and S. P. Nolan, Chem.–Eur. J., 2014, 20, 17272–17276 CrossRef CAS PubMed; (f) B. Zheng, T. Jia and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 165–178 CrossRef CAS PubMed.
- (a) F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269–10310 CrossRef CAS PubMed; (b) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673–714 CrossRef CAS PubMed; for recent examples, see: (c) L. Ackermann and S. Fenner, Chem. Commun., 2011, 47, 430–432 RSC; (d) L. Ackermann, C. Kornhaass and Y. Zhu, Org. Lett., 2012, 14, 1824–1826 CrossRef CAS PubMed; (e) S. I. Kozhushkov, H. K. Potukuchi and L. Ackermann, Catal. Sci. Technol., 2013, 3, 562–571 RSC; (f) D.-T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450–7453 CrossRef CAS PubMed.
- Directing group assisted CH activation at the benzylic position: (a) L.-C. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266–3267 CrossRef CAS PubMed; (b) J. J. Mousseau, A. Larivée and A. B. Charette, Org. Lett., 2008, 10, 1641–1643 CrossRef CAS PubMed; (c) N. Dastbaravardeh, M. Schnürch and M. D. Mihovilovic, Org. Lett., 2012, 14, 1930–1933 CrossRef CAS PubMed; (d) N. Dastbaravardeh, M. Schnürch and M. D. Mihovilovic, Eur. J. Org. Chem., 2013, 2013, 2878–2890 CrossRef CAS PubMed.
- Metal-mediated CH activation at the benzylic position: (a) V. N. Kalinin, I. A. Cherepanov and S. K. Moiseev, J. Organomet. Chem., 1997, 536–537, 437–455 CrossRef; (b) G. I. McGrew, J. Temaismithi, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2010, 49, 5541–5544 CrossRef CAS PubMed; (c) G. I. McGrew, C. Stanciu, J. Zhang, P. J. Carroll, S. D. Dreher and P. J. Walsh, Angew. Chem., Int. Ed., 2012, 51, 11510–11513 CrossRef CAS PubMed.
- (a) J.-I. Inoh, T. Satoh, S. Pivsa-Art, M. Miura and M. Nomura, Tetrahedron Lett., 1998, 39, 4673–4676 CrossRef CAS; (b) G. Dyker, J. Heiermann, M. Miura, J.-I. Inoh, S. Pivsa-Art, T. Satoh and M. Nomura, Chem.–Eur. J., 2000, 6, 3426–3433 CrossRef CAS; (c) T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 2373–2375 CrossRef CAS PubMed; (d) J. Zhang, A. Bellomo, A. D. Creamer, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2012, 134, 13765–13772 CrossRef CAS PubMed; (e) N. Hussain, G. Frensch, J. Zhang and P. J. Walsh, Angew. Chem., Int. Ed., 2014, 53, 3693–3697 CrossRef CAS PubMed.
- (a) T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 4689–4691 CrossRef CAS PubMed; (b) T. Niwa, T. Suehiro, H. Yorimitsu and K. Oshima, Tetrahedron, 2009, 65, 5125–5131 CrossRef CAS PubMed.
- (a) Y. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 4500–4503 CrossRef CAS PubMed; (b) J. Zhang, A. Bellomo, A. D. Creamer, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2012, 134, 13765–13772 CrossRef CAS PubMed; (c) M. Li, B. Yucel, J. Adrio, A. Bellomo and P. J. Walsh, Chem. Sci., 2014, 5, 2383–2391 RSC.
- (a) C. Gosmini, J.-M. Begouin and A. Moncomble, Chem. Commun., 2008, 3221–3233 RSC; (b) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13–31 RSC; (c) G. Evano, C. Theunissen and A. Pradal, Nat. Prod. Rep., 2013, 30, 1467–1489 RSC; (d) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed; (e) W. M. Czaplik, M. Mayer, J. Cvengroš and A. J. von Wangelin, ChemSusChem, 2009, 2, 396–417 CrossRef CAS PubMed.
- (a) W. Keim, Angew. Chem., Int. Ed., 1990, 29, 235–244 CrossRef PubMed; (b) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- (a) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC; (b) B. M. Rosen, K. W. Quasdorf, D. A Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (c) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC; (d) M. Henrion, V. Ritleng and M. J. Chetcuti, ACS Catal., 2015, 5, 1283–1302 CrossRef CAS; (e) S. Son and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 2756–2757 CrossRef CAS PubMed; (f) F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 8347–8349 CrossRef CAS PubMed; (g) C. Lohre, T. Dröge, C. Wang and F. Glorius, Chem.–Eur. J., 2011, 17, 6052–6055 CrossRef CAS PubMed; (h) A. R. Martin, D. J. Nelson, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Eur. J. Org. Chem., 2014, 3127–3131 CrossRef CAS PubMed; (i) S. Z. Tasker, A. C. Gutierrez and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 1858–1861 CrossRef CAS PubMed; (j) E. A. Standley, S. J. Smith, P. Müller and T. F. Jamison, Organometallics, 2014, 33, 2012–2018 CrossRef CAS PubMed; (k) K. Nakai, Y. Yoshida, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2014, 136, 7797–7800 CrossRef CAS PubMed; (l) J. Cornella, E. P. Jackson and R. Martin, Angew. Chem., Int. Ed., 2015, 54, 4075–4078 CrossRef CAS PubMed; (m) J. A. Fernández-Salas, E. Marelli, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2015, 21, 3906–3909 CrossRef PubMed; (n) S. Ge, R. A. Green and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 1617–1627 CrossRef CAS PubMed; (o) S. Ge and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 16330–16333 CrossRef CAS PubMed; (p) S. Ge and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 12837–12841 CrossRef CAS PubMed; (q) R. A. Green and J. F. Hartwig, Angew. Chem., Int. Ed., 2015, 54, 3768–3772 CrossRef CAS PubMed; (r) A. Borzenko, N. L. Rotta-Loria, P. M. MacQueen, C. M. Lavoie, R. McDonald and M. Stradiotto, Angew. Chem., Int. Ed., 2015, 54, 3773–3777 CrossRef CAS PubMed; (s) A. M. Whittaker and V. M. Dong, Angew. Chem., Int. Ed., 2015, 54, 1312–1315 CrossRef CAS PubMed; (t) M. Grigalunas, T. Ankner, P.-O. Norrby, O. Wiest and P. Helquist, J. Am. Chem. Soc., 2015, 137, 7019–7022 CrossRef CAS PubMed.
- (a) H. Shiota, Y. Ano, Y. Aihara, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 14952–14955 CrossRef CAS PubMed; (b) I. Hyodo, M. Tobisu and N. Chatani, Chem.–Asian J., 2012, 7, 1357–1365 CrossRef CAS PubMed; (c) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789–1792 CrossRef CAS PubMed; (d) M. Li, J. Dong, X. Huang, K. Li, Q. Wu, F. Song and J. You, Chem. Commun., 2014, 50, 3944–3946 RSC; (e) X. Wu, Y. Zhao and H. Ge, Chem.–Eur. J., 2014, 20, 9530–9533 CrossRef CAS PubMed; (f) C. Lin, W. Yu, J. Yao, B. Wang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 1340–1343 CrossRef CAS PubMed; (g) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 4069–4072 RSC; (h) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098–13101 CrossRef CAS PubMed; (i) L. Ackermann, B. Punji and W. Song, Adv. Synth. Catal., 2011, 353, 3325–3329 CrossRef CAS PubMed; (j) W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477–2480 CrossRef CAS PubMed.
- (a) S. Yao and M. Driess, Acc. Chem. Res., 2011, 45, 276–287 CrossRef PubMed; (b) T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106–9109 CrossRef CAS PubMed; (c) T. León, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221–1224 CrossRef PubMed; (d) Y. Makida, E. Marelli, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 8010–8013 RSC; (e) C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826–7827 CrossRef CAS PubMed; (f) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2013, 136, 1062–1069 CrossRef PubMed; (g) Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212–11215 CrossRef CAS PubMed; (h) T. Moragas, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 17702–17705 CrossRef CAS PubMed.
- (a) Y. Ko, D. C. Malone and E. P. Armstrong, Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 2006, 26, 1694–1702 CrossRef CAS PubMed; (b) S. A. Doggrell and L. C. Liang, Naunyn-Schmiedeberg's Arch. Pharmacol., 1998, 357, 126–132 CrossRef CAS; (c) N. Plobeck, D. Delorme, Z.-Y. Wei, H. Yang, F. Zhou, P. Schwarz, L. Gawell, H. Gagnon, B. Pelcman, R. Schmidt, S. Y. Yue, C. Walpole, W. Brown, E. Zhou, M. Labarre, K. Payza, S. St-Onge, A. Kamassah, P.-E. Morin, D. Projean, J. Ducharme and E. Roberts, J. Med. Chem., 2000, 43, 3878–3894 CrossRef CAS PubMed.
- Walsh also observed a need for a very strong base to permit deprotonative cross couplings (see ref. 8d).
- F. Izquierdo, S. Manzini and S. P. Nolan, Chem. Commun., 2014, 50, 14926–14937 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and optimization details, characterization of all the synthesized products. See DOI: 10.1039/c5sc01589h |
| This journal is © The Royal Society of Chemistry 2015 |