
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to enantioenriched 2,3- and 2,5-dihydrofurans with a fully substituted C2 stereocenter by Pd-catalyzed asymmetric intermolecular Heck reaction†
Gustavo M.
Borrajo-Calleja
a,
Vincent
Bizet
a,
Thomas
Bürgi
b and
Clément
Mazet
*a
aDepartment of Organic Chemistry, University of Geneva, 30 quai Ernest Ansermet, 1211 Geneva-4, Switzerland. E-mail: clement.mazet@unige.ch
bDepartment of Physical Chemistry, University of Geneva, 30 quai Ernest Ansermet, 1211 Geneva-4, Switzerland
First published on 3rd June 2015
Abstract
A palladium-catalyzed intermolecular asymmetric Heck reaction with dihydrofurans with a trisubstituted double bond is reported. The use of two different chiral ligands provides access to valuable 2,3- and 2,5-dihydrofurans with a fully substituted C2 stereocenter with high levels of regio- and enantiocontrol.
Introduction
During the past decades, building on the seminal contributions of Overman and Shibasaki, the asymmetric intramolecular Heck reaction has been successfully applied in natural product synthesis to install tertiary and quaternary stereocenters.1,2 In stark contrast, the intermolecular version of this asymmetric transformation has not reached the same level of development and its synthetic utility remains limited (Fig. 1).3 A survey of the literature following Hayashi's pioneering contribution indicates that this transformation has been used essentially as a benchmark reaction to validate the design of novel homotopic and heterotopic chiral ligands.4,5 While variations of the electronic and steric nature of the aryl halides and pseudo-halides has been investigated in detail, most of these studies have been focused on the use of dihydrofurans or N-protected dihydropyrroles to afford the corresponding coupling products with a C2 tertiary center. Systematic studies where emphasis has been placed on exploring the diversity of the olefinic coupling partner are scarce, in particular for the construction of congested quaternary centers or related fully saturated centers. Very recently, Sigman has reported a series of elegant contributions for the redox-relay Heck arylations of acyclic alkenyl alcohols enabling the installation of remote tertiary and quaternary stereogenic centers.6,7 Despite these recent breakthroughs, it is apparent that the development of an intermolecular asymmetric Heck reaction to enable the construction of fully saturated stereocenters in cyclic systems constitutes an important synthetic challenge.8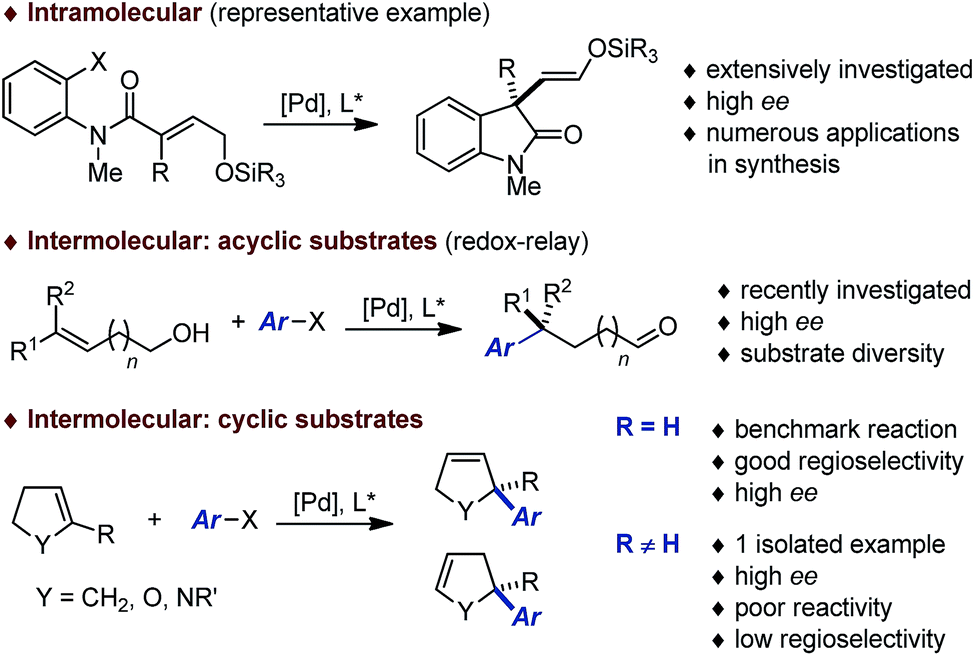 | ||
| Fig. 1 Access to quaternary and related fully saturated stereocenters by asymmetric Heck reactions. | ||
In this report, we disclose a methodology that gives access to chiral 2,3- and 2,5-dihydrofurans (dhfs) with a fully substituted C2 stereocenter. Our study highlights the complementarity in product selectivity between two distinct chiral ligands while it also delineates the scope of trisubstituted dihydrofurans compatible with the catalytic systems.
Results and discussion
The choice of our model reaction was inspired by a report from Pregosin and co-workers where 5-methyl-2,3-dihydrofuran 1a was cross-coupled with phenyl triflate 2a using (R)-DTB-MeO-BIPHEP (i.e. (R)-(+)-2,2′-bis[di(3,5-di-tert-butylphenyl)phosphino]-6,6′-dimethoxy-1,1′-biphenyl).9 To the best of our knowledge this represents the only precedent of an asymmetric intermolecular Heck reaction employing a cyclic substrate with a trisubstituted olefinic moiety. Despite an excellent enantioselectivity (4aa: 98% ee), the low yield (38%), the moderate regioselectivity (1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.0) and the impractical reaction time (7 days) certainly precluded further developments. We began our investigations by evaluating an array of chiral (P,N) and (P,P) ligands as to their complementarity in providing access to the product of direct cross-coupling 3 and the product of further isomerization 4, respectively, as is well documented for unsubstituted cyclic substrates.4,5,10
:6.0) and the impractical reaction time (7 days) certainly precluded further developments. We began our investigations by evaluating an array of chiral (P,N) and (P,P) ligands as to their complementarity in providing access to the product of direct cross-coupling 3 and the product of further isomerization 4, respectively, as is well documented for unsubstituted cyclic substrates.4,5,10
With an initial set of reaction conditions (Pd(OAc)2, toluene, i-Pr2NEt, 110 °C, 62 h), only traces of products 3aa and 4aa were observed in the coupling between 1a and 2a using L1 (Table 1, entry 1). Evaluation of two of our home-made chiral (P,N) ligands did not significantly enhance the reactivity, even though 3aa was the only detectable product of cross-coupling.11 While 3aa was obtained in 76% ee with L3, L4 gave an excellent 94% ee value (entry 2 and 3). A slightly improved reactivity along with similar regio- and enantioselectivities were obtained with the electron-rich 4-methoxyphenyl triflate 2b (entry 4). An extensive optimization of all reaction parameters was then conducted. Multiple combinations of palladium sources, solvents and bases along with adjustment of the temperature, stoichiometry, concentration and reaction time led to the identification of a very efficient catalytic system (Pd2(dba)3, 2-Me-THF, i-Pr2NEt, 100 °C, 48 h; see ESI† for details) affording 3ab in 64% and 93% ee with L4 (entry 5).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entryb | 2 | Ligand |
3:4c |
3 | 4 | ||
| Yieldd (%) | eee (%) | Yieldd (%) | eee (%) | ||||
| a Reaction conditions: 1a (1 mmol), 2a–c (0.2 mmol). b Entries 1–4: Pd(OAc)2 (3 mol%), ligand (6 mol%), toluene, 110 °C, 62 h. Entries 5–16: Pd2(dba)3 (2.5 mol%), ligand (10 mol%), 2-Me-THF, 100 °C, 48 h. c Determined by 1H NMR of the crude reaction mixture. d Yield of pure compound after chromatography. e Determined by GC or HPLC with a chiral column. f Not determined. g No reaction. | |||||||
| 1 | 2a | L1 | 22:78 |
<5 | ndf | 5 | 9 |
| 2 | 2a | L3 | 99:1 |
12 | 76 | — | — |
| 3 | 2a | L4 | 99:1 |
16 | 94 | — | — |
| 4 | 2b | L4 | 99:1 |
11 | 92 | — | — |
| 5 | 2b | L4 | 99:1 |
64 | 93 | — | — |
| 6 | 2c | L4 | — | nrg | — | nr | — |
| 7 | 2b | L5 | 98:2 |
34 | 98 | — | — |
| 8 | 2c | L5 | — | nr | — | nr | — |
| 9 | 2b | L2 | 44:56 |
21 | rac | 28 | 13 |
| 10 | 2c | L2 | 5:95 |
— | — | 55 | 93 |
| 11 | 2b | L6 | 5:95 |
— | — | 30 | 56 |
| 12 | 2c | L6 | 5:95 |
— | — | 32 | 62 |
| 13 | 2b | L7 | 5:95 |
— | — | 39 | 45 |
| 14 | 2c | L7 | 5:95 |
— | — | 34 | 51 |
| 15 | 2b | L8 | 57:43 |
<5 | — | <5 | — |
| 16 | 2c | L8 | 5:95 |
— | — | 51 | 97 |
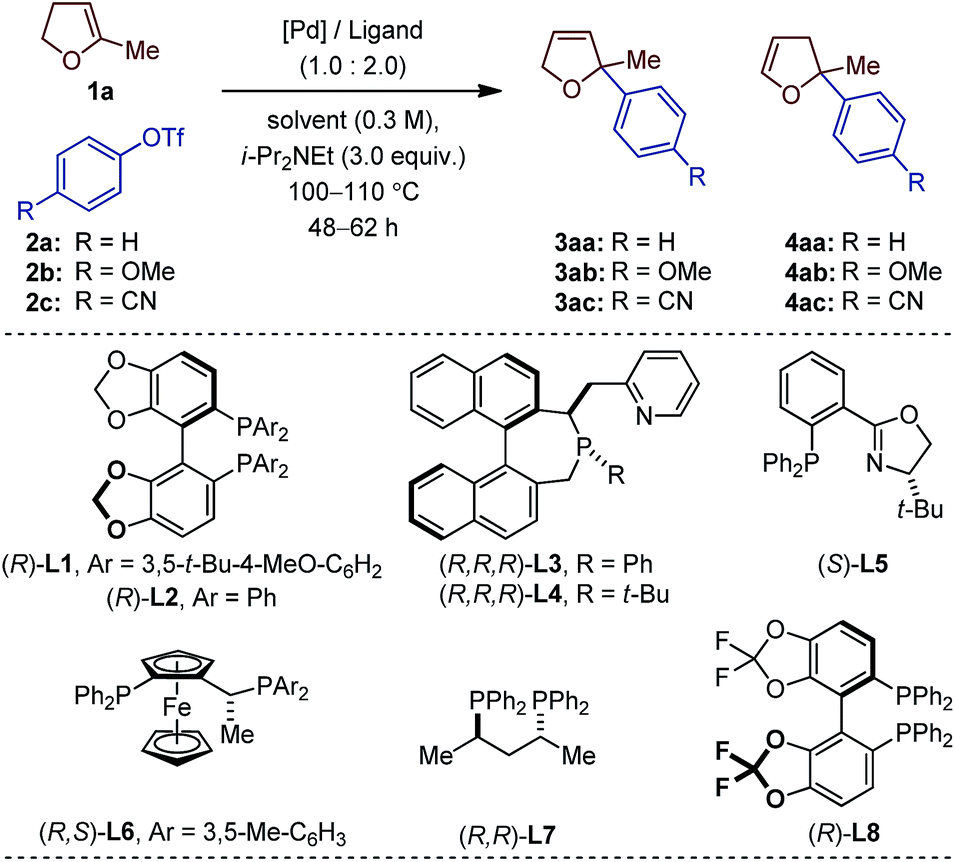
Of important note, while the catalyst loading, the relative stoichiometry between the coupling partners and the yields obtained (ca. 40–65%) are typical of intermolecular asymmetric Heck reactions, 48 h is unusually short for such a process involving cyclic olefins. Under classical thermal conditions, reaction times often range from 4 to 7 days.12 Unexpectedly, when the electron-deficient aryl triflate 2c was employed, no product of cross-coupling was observed.13 The prototypical (P,N) ligand L5 for intermolecular Heck reactions using unsubstituted dhf did not display better performances (entry 7 and 8). For sake of operational simplicity, the same protocol was used in the survey of several (P,P) ligands (entry 9–16). Consistent with literature precedents for the coupling of unsubstituted dhf, a marked regioselectivity switch in favor of the isomerized products 4 was observed with most ligands. Noticeably, while poor results were obtained in the coupling reactions with the electron-rich triflate 2b, the electron-deficient aryl triflate 2c permitted to reach excellent enantioselectivity levels with L2, and L8 (entry 10 and 16).14
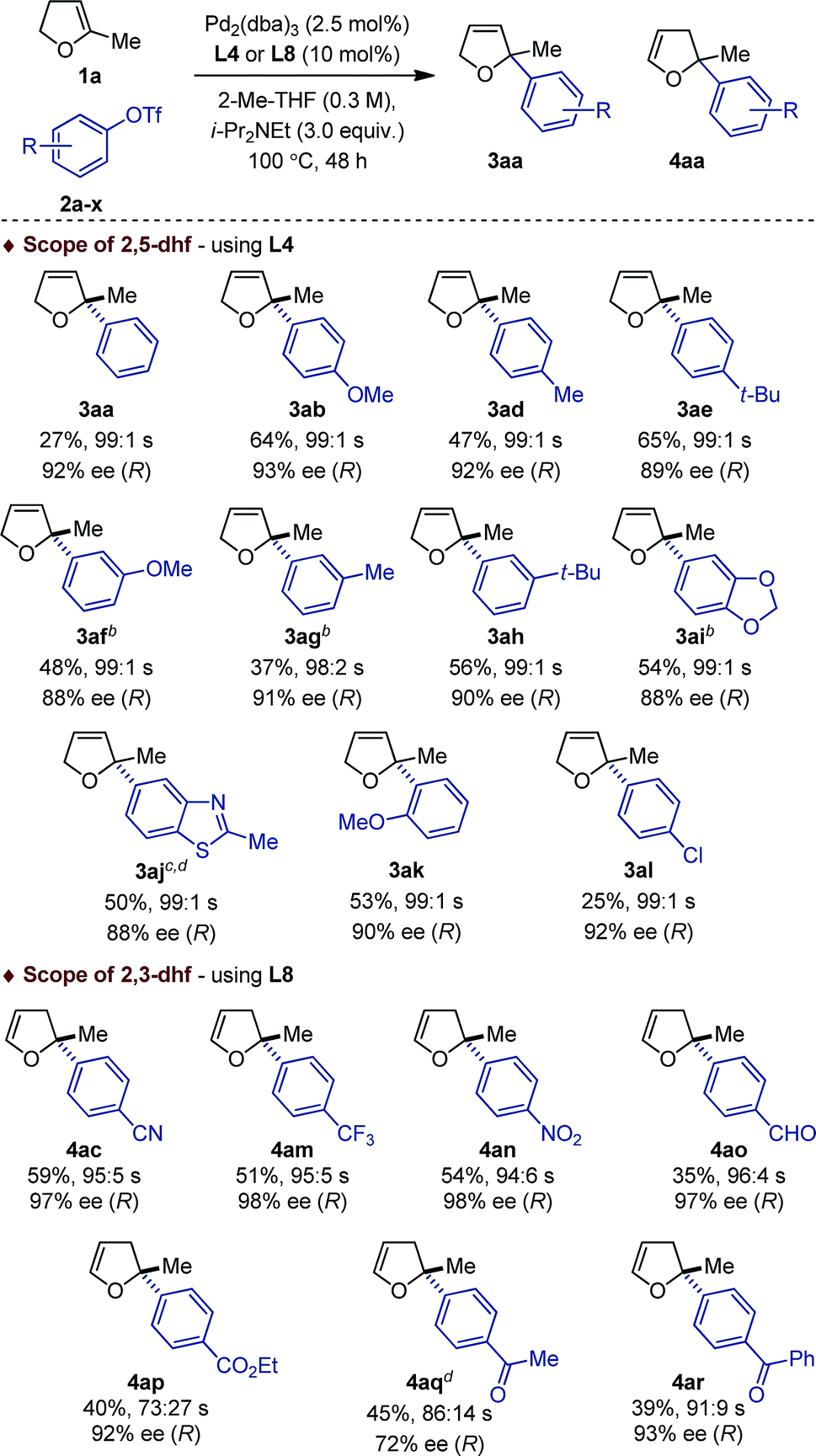
This dichotomous situation was further confirmed when the scope or aryl triflates compatible with both L4 and L8 was delineated (Table 2). With the (P,N) ligand L4, a wide variety of electron-rich aryl triflates afforded quasi-exclusively 2,5-dihydrofurans (98:2–99:1) in consistently very high enantioselectivity (88–93% ee) and in practical yields. Remarkably, even the sterically demanding o-methoxyphenyl triflate 2k was cross-coupled efficiently (3ak: 53%, 99:1, 90% ee). Reduced yields were obtained with 2a and 2l, albeit the regio- and enantioselectivities remained excellent. Attempts to couple electron-deficient aryl triflates were unsuccessful. In contrast, with the (P,P) ligand L8, only electron-deficient aryl triflates 2c, 2m–r were found to be compatible electrophilic coupling partners. This afforded 2,3-dihydrofurans with a saturated C2 stereocenter in excellent enantioselectivity and allowed functional groups such as cyano, nitro, trifluoromethyl, ester, aldehyde, aryl and alkyl ketones to be introduced.
|
a
Reaction conditions: 1a (1 mmol), 2a–c (0.2 mmol). Absolute configurations determined by VCD. The regioselectivity 3:4 is denoted by s.
b
3 days. c4 days. d95% purity. |
|---|
|
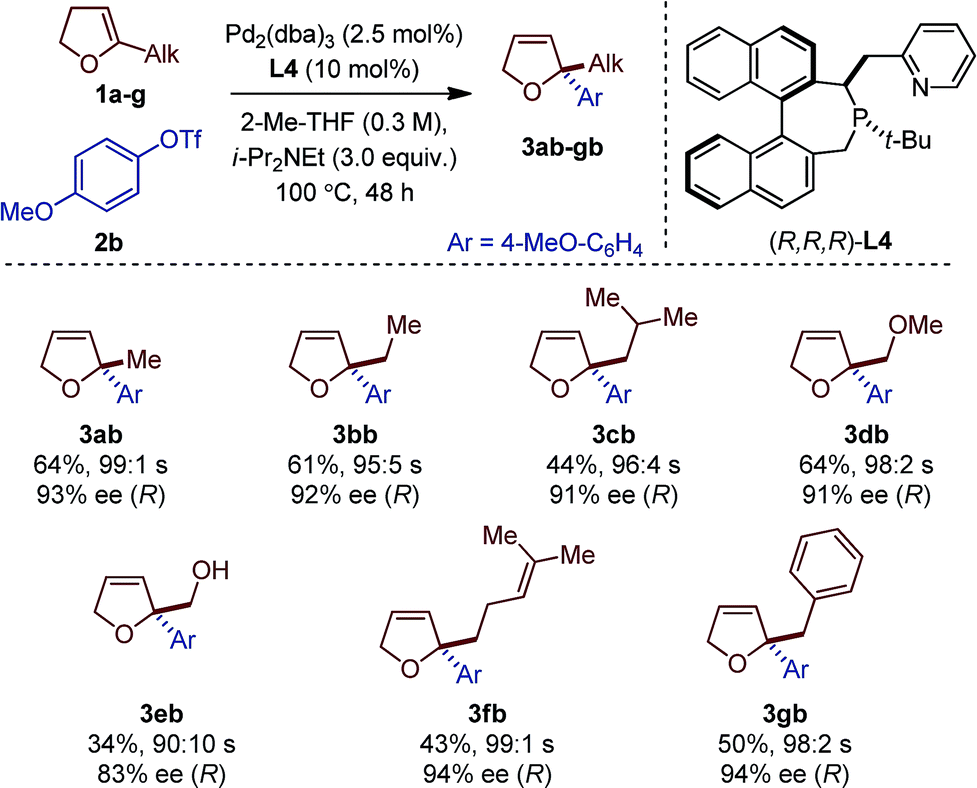
Seven different 5-substituted 2,3-dhfs were found to be suitable substrates with both methodologies (Table 3 and 4). These compounds proved particularly unstable and presented challenges for study because of the difficulty associated with their preparation and purification (see ESI†).
| a Reaction conditions: 1a (1 mmol), 2a–c (0.2 mmol). Absolute configurations determined by VCD. |
|---|
|
| a Reaction conditions: 1a (1 mmol), 2a–c (0.2 mmol). Absolute configurations determined by VCD. |
|---|
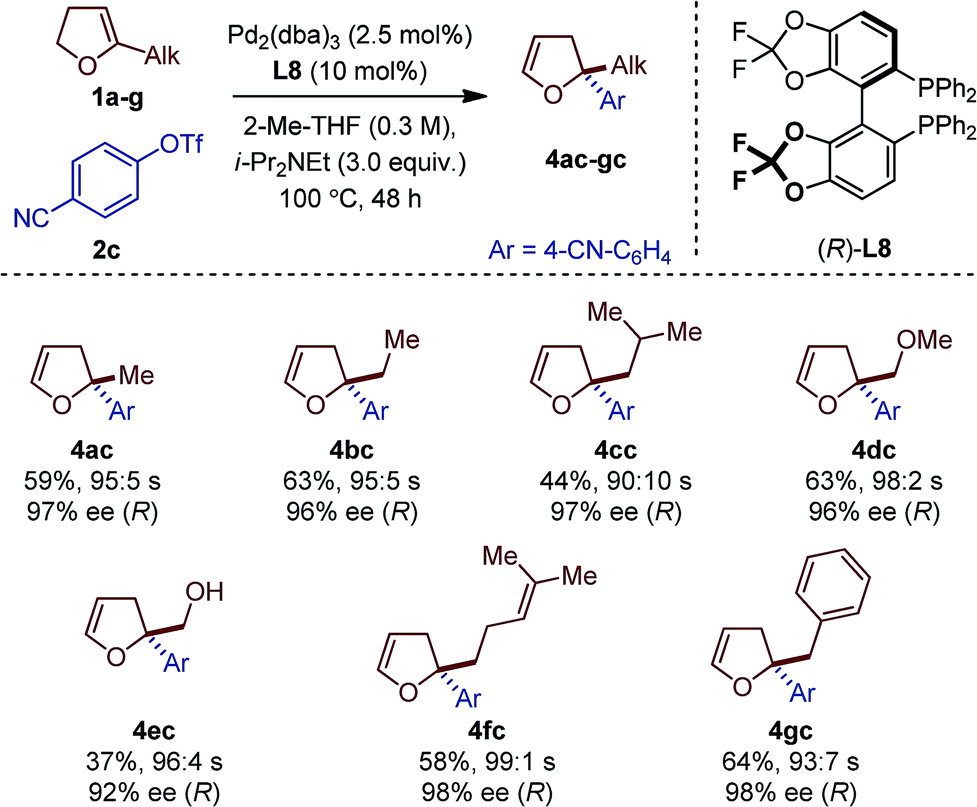
|
The C1-symmetric (P,N) ligand L4 and the C2-symmetric (P,P) ligand L8 gave access to 2,2-disubstituted 2,5-dhf and 2,2-disubstituted 2,3-dhf, respectively, in usually practical yields and with excellent regio- and enantioselectivity. Both catalytic systems behaved similarly and overall excellent results were obtained. The yield range obtained for these reactions are consistent with literature precedents.2,3,6 They do not only reflect the entropic cost inherent to an intermolecular cross-coupling reaction but also the difficulty associated with creating saturated stereocenters. In addition to primary alkyl substituents, a methyl ether, a free alcohol, a remote olefin and a benzyl substituent were compatible. Whereas the yields were slightly reduced in the reactions with 3e, it is remarkable that no competing Heck arylation was observed in the reactions with 1f which bears a remote olefinic moiety.6 Finally, a large-scale experiment (8.5 mmol) was conducted for the cross-coupling between 1a and 2c (Fig. 2). Using (R)-L2, the isomerized product (R)-4ac was isolated with excellent regioselectivity, good enantioselectivity and in 46% yield (0.725 g).15
 | ||
| Fig. 2 Large-scale experiment for the intermolecular asymmetric Heck reaction. | ||
The absolute configuration of the cross-coupling products was established by vibrational circular dichroism (VCD).16 The most stable conformer of 3ab (obtained with (R,R,R)-L4) and 4ap (obtained with (R)-L8) were compared to the experimental IR absorption and VCD spectra recorded in CD2Cl2 solutions (see ESI† for details). The good agreement between the predicted and measured data enabled the assignment of a (R) configuration to both compounds. The absolute configuration of all other cross-coupling products was assigned by analogy, on the basis of their optical activity.
Conclusions
In summary, we have developed an efficient protocol for the enantioselective intermolecular Heck reaction using a variety of 5-substituted 2,3-dihydrofurans. We found that electron-rich aryl triflates can be coupled with various trisubstituted olefinic substrates when a chiral (P,N) ligand is used. The corresponding 2,5-dihydrofurans with fully saturated C2 stereocenters were obtained in excellent regio- and enantioselectivities. Electron-deficient aryl triflates were compatible with the method when commercial chiral (P,P) ligands were employed, leading to 2,2-disubstituted 2,3-dihydrofurans with excellent stereocontrol. The diversity, functional group tolerance and scalability of the approach have been also demonstrated and provide access to enantioenriched dihydrofurans that would be difficult to prepare otherwise.17 The underlying mechanistic features responsible for the marked electronic dichotomy observed between the two ligand systems, along with extension of the method to other substrate classes, are currently being investigated in our laboratories.Acknowledgements
This work was supported by the University of Geneva. Stéphane Rosset is acknowledged for his expertise on analytical instruments and for assistance in ligand synthesis. Johnson-Matthey is acknowledged for a generous gift of palladium precursors.Notes and references
- (a) N. E. Carpenter, D. J. Kucera and L. E. Overman, J. Org. Chem., 1989, 54, 5846 CrossRef CAS; (b) Y. Sato, M. Sodeoka and M. Shibasaki, J. Org. Chem., 1989, 54, 4738 CrossRef CAS.
- (a) M. Oestreich, The Mizoroki–Heck Reaction, Wiley, New York, 2009 Search PubMed; (b) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (c) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS PubMed; (d) L. F. Tietze, H. Ila and H. P. Bell, Chem. Rev., 2004, 104, 3453 CrossRef CAS PubMed; (e) M. Diéguez and O. Pàmies, Isr. J. Chem., 2012, 52, 572 CrossRef PubMed; (f) M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 2282 CrossRef CAS PubMed.
- For a specific review on the intermolecular Heck reaction, see: D. Mc Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC.
- (a) F. Ozawa, A. Kubo and T. Hayashi, J. Am. Chem. Soc., 1991, 113, 1417 CrossRef CAS; (b) F. Ozawa and T. Hayashi, J. Organomet. Chem., 1992, 428, 267 CrossRef CAS.
- For selected leading examples, see: (a) F. Ozawa, Y. Kobatake and T. Hayashi, Tetrahedron Lett., 1993, 34, 2505 CrossRef CAS; (b) Y. Kurihara, M. Sodeoka and M. Shibasaki, Chem. Pharm. Bull., 1994, 42, 2357 CrossRef CAS; (c) O. Loiseleur, M. Hayashi, N. Schmees and A. Pfaltz, Synthesis, 1997, 1338 CrossRef CAS PubMed; (d) A. J. Hennessy, D. J. Connolly, Y. M. Malone and P. J. Guiry, Tetrahedron Lett., 2000, 41, 7757 CrossRef CAS; (e) S. T. Henriksen, P.-O. Norrby, P. Kaukoranta and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 10414 CrossRef CAS PubMed; (f) T. H. Wöste and M. Oestreich, Chem.–Eur. J., 2011, 17, 11914 CrossRef PubMed; (g) J. Hu, H. Hirao, Y. Li and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 8676 CrossRef CAS PubMed; (h) S. Liu and J. Zhou, Chem. Commun., 2013, 49, 11758 RSC; (i) C. Wu and J. Zhou, J. Am. Chem. Soc., 2014, 136, 650 CrossRef CAS PubMed.
- (a) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed; (b) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (c) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed; (d) L. Xu, M. J. Hilton, X. Zhang, P.-O. Norrby, Y.-D. Wu, M. S. Sigman and O. Wiest, J. Am. Chem. Soc., 2014, 136, 1960 CrossRef CAS PubMed; (e) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 3462 CrossRef CAS PubMed.
- Correia and co-workers reported a related enantioselective intermolecular Heck–Matsuda reaction employing (Z)-buten-1,4-diols or 3-cyclopentenol. We note that only tertiary centers were generated by this approach. For relevant references, see: (a) C. R. D. Correia, C. C. Oliveira, A. G. Salles Jr and E. A. F. Santos, Tetrahedron Lett., 2012, 53, 3325 CrossRef CAS PubMed; (b) C. C. Oliveira, R. A. Angnes and C. R. D. Correia, J. Org. Chem., 2013, 78, 4373 CrossRef CAS PubMed; (c) R. A. Angnes, J. M. Oliveira, C. C. Oliveira, N. C. Martins and C. R. D. Correia, Chem.–Eur. J., 2014, 20, 13117 CrossRef CAS PubMed.
- In the strict sense, the stereogenic centers generated in this study are not quaternary, even though they are very congested. We therefore use the terms ‘fully saturated stereocenters’ to highlight this notion. A fully saturated center is defined by a tetrasubstituted C atom with at least one substituent different from H or C. For relevant reviews on the challenges associated with installing congested stereocenters, see: (a) J. Christoffers and A. Baro, Quaternary Stereocentres: Challenges and Solutions for Organic Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) K. Fuji, Chem. Rev., 1993, 93, 2037 CrossRef CAS; (c) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS PubMed; (d) I. Marek and G. Sklute, Chem. Commun., 2007, 1683 RSC; (e) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC.
- M. Tschoerner, P. S. Pregosin and A. Albinati, Organometallics, 1999, 18, 670 CrossRef CAS.
- (a) R. Grigg, V. Loganathan, V. Santhakumar, V. Sridharan and A. Teasdale, Tetrahedron Lett., 1991, 32, 687 CrossRef CAS; (b) Y. Fall, F. Berthiol, H. Doucet and M. Santelli, Synthesis, 2007, 1683 CAS; (c) B. M. M. Wheatley and B. A. Keay, J. Org. Chem., 2007, 72, 7253 CrossRef CAS PubMed; (d) M. G. Lauer, M. K. Thompson and K. H. Shaughnessy, J. Org. Chem., 2014, 79, 10837 CrossRef CAS PubMed.
- (a) P. Nareddy, L. Mantilli, L. Guénée and C. Mazet, Angew. Chem., Int. Ed., 2012, 51, 3826 CrossRef CAS PubMed; (b) N. Humbert, E. Larionov, L. Mantilli, P. Nareddy, C. Besnard, L. Guénée and C. Mazet, Chem.–Eur. J., 2014, 20, 745 CrossRef CAS PubMed.
- Asymmetric intermolecular Heck reactions can be accelerated using microwave irradiation. However, in some cases a drop in enantioselectivity has been observed. When the reaction reported in Table 1 (entry 5) was conducted under microwave irradiation (150 °C, 2 h), 3ab was isolated in 38% yield and 87% ee. For relevant examples, see: (a) P. Nilsson, H. Gold, M. Larhed and A. Hallberg, Synthesis, 2002, 1611 CAS; (b) P. Kaukoranta, K. Källstrom and P. G. Andersson, Adv. Synth. Catal., 2007, 349, 2595 CrossRef CAS PubMed; (c) Y. Mata, O. Pàmies and M. Diéguez, Chem.–Eur. J., 2007, 13, 3296 CrossRef CAS PubMed; (d) J. Mazuela, O. Pàmies and M. Diéguez, Chem.–Eur. J., 2010, 16, 3434 CrossRef CAS PubMed.
- Analysis of the crude reaction mixture by 19F NMR indicates detriflation of 2c, suggesting that oxidative addition into the C(sp2)–O bond of the aryl triflate is not problematic.
- J. P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Chem. Rev., 2014, 114, 2824 CrossRef CAS PubMed.
- Ligand L2 is ca. 8-fold less-expensive than L8. This aspect and the relatively similar performances reported in Table 1 (entries 10 and 16) justify the use of L2 for large-scale experiments.
- (a) L. A. Nafie, T. A. Keiderling and P. J. Stephens, J. Am. Chem. Soc., 1976, 98, 2715 CrossRef CAS; (b) G. Holzwarth, E. C. Hsu, H. S. Mosher, T. R. Faulkner and A. Moscowitz, J. Am. Chem. Soc., 1974, 96, 251 CrossRef CAS; (c) T. B. Freedman, X. Cao, R. K. Dukor and L. A. Nafie, Chirality, 2003, 15, 743 CrossRef CAS PubMed.
- For potential use and derivatization of the products obtained in this work, see: J. Cornella and R. Martin, Org. Lett., 2013, 15, 6298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of all new compounds, VCD studies and spectral data. See DOI: 10.1039/c5sc01460c |
| This journal is © The Royal Society of Chemistry 2015 |