
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophilic sulfonated bis-1,2,4-triazine ligands are highly effective reagents for separating actinides(III) from lanthanides(III) via selective formation of aqueous actinide complexes†
Frank W.
Lewis
*ab,
Laurence M.
Harwood
*a,
Michael J.
Hudson
a,
Andreas
Geist
c,
Valery N.
Kozhevnikov
b,
Petr
Distler
d and
Jan
John
d
aDepartment of Chemistry, The University of Reading, Whiteknights, Reading RG6 6AD, UK. E-mail: l.m.harwood@reading.ac.uk
bDepartment of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne NE1 8ST, UK. E-mail: frank.lewis@northumbria.ac.uk
cKarlsruher Institut für Technologie (KIT-INE), Institut für Nukleare Entsorgung, Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany. E-mail: andreas.geist@kit.edu
dDepartment of Nuclear Chemistry, Czech Technical University in Prague, Břehová 7, 115 19 Prague 1, Czech Republic. E-mail: jan.john@fjfi.cvut.cz
First published on 28th May 2015
Abstract
We report the first examples of hydrophilic 6,6′-bis(1,2,4-triazin-3-yl)-2,2′-bipyridine (BTBP) and 2,9-bis(1,2,4-triazin-3-yl)-1,10-phenanthroline (BTPhen) ligands, and their applications as actinide(III) selective aqueous complexing agents. The combination of a hydrophobic diamide ligand in the organic phase and a hydrophilic tetrasulfonated bis-triazine ligand in the aqueous phase is able to separate Am(III) from Eu(III) by selective Am(III) complex formation across a range of nitric acid concentrations with very high selectivities, and without the use of buffers. In contrast, disulfonated bis-triazine ligands are unable to separate Am(III) from Eu(III) in this system. The greater ability of the tetrasulfonated ligands to retain Am(III) selectively in the aqueous phase than the corresponding disulfonated ligands appears to be due to the higher aqueous solubilities of the complexes of the tetrasulfonated ligands with Am(III). The selectivities for Am(III) complexation observed with hydrophilic tetrasulfonated bis-triazine ligands are in many cases far higher than those found with the polyaminocarboxylate ligands previously used as actinide-selective complexing agents, and are comparable to those found with the parent hydrophobic bis-triazine ligands. Thus we demonstrate a feasible alternative method to separate actinides from lanthanides than the widely studied approach of selective actinide extraction with hydrophobic bis-1,2,4-triazine ligands such as CyMe4-BTBP and CyMe4-BTPhen.
Introduction
In recent years there has been a renewed global interest in electricity production through nuclear power as many countries seek to satisfy their future energy needs while reducing their dependence on fossil fuels and their associated greenhouse gas emissions. As a result, nuclear power generation is expected to expand significantly in the next few decades, with several countries announcing plans for new reactor construction.1 The used nuclear fuel produced by the current light water reactors is comprised mainly of uranium, plutonium, the lanthanides (>98.5 wt%) and less than 1 wt% of the minor actinides Am(III), Cm(III) and Np(III). Currently, the uranium and plutonium are recovered and recycled for re-use as mixed-oxide (MOX) fuel in the PUREX process,2 but the remaining used fuel still contains the minor actinides, which are responsible for much of the long-term radiotoxicity (t1/2 = 103 to 106 years) and heat load of used fuel.One approach currently being studied for the long-term management of used fuel is the ‘partitioning and transmutation’ strategy.3 In this strategy, plutonium and the minor actinides will first be separated from fission products (including the lanthanides) by solvent extraction, and then used as fuel in the next generation of nuclear reactor designs. This separation is essential since some of the fission products and the lanthanides will absorb neutrons instead of the transmutable actinides. The separation of the actinides americium and curium from the lanthanides is considered a key step in increasing the safety and sustainability of nuclear energy,4 but is nevertheless a challenging goal as the chemical properties of the two groups of elements are very similar.5
There is believed to be a more covalent contribution to the metal-ligand bonding with the actinides than with the lanthanides, although the exact origins of this covalency are still not fully understood.6 Recent evidence from structural, spectroscopic and theoretical studies on a range of f-element complexes reinforce this view, although the extent and the nature of this covalent interaction appears to vary across the actinide series.7,8 Consequently, many soft N- and S-donor ligands have been extensively studied9 to perform the actinide–lanthanide separation by direct and selective extraction of the actinides from PUREX waste solutions (known as the SANEX process).10 N-donor ligands containing 1,2,4-triazine11 moieties have emerged as the most promising class of ligands to perform this separation. The tridentate 2,6-bis(1,2,4-triazin-3-yl)pyridines (BTPs)12 and the tetradentate 6,6′-bis(1,2,4-triazin-3-yl)-2,2′-bipyridines (BTBPs)13 have been extensively studied for this purpose in recent years. It has been shown that the annulated BTBP ligand 1 (Fig. 1) is capable of performing the selective extraction of the minor actinides directly from nitric acid solutions into an organic solvent,14 and various laboratory demonstrations of this separation have been successfully carried out on both simulated and genuine waste solutions.15 The more pre-organized 2,9-bis(1,2,4-triazin-3-yl)-1,10-phenanthroline (BTPhen) ligand 2 was recently reported as a highly efficient and selective minor actinide extraction agent with greatly improved properties compared to 1.16 Very recently, magnetic nanoparticles functionalized with ligand 2 were shown to quantitatively separate Am(III) from Eu(III),17 paving the way for the application of ligands such as 2 in solid-phase separations. Moreover, it has been shown that two 1,2,4-triazine moieties are required for efficient and selective extractions by polypyridine N-donor ligands.18
 | ||
| Fig. 1 Structures of the ligands CyMe4-BTBP 1, CyMe4-BTPhen 2, di(2-ethylhexyl)phosphoric acid 3 and diethylenetriaminepentaacetic acid (DTPA) 4. | ||
An alternative method for carrying out the actinide–lanthanide separation has been proposed in several countries. This approach involves the non-selective co-extraction of actinides and lanthanides into an organic phase, followed by selective actinide back-extraction (or stripping) into an aqueous phase using a hydrophilic actinide-selective aqueous complexing agent. This is illustrated by the TALSPEAK process which was developed in the 1960s at Oak Ridge National Laboratory in the USA.19 In this process, an acidic organophosphorus reagent such as di(2-ethylhexyl)phosphoric acid 3 is employed as the extractant and a polyaminocarboxylate ligand such as diethylenetriaminepentaacetic acid (DTPA) 4 (Fig. 1) is used as the actinide-selective hydrophilic complexing agent.
Unfortunately, this process requires the use of carboxylic acid buffers such as lactic acid or citric acid, which would result in additional secondary waste generation, and only operates within a narrow range of pH (pH 2–3) which is not compatible with that typically found in genuine PUREX waste solutions (pH ≤ 0). Despite extensive studies involving different combinations of hydrophobic extractants and hydrophilic aqueous complexant/buffer systems, as well as studies examining the influence of various operational parameters (e.g.: nature of the organic diluent, pH, temperature),20 the TALSPEAK process has not yet reached the level of maturity required for industrial implementation.
In order to overcome the limitations of these processes, we sought to develop water-soluble hydrophilic derivatives of the highly effective bis-1,2,4-triazine N-donor ligands developed to date.21 Furthermore, it is worth noting that the highly selective BTP and BTBP ligands retain their actinide binding selectivity when dissolved in aqueous solutions.22 We therefore reasoned that hydrophilic sulfonated bis-1,2,4-triazine ligands would be promising reagents for selective actinide complexation even at the high nitric acid concentrations usually found in genuine waste solutions without the need for additional buffers. Indeed, a sulfonated BTP ligand was found to have excellent selectivity for actinides over lanthanides under these conditions.23,24 In this article, we report the results of our further studies on sulfonated bis-1,2,4-triazine ligands as highly effective reagents for carrying out actinide–lanthanide separations via selective actinide aqueous complex formation.
Results and discussion
Ligand synthesis
The sulfonated bis-triazine ligands were synthesized by the sulfonation of the phenyl rings of both di- and tetraphenyl bis-1,2,4-triazine ligands.25 The di- and tetraphenyl bis-1,2,4-triazine ligands were obtained by the condensation reactions of diamide dihydrazides with either benzil or phenylglyoxal.26 The synthesis of disulfonated BTBP ligands (DS-BTBP) and tetrasulfonated BTBP ligands (TS-BTBP) is shown in Scheme 1. The reactions of diamide dihydrazide 5 with benzil 6 and phenylglyoxal 7 afforded novel BTBPs 8 and 9, respectively. In the synthesis of 9, a single regioisomer was obtained, which was assigned as BTBP 9 based on literature precedent.27 The novel sodium sulfonate BTBPs TS-BTBP 1 and DS-BTBP 1 were synthesized using two different approaches. The sulfonation of 8 and 9 with oleum at 170 °C, followed by base treatment (NaHCO3) generated sodium sulfonates TS-BTBP 1 and DS-BTBP 1 directly. Alternatively, these ligands were synthesized in a two-step procedure. Treatment of 8 and 9 with chlorosulfonic acid at 170 °C generated the di- and tetrasulfonyl chlorides 10 and 11, respectively. Hydrolysis of 10 and 11 with sodium hydroxide in refluxing methanol furnished the sodium sulfonates TS-BTBP 1 and DS-BTBP 1, respectively. We found that optimization of this latter two-step route to TS-BTBP 1 and DS-BTBP 1 minimized the contamination of the ligands with inorganic salts. | ||
| Scheme 1 Synthesis of disulfonated BTBP (DS-BTBP) ligands DS-BTBP 1 and DS-BTBP 2, and tetrasulfonated BTBP (TS-BTBP) ligands TS-BTBP 1 and TS-BTBP 2. | ||
To probe the effect of the counterion on the selective complexation properties of the sulfonated ligands, we also synthesized the di- and tetrasulfonated BTBPs as their corresponding free acids TS-BTBP 2 and DS-BTBP 2 (Scheme 1). These were synthesized either by hydrolysis of the sulfonyl chlorides 10 and 11 with water at reflux, or, more preferably, by direct sulfonation of BTBPs 8 and 9 with oleum and subsequent precipitation of the ligands with acetone.
The regioselectivity of the sulfonation reactions of 8 and 9 was established by 1H NMR spectroscopy. The 1H NMR spectrum of the disulfonated BTBP ligand DS-BTBP 1 in deuterated DMSO (Fig. 2) shows the expected spin–spin coupling pattern of a meta-disubstituted phenyl ring. As well as the expected resonances for the pyridine protons H1–H3, the spectrum displays a triplet for H8 at 8.74 ppm with very weak (J = 1.4 Hz) meta-coupling to H7/H5. Proton H6 appears as a triplet at 7.67 ppm with strong (J = 7.7 Hz) ortho-coupling to H7/H5, while protons H7 and H5 appear as a pair of double-triplets. Thus the sulfonation reactions of 8 and 9 occurred in the meta-position, as anticipated based on the electronic deactivation of the ortho- and para-positions of the phenyl rings of 8 and 9 by the electron withdrawing triazine rings. Regioselective meta-sulfonation was previously reported in the chlorosulfonation reactions of some 5,6-diphenylpyrazines,28 which are electronically similar to the 5,6-diphenyl-1,2,4-triazine moiety of ligand 8.
 | ||
| Fig. 2 Aromatic region of the 1H NMR spectrum of disulfonated BTBP ligand DS-BTBP 1 in deuterated DMSO with peak assignments (H4 appears as a singlet at 10.18 ppm and is omitted for clarity). | ||
We also synthesized the disulfonated BTP (DS-BTP) ligands DS-BTP 1 and DS-BTP 2 as shown in Scheme 2. These ligands have a lower sulfur content that the previously reported tetrasulfonated BTP,23,24 and thus would generate less solid waste after incineration of the spent solvent streams from used fuel reprocessing. Diphenyl-BTP 13 was obtained as a single regioisomer by treatment of diamide dihydrazide 12 with phenylglyoxal in hot dioxane. Disodium sulfonate BTP DS-BTP 1 was synthesized by the chlorosulfonation of 13 with chlorosulfonic acid, followed by hydrolysis of the resulting disulfonyl chloride 14. Disulfonic acid BTP DS-BTP 2 was also synthesized by the direct sulfonation of 13 with oleum, followed by precipitation with acetone (Scheme 2).
 | ||
| Scheme 2 Synthesis of disulfonated BTP (DS-BTP) ligands DS-BTP 1 and DS-BTP 2. | ||
In order to establish if the point of attachment of the sulfonated phenyl rings on the triazine rings of disulfonated BTPs DS-BTP 1 and DS-BTP 2 had any significant influence on its complexation properties, we also synthesized and screened the regioisomeric BTPs DS-BTP 3 and DS-BTP 4 in which the sulfonated phenyl rings are attached at C-6 of the triazine ring (Scheme 3). Diphenyl-BTP 15 (the opposite regioisomer of 13) was thus synthesized from acetophenone as previously described in the literature,29 and sulfonated as before to yield DS-BTP 3 and DS-BTP 4.
 | ||
| Scheme 3 Synthesis of disulfonated BTP (DS-BTP) ligands DS-BTP 3 and DS-BTP 4. | ||
The hydrophobic BTPhen ligand 2 was found to be an improved ligand for the selective extraction of actinide(III) over lanthanide(III) than the related BTBP 1 (Fig. 1).16 We therefore reasoned that a hydrophilic tetrasulfonated BTPhen ligand might be a more selective actinide(III) aqueous complexant than its BTBP counterparts TS-BTBP 1 and TS-BTBP 2 (Scheme 1), and could be capable of preventing the extraction of actinides(III) by the non-selective hydrophobic ligand N,N,N′,N′-tetraoctyldiglycolamide (TODGA) at higher nitric acid concentrations. We thus synthesized the tetrasulfonated BTPhen (TS-BTPhen) ligands TS-BTPhen 1 and TS-BTPhen 2 from the novel tetraphenyl-BTPhen 18 as shown in Scheme 4.
 | ||
| Scheme 4 Synthesis of tetrasulfonated BTPhen (TS-BTPhen) ligands TS-BTPhen 1 and TS-BTPhen 2. | ||
Numerous attempts to grow suitable crystals of the sulfonated ligands TS-BTBP 1, DS-BTBP 1, DS-BTP 1 and TS-BTPhen 1 for X-ray crystallographic analysis by slow evaporation from water or water/methanol mixtures were made without success. In order to aid the isolation of crystals suitable for X-ray analysis, lipophilic derivatives of TS-BTBP 1 and DS-BTP 1 were synthesized by cation metathesis reactions of TS-BTBP 1 and DS-BTP 1 with tetraphenylphosphonium chloride in water (Scheme 5). The resulting tetraphenylphosphonium salts 20 and 21 were obtained in high yields, and were soluble in most organic solvents. However, our attempts to obtain crystals of 20 and 21 suitable for X-ray analysis by slow evaporation from organic solvents met with no success.
 | ||
| Scheme 5 Synthesis of tetraphenylphosphonium sulfonate ligands 20 and 21via cation metathesis reactions of ligands TS-BTBP 1 and DS-BTP 1. | ||
Solvent extraction studies
The solubilities of the sulfonated bis-triazine ligands in 0.5 M nitric acid are presented in the ESI.† The tetrasulfonated ligands TS-BTBP 1, TS-BTBP 2, TS-BTPhen 1, and TS-BTPhen 2 all showed high aqueous solubilities (>0.11 M). Surprisingly, the solubilities of disulfonated BTPs DS-BTP 1 and DS-BTP 2 were similar to those of the tetrasulfonated BTBP and BTPhen ligands, despite only having half the number of sulfonate groups. In contrast, disulfonated BTBPs DS-BTBP 1 and DS-BTBP 2 were significantly less soluble in water than their tetrasulfonated counterparts TS-BTBP 1 and TS-BTBP 2, and formed turbid solutions in water and nitric acid. Disulfonated BTP DS-BTP 4 was significantly less soluble than its regioisomer DS-BTP 2, while disodium sulfonate BTP DS-BTP 3 was not sufficiently soluble to be used in the extraction tests (<0.005 M).The sulfonated ligands were tested for their ability to suppress selectively (or mask) the extraction of Am(III) from nitric acid solutions by the hydrophobic O-donor ligand N,N,N′,N′-tetraoctyldiglycolamide (TODGA). This ligand is the preferred ligand for the non-selective co-extraction of An(III) and Ln(III) from high level waste solutions; the essential first step in the reprocessing of used nuclear fuel (known in Europe as the DIAMEX process).30 Each of the sulfonated ligands (0.01 M) was added to 0.5 M HNO3 spiked with Am(III) and Eu(III) tracers, and the distribution ratios and separation factors were measured after contacting these aqueous phases with organic solutions containing TODGA (0.2 M) in kerosene/octanol (volume ratio 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). These results were compared to that of a blank sample, which did not contain any sulfonated ligand in the aqueous phase. The results for the tetrasulfonated bis-triazine ligands TS-BTBP 1, TS-BTBP 2, TS-BTPhen 1 and TS-BTPhen 2 are shown in Fig. 3.
:5). These results were compared to that of a blank sample, which did not contain any sulfonated ligand in the aqueous phase. The results for the tetrasulfonated bis-triazine ligands TS-BTBP 1, TS-BTBP 2, TS-BTPhen 1 and TS-BTPhen 2 are shown in Fig. 3.
 | ||
| Fig. 3 Extraction of Am(III) and Eu(III) from 0.5 M nitric acid by TODGA (0.2 M dissolved in 5 vol% octanol in kerosene) in the absence and presence of tetrasulfonated BTBP and BTPhen ligands (0.01 M) in the aqueous phase (D = distribution ratio, SF = separation factor, blue bar = DAm, red bar = DEu, ● = SFEu/Am, mixing time: 360 min, temperature: 22 °C ± 1 °C). | ||
As shown, all the tetrasulfonated ligands are able to suppress the extraction of Am(III) from the aqueous phase by TODGA, while the extraction of Eu(III) by TODGA is far less suppressed. The net result is that Eu(III) is selectively extracted. In the case of TS-BTBP 1, the distribution ratio for Am(III) decreases from 46.0 ± 4 in the absence of TS-BTBP 1 in the aqueous phase to 0.121 ± 0.009 in the presence of TS-BTBP 1 in the aqueous phase. The resulting separation factor for Eu(III) over Am(III) increases from 3.5 ± 0.9 in the absence of TS-BTBP 1 to 616 ± 178 in the presence of TS-BTBP 1 in the aqueous phase. When any of the four tetrasulfonated BTBP or BTPhen ligands are used, the distribution ratios for Am(III) are below 1, while those for Eu(III) remain above 50. The separation factors for Eu(III) over Am(III) (SFEu/Am) for all four ligands are in the range 256–616. The decrease in DAm and increase in SFEu/Am on adding a tetrasulfonated bis-triazine ligand to the aqueous phase is an indication that these sulfonated ligands are complexing Am(III) over Eu(III) in a highly selective manner. The results for the free acids TS-BTBP 2 and TS-BTPhen 2 are comparable to those for the corresponding sodium salts TS-BTBP 1 and TS-BTPhen 1, indicating that the counterions play very little role in the selective complexation of Am(III) as expected. Interestingly, the results for the tetrasulfonated BTBPs are comparable to those of the corresponding BTPhens, with the SFEu/Am values for the tetrasulfonated BTPhens TS-BTPhen 1 and TS-BTPhen 2 being slightly lower than those of the corresponding BTBPs TS-BTBP 1 and TS-BTBP 2.
Thus, in contrast to ligands 3 and 4 employed in the TALSPEAK process, the combination of a tetrasulfonated bis-1,2,4-triazine ligand TS-BTBP 1, TS-BTBP 2, TS-BTPhen 1 or TS-BTPhen 2 in the aqueous phase and TODGA in the organic phase is able to separate An(III) from Ln(III) in nitric acid solutions of low pH (0.5 M HNO3) with very high selectivity. It should also be emphasized that, in contrast to the TALSPEAK process, there is no need for additional buffers or salting out agents when one of these tetrasulfonated bis-triazine ligands is used.
The disulfonated ligands DS-BTBP 1, DS-BTBP 2, DS-BTP 1 and DS-BTP 2 were tested under identical conditions to those of the tetrasulfonated BTBP and BTPhen ligands, and the results are presented in Fig. 4. In these cases, the extraction of Am(III) from the aqueous phase by TODGA is only slightly suppressed, and the separation factor for Eu(III) over Am(III) increases only slightly. The highest separation factor was found with disulfonated BTBP DS-BTBP 1 (SFEu/Am = 7.75 ± 1.1). Clearly, these disulfonated ligands are less able to suppress the extraction of Am(III) from the aqueous phase by TODGA. In the case of disulfonated BTBPs DS-BTBP 1 and DS-BTBP 2, this could be due to their low aqueous solubilities. However, the results are no better for disulfonated BTPs DS-BTP 1 and DS-BTP 2 despite their higher aqueous solubilities (see ESI†). These ligands also fail to suppress the extraction of Am(III) by TODGA. The results for ligands DS-BTP 1 and DS-BTP 2 are inferior to those of their tetrasulfonated BTP counterpart.24 Likewise, 5 mM solutions of disulfonated BTP DS-BTP 4 were unable to suppress Am(III) extraction by TODGA, and showed similar extraction results to its regioisomeric BTP DS-BTP 2 (see ESI†). These results demonstrate that four sulfonate groups are required for the highly selective complexation of Am(III) over Eu(III) by bis-triazine ligands in these TALSPEAK-type separation processes.
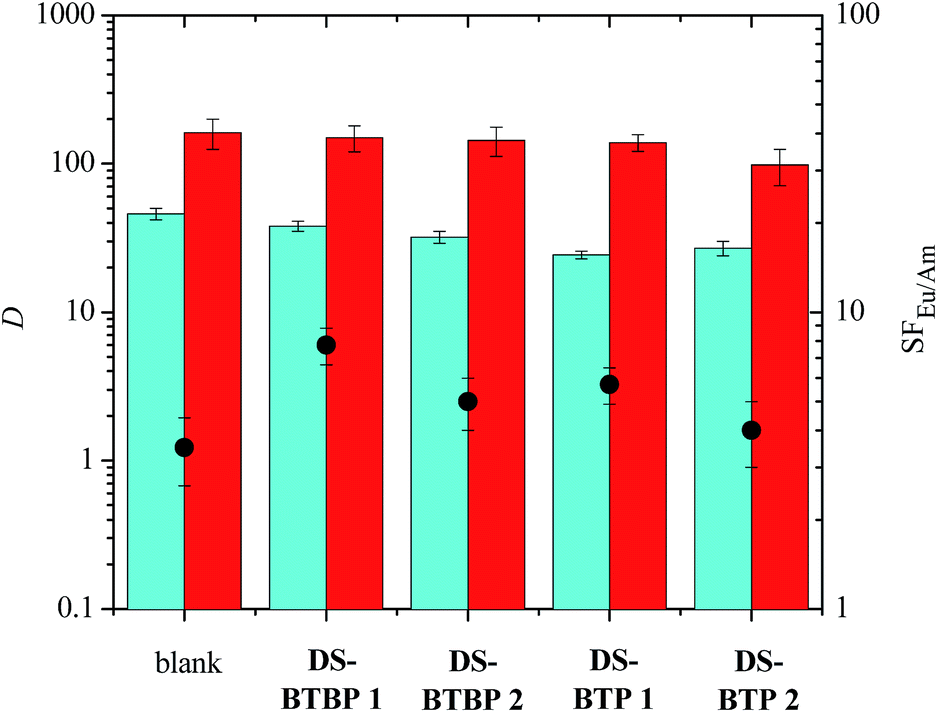 | ||
| Fig. 4 Extraction of Am(III) and Eu(III) from 0.5 M nitric acid by TODGA (0.2 M dissolved in 5 vol% octanol in kerosene) in the absence and presence of disulfonated BTBP and BTP ligands (0.01 M) in the aqueous phase (D = distribution ratio, SF = separation factor, blue bar = DAm, red bar = DEu, ● = SFEu/Am, mixing time: 360 min, temperature: 22 °C ± 1 °C). | ||
We next examined the ability of each sulfonated bis-triazine ligand to suppress the extraction of Am(III) at different nitric acid concentrations to probe the effect of pH on the separation process. Each of the sulfonated bis-triazine ligands followed a trend of increasing distribution ratios for Am(III) and decreasing separation factors of Eu(III) over Am(III) with increasing nitric acid concentration of the aqueous phase (see ESI†). The results for TS-BTBP 1 are shown in Fig. 5. For the tetrasulfonated BTBP TS-BTBP 1, SFEu/Am decreases from 707 ± 312 in 0.28 M HNO3 to 127 ± 73 in 1.04 M HNO3, and the D values for both Am(III) and Eu(III) increase as the nitric acid concentration increases. However, DAm increases somewhat more rapidly than DEu as [HNO3] increases, leading to lower selectivities for Eu(III) over Am(III) at higher acid concentrations. However, TS-BTBP 1 still complexes Am(III) in a selective manner (SFEu/Am = 127 ± 73) even in 1.04 M HNO3 (Fig. 5), indicating that effective separations of Eu(III) over Am(III) are possible across a wide pH range.
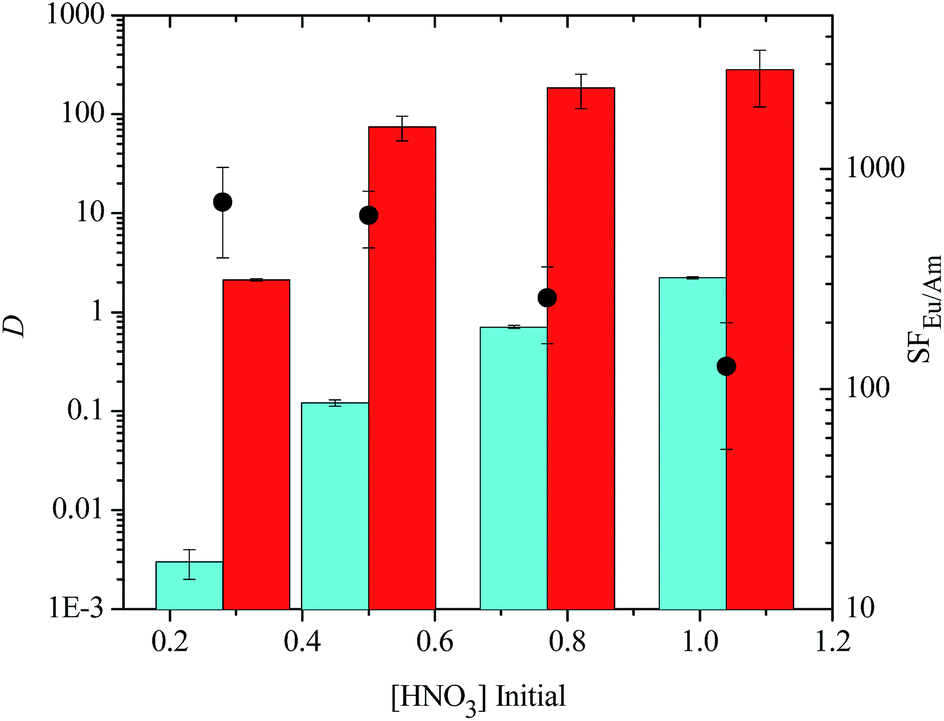 | ||
| Fig. 5 Extraction of Am(III) and Eu(III) from nitric acid by TODGA (0.2 M dissolved in 5 vol% octanol in kerosene) in the presence of tetrasulfonated BTBP ligand TS-BTBP 1 (0.01 M) in the aqueous phase as a function of initial nitric acid concentration (D = distribution ratio, SF = separation factor, blue bars = DAm, red bars = DEu, ● = SFEu/Am, mixing time: 360 min, temperature: 22 °C ± 1 °C). | ||
Similar results were observed for TS-BTPhen 2 (Fig. 6). For this ligand, SFEu/Am decreased from 934 ± 233 in 0.28 M HNO3 to 65 ± 22 in 1.04 M HNO3. For all the tetrasulfonated ligands, good separations (DAm < 1, DEu > 1) of Eu(III) over Am(III) were observed at [HNO3] ≤ 0.5 M. In the case of TS-BTBP 1 and TS-BTBP 2, the distribution ratios for Am(III) remained below 1 even in 0.77 M HNO3. However, at higher nitric acid concentrations, Am(III) extraction by TODGA was no longer suppressed by the sulfonated ligand, and both elements were extracted from the aqueous phase. This is consistent with the observation that the D values for the extraction of Am(III) and Eu(III) by TODGA increase as [HNO3] increases.30 These results show that the tetrasulfonated bis-triazine ligands can selectively complex Am(III) and prevent its extraction by TODGA across a wide range of nitric acid concentrations without the need for additional buffers such as lactic acid or citric acid. This is in contrast to the TALSPEAK process which operates within a very restricted pH range (pH = 2–3) that has to be maintained with the aid of buffers. None of the disulfonated BTBP or BTP ligands were able to complex Am(III) selectively and suppress its extraction by TODGA regardless of the nitric acid concentration of the aqueous phase (i.e.: DAm > 1, see ESI†).
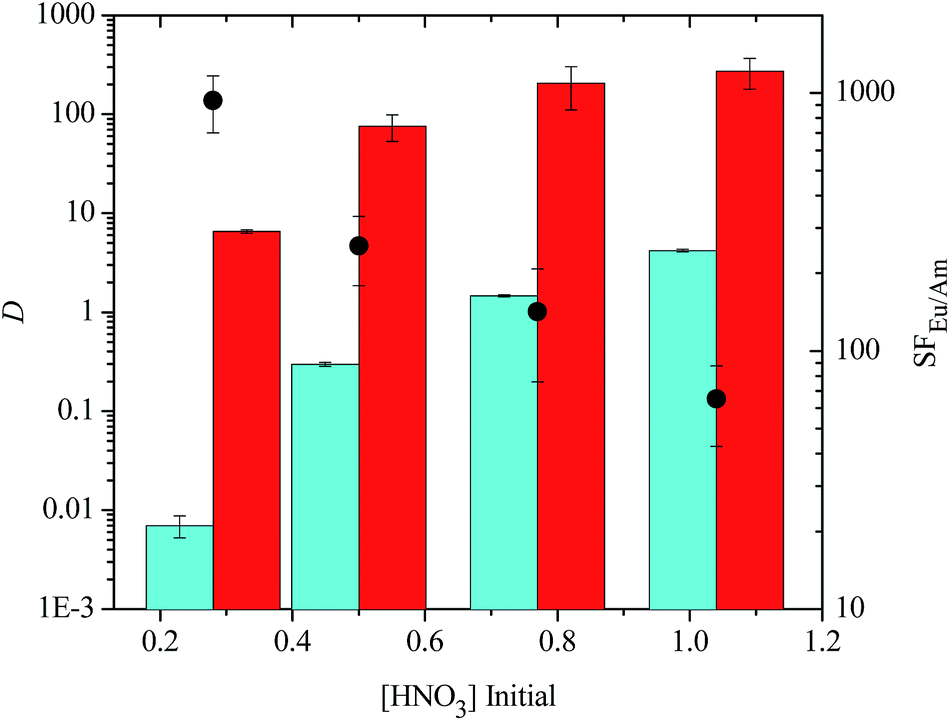 | ||
| Fig. 6 Extraction of Am(III) and Eu(III) from nitric acid by TODGA (0.2 M dissolved in 5 vol% octanol in kerosene) in the presence of tetrasulfonated BTPhen ligand TS-BTPhen 2 (0.01 M) in the aqueous phase as a function of initial nitric acid concentration (D = distribution ratio, SF = separation factor, blue bars = DAm, red bars = DEu, ● = SFEu/Am, mixing time: 360 min, temperature: 22 °C ± 1 °C). | ||
The selectivities of the tetrasulfonated bis-triazine ligands for Am(III) complexation are in general higher than those observed with the polyaminocarboxylate ligands used in the TALSPEAK process. The separation factors for Eu(III) over Am(III) observed with the polyaminocarboxylate ligands used in the TALSPEAK process are shown below in Table 1. Diethylenetriaminepentaacetic acid (DTPA) 4 generally gives the highest selectivities for the complexation of Am(III) over Eu(III), with a maximum separation factor for Eu(III) over Am(III) (SFEu/Am) of 105 being observed in a 1 M citric acid-buffered aqueous phase at pH 3. Other polyaminocarboxylates such as hydroxyethylethylenediaminetriacetic acid (HEDTA), ethylenediaminetetraacetic acid (EDTA) and trans-1,2-diaminocyclohexanetetraacetic acid (DCTA) give lower separation factors for Eu(III) over Am(III).
| Hydrophilic ligand | SFEu/Am | Aqueous phase | Organic phase | Ref. |
|---|---|---|---|---|
| a HEDTA = hydroxyethylethylenediaminetriacetic acid, EDTA = ethylenediaminetetraacetic acid, DCTA = trans-1,2-diaminocyclohexanetetraacetic acid, DIPB = 1,4-diisopropylbenzene. b 1 M. c 0.2 M. d 0.5 M. | ||||
| DTPA 4 (0.05 M) | 84 | Glycolic acid,b pH 3 | 3 In DIPBc | 19c,20a |
| DTPA 4 (0.05 M) | 91 | Lactic acid,b pH 3 | 3 In DIPBc | 19c,20a |
| DTPA 4 (0.05 M) | 105 | Citric acid,b pH 3 | 3 In DIPBc | 19c,20a |
| DTPA 4 (0.05 M) | 100 | Lactic acid,b Na+,b pH 4.27 | 3 In DIPBd | 20b |
| DTPA 4 (0.05 M) | 84 | Lactic acid,b Na+,b pH 2.48 | 3 In DIPBd | 20b |
| HEDTA (0.005 M) | 62 | Lactic acid,b pH 3 | 3 In DIPBc | 19c |
| EDTA (0.005 M) | 59 | Lactic acid,b pH 3 | 3 In DIPBc | 19c |
| DCTA (0.005 M) | 32 | Lactic acid,b pH 3 | 3 In DIPBc | 19c |
In contrast, the separation factors for Eu(III) over Am(III) observed with the tetrasulfonated ligands TS-BTBP 1 and TS-BTPhen 2 shown in Table 2 are in many cases significantly higher than those found with the polyaminocarboxylate ligands used in TALSPEAK separations. For TS-BTBP 1, higher separation factors (SFEu/Am) are found in nitric acid concentrations ranging from 0.28 M to 1.04 M, with a maximum separation factor of 707 ± 312 in 0.28 M HNO3. For TS-BTPhen 2, higher separation factors (SFEu/Am) are found between 0.28 and 0.77 M HNO3, and the highest separation factor (SFEu/Am) is 934 ± 233 in 0.28 M HNO3. Only in the case of TS-BTPhen 2 and TS-BTPhen 1 in 1.04 M HNO3 are lower separation factors for Eu(III) over Am(III) found than with DTPA 4 in the TALSPEAK system (SFEu/Am = 65 ± 22 and 31 ± 13, respectively).
| Hydrophilic ligand | SFEu/Am | Aqueous phase | Organic phase |
|---|---|---|---|
| a TODGA = N,N,N′,N′-tetraoctyldiglycolamide. b 0.2 M. | |||
| TS-BTBP 1 | 707 ± 312 | 0.28 M HNO3 | TODGAb in 5% octanol in kerosene |
| TS-BTBP 1 | 616 ± 178 | 0.5 M HNO3 | TODGAb in 5% octanol in kerosene |
| TS-BTBP 1 | 260 ± 99 | 0.77 M HNO3 | TODGAb in 5% octanol in kerosene |
| TS-BTBP 1 | 127 ± 73 | 1.04 M HNO3 | TODGAb in 5% octanol in kerosene |
| TS-BTPhen 2 | 934 ± 233 | 0.28 M HNO3 | TODGAb in 5% octanol in kerosene |
| TS-BTPhen 2 | 256 ± 77 | 0.5 M HNO3 | TODGAb in 5% octanol in kerosene |
| TS-BTPhen 2 | 142 ± 66 | 0.77 M HNO3 | TODGAb in 5% octanol in kerosene |
| TS-BTPhen 2 | 65 ± 22 | 1.04 M HNO3 | TODGAb in 5% octanol in kerosene |
The selectivities of the tetrasulfonated ligands TS-BTBP 1, TS-BTBP 2, TS-BTPhen 1 and TS-BTPhen 2 for Am(III) complexation over Eu(III) are also similar to those of the parent hydrophobic ligands BTBP 1 and BTPhen 2 (Fig. 1). The overall separation factor for Eu(III) over Am(III) observed herein with the combination of TODGA in the organic phase and a tetrasulfonated bis-triazine ligand in the aqueous phase is approximately equal to the product of that found for TODGA and a typical hydrophobic BTBP or BTPhen ligand such as BTBP 1 or BTPhen 2. Thus, in the case of TS-BTBP 1, the overall SFEu/Am of 707 ± 312 in 0.28 M HNO3 is comparable to the product of that typically found with TODGA (SFEu/Am = 6.2, see ESI†) and BTBP 1 (SFAm/Eu = approx. 150).14 Similarly, with TS-BTPhen 2, the overall SFEu/Am of 934 ± 233 is comparable to the product of that found with TODGA (SFEu/Am = 6.2) and BTPhen 2 (SFAm/Eu = approx. 150–200).16 This shows that the selectivities of the parent hydrophobic ligands 1 and 2 for Am(III) complexation over Eu(III) are largely preserved when they are made water-soluble by sulfonation.
Conclusions
In summary, we report that tetrasulfonated bis-triazine ligands are highly promising reagents for the separation of trivalent actinides from trivalent lanthanides via selective aqueous complexation of actinides in new actinide–lanthanide separation processes based on the TALSPEAK system. Tetrasulfonated bis-triazine ligands are able to selectively complex Am(III) over Eu(III) across a range of nitric acid concentrations (0.28–0.77 M HNO3) with very high selectivities (SFEu/Am = 138–934) and without the use of buffers. The selectivities of the tetrasulfonated ligands for Am(III) complexation over Eu(III) are in many cases far higher than those found with the polyaminocarboxylate ligands used in TALSPEAK separations, and are comparable to those of the parent hydrophobic BTBP and BTPhen ligands being studied for selective actinide extraction. Tetrasulfonated bis-triazine ligands thus represent a considerable improvement over the hydrophilic ligands used in the TALSPEAK process. In contrast, disulfonated bis-triazine ligands are unable to selectively complex Am(III) in nitric acid, indicating that four sulfonate groups are required for selective Am(III) complex formation in nitric acid. The number of sulfonate groups was found to be more important for the separation of Am(III) from Eu(III) than the type of ligand used (BTP/BTBP/BTPhen), the location of the sulfonated phenyl ring(s) in the molecule (attached to C-5 or C-6 of the triazine rings) or the counterion used (H+/Na+).Acknowledgements
We thank the Nuclear Fission Safety Program of the European Union for support under the ACSEPT (FP7-CP-2007-211 267) contract. Additional support was provided by the Czech Ministry of Education, Youth and Sports grant MSM 6840770020. Use of the Chemical Analysis Facility at the University of Reading is gratefully acknowledged.Notes and references
- (a) B. J. Mincher, Nuclear Energy and the Environment, ed. C. M. Wai and B. J. Mincher, ACS Symposium Series, ACS, Washington DC, 2010, vol. 1046, ch. 1, pp. 3–10 Search PubMed; (b) Nuclear Fission, The Energy Research Partnership Technology Report, The Energy Research Partnership, London, 2010, www.energyresearchpartnership.org.uk; (c) Fuel Cycle Stewardship in a Nuclear Renaissance, The Royal Society Science Policy Centre Report 10/11, The Royal Society, London, 2011 (ISBN: 978-0-85403-891-6); (d) Assessing the Sustainability of Nuclear Power in the UK, Sustainability Assessment of Nuclear Power: An Integrated Approach, SPRING Report, The University of Manchester, 2011, www.springsustainability.org.
- (a) J. M. McKibben, Radiochim. Acta, 1984, 36, 3–16 CrossRef CAS; (b) D. D. Sood and S. K. Patil, J. Radioanal. Nucl. Chem., 1996, 203, 547–573 CrossRef CAS; (c) C. Musikas, W. Schultz and J.-O. Liljenzin, Solvent Extraction Principles and Practice, ed J. Rydberg, M. Cox, C. Musikas and G. Choppin, Marcel Dekker, Inc., New York, 2nd edn, 2004, pp. 507–557 Search PubMed; (d) K. L. Nash, C. Madic, J. Mathur and J. Lacquement, The Chemistry of the Actinide and Transactinide Elements, ed. J. J. Katz, L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Dordrecht, 2006, vol. 1, pp. 2622–2798 Search PubMed.
- (a) Actinide and fission product partitioning and transmutation status and assessment report, OECD/NEA, Paris, 1999 Search PubMed; (b) J. Magill, V. Berthou, D. Haas, J. Galy, R. Schenkel, H.-W. Wiese, G. Heusener, J. Tommasi and G. Youinou, Nucl. Energy, 2003, 42, 263–277 CAS; (c) M. Salvatores, Nucl. Eng. Des., 2005, 235, 805–816 CrossRef CAS PubMed; (d) M. Salvatores and G. Palmiotti, Prog. Part. Nucl. Phys., 2011, 66, 144–166 CrossRef CAS PubMed.
- (a) K. L. Nash, Solvent Extr. Ion Exch., 1993, 11, 729–768 CrossRef CAS PubMed; (b) J. N. Mathur, M. S. Murali and K. L. Nash, Solvent Extr. Ion Exch., 2001, 19, 357–390 CrossRef CAS; (c) G. J. Lumetta, A. V. Gelis and G. F. Vandegrift, Solvent Extr. Ion Exch., 2010, 28, 287–312 CrossRef CAS PubMed; (d) C. Hill, Ion Exchange and Solvent Extraction: A Series of Advances, ed. B. A. Moyer, CRC Press, Boca Raton, 2010, vol. 19, pp. 119–193 Search PubMed; (e) G. Modolo, A. Wilden, A. Geist, D. Magnusson and R. Malmbeck, Radiochim. Acta, 2012, 100, 715–725 CrossRef CAS.
- (a) G. T. Seaborg, Radiochim. Acta, 1993, 61, 115–122 CrossRef CAS; (b) S. Cotton, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 3, pp. 93–188 Search PubMed; (c) C. J. Burns, M. P. Neu, H. Boukhalfa, K. E. Gutowski, N. J. Bridges and R. D. Rogers, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier: Oxford, 2004, vol. 3, pp. 189–332 Search PubMed; (d) J. J. Katz, L. R. Morss, N. M. Edelstein and J. Fuger, The Chemistry of the Actinide and Transactinide Elements, ed. J. J. Katz, L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Dordrecht, 2006, vol. 1, pp. 1–17 Search PubMed.
- (a) G. R. Choppin, J. Alloys Compd., 1995, 223, 174–179 CrossRef CAS; (b) V. Alexander, Chem. Rev., 1995, 95, 273 CrossRef CAS; (c) G. R. Choppin, J. Alloys Compd., 2002, 344, 55–59 CrossRef CAS; (d) A. J. Gaunt and M. P. Neu, C. R. Chim., 2010, 13, 821–831 CrossRef CAS PubMed.
- For reviews, see: (a) N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed; (b) M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS PubMed.
- For recent examples, see: (a) S. A. Kozimor, P. Yang, E. R. Batista, K. S. Boland, C. J. Burns, D. L. Clark, S. D. Conradson, R. L. Martin, M. P. Wilkerson and L. E. Wolfsberg, J. Am. Chem. Soc., 2009, 131, 12125–12136 CrossRef CAS PubMed; (b) I. Kirker and N. Kaltsoyannis, Dalton Trans., 2011, 40, 124–131 RSC; (c) S. R. Daly, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor and R. L. Martin, J. Am. Chem. Soc., 2012, 134, 14408–14422 CrossRef CAS PubMed; (d) M. B. Jones, A. J. Gaunt, J. C. Gordon, N. Kaltsoyannis, M. P. Neu and B. L. Scott, Chem. Sci., 2013, 4, 1189–1203 RSC; (e) L. P. Spencer, P. Yang, S. G. Minasian, R. E. Jilek, E. R. Batista, K. S. Boland, J. M. Boncella, S. D. Conradson, D. L. Clark, T. W. Hayton, S. A. Kozimor, R. L. Martin, M. M. MacInnes, A. C. Olson, B. L. Scott, D. K. Shuh and M. P. Wilkerson, J. Am. Chem. Soc., 2013, 135, 2279–2290 CrossRef CAS PubMed; (f) S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor, R. L. Martin, D. K. Shuh and T. Tyliszczak, Chem. Sci., 2014, 5, 351–359 RSC.
- (a) C. Ekberg, A. Fermvik, T. Retegan, G. Skarnemark, M. R. S. Foreman, M. J. Hudson, S. Englund and M. Nilsson, Radiochim. Acta, 2008, 96, 225–233 CrossRef CAS; (b) Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS PubMed; (c) F. W. Lewis, M. J. Hudson and L. M. Harwood, Synlett, 2011, 2609–2632 CAS; (d) M. J. Hudson, L. M. Harwood, D. M. Laventine and F. W. Lewis, Inorg. Chem., 2013, 52, 3414–3428 CrossRef CAS PubMed; (e) P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS PubMed.
- (a) C. Madic, M. J. Hudson, J.-O. Liljenzin, J.-P. Glatz, R. Nannicini, A. Facchini, Z. Kolarik and R. Odoj, Prog. Nucl. Energy, 2002, 40, 523–526 CrossRef CAS; (b) C. Madic, B. Boullis, P. Baron, F. Testard, M. J. Hudson, J.-O. Liljenzin, B. Christiansen, M. Ferrando, A. Facchini, A. Geist, G. Modolo, A. G. Espartero and J. De Mendoza, J. Alloys Compd., 2007, 444–445, 23–27 CrossRef CAS PubMed; (c) S. Bourg, C. Hill, C. Caravaca, C. Rhodes, C. Ekberg, R. Taylor, A. Geist, G. Modolo, L. Cassayre, R. Malmbeck, M. Harrison, G. de Angelis, A. Espartero, S. Bouvet and N. Ouvrier, Nucl. Eng. Des., 2011, 241, 3427–3435 CrossRef CAS PubMed.
- For a review on the chemistry of 1,2,4-triazines, see: S. A. Raw and R. J. K. Taylor, Advances in Heterocyclic Chemistry, ed. A. R. Katritzky, Elsevier, Amsterdam, 2010, vol. 100, pp. 75–100 Search PubMed.
- For leading references, see: (a) Z. Kolarik, U. Müllich and F. Gassner, Solvent Extr. Ion Exch., 1999, 17, 1155–1170 CrossRef CAS PubMed; (b) M. G. B. Drew, D. Guillaneux, M. J. Hudson, P. B. Iveson, M. L. Russell and C. Madic, Inorg. Chem. Commun., 2001, 4, 12–15 CrossRef CAS; (c) P. B. Iveson, C. Riviére, D. Guillaneux, M. Nierlich, P. Thuéry, M. Ephritikhine and C. Madic, Chem. Commun., 2001, 1512–1513 RSC; (d) C. Boucher, M. G. B. Drew, P. Giddings, L. M. Harwood, M. J. Hudson, P. B. Iveson and C. Madic, Inorg. Chem. Commun., 2002, 5, 596–599 CrossRef CAS; (e) G. Ionova, C. Rabbe, R. Guillaumont, S. Ionov, C. Madic, J.-C. Krupa and D. Guillaneux, New J. Chem., 2002, 26, 234–242 RSC; (f) J.-C. Berthet, Y. Miquel, P. B. Iveson, M. Nierlich, P. Thuéry, C. Madic and M. Ephritikhine, J. Chem. Soc., Dalton Trans., 2002, 3265–3272 RSC; (g) S. Colette, B. Amekraz, C. Madic, L. Berthon, G. Cote and C. Moulin, Inorg. Chem., 2002, 41, 7031–7041 CrossRef CAS PubMed; (h) S. Colette, B. Amekraz, C. Madic, L. Berthon, G. Cote and C. Moulin, Inorg. Chem., 2003, 42, 2215–2226 CrossRef CAS PubMed; (i) S. Colette, B. Amekraz, C. Madic, L. Berthon, G. Cote and C. Moulin, Inorg. Chem., 2004, 43, 6745–6751 CrossRef CAS PubMed; (j) M. A. Denecke, A. Rossberg, P. J. Panak, M. Weigl, B. Schimmelpfennig and A. Geist, Inorg. Chem., 2005, 44, 8418–8425 CrossRef CAS PubMed; (k) M. G. B. Drew, M. R. S. J. Foreman, A. Geist, M. J. Hudson, F. Marken, V. Norman and M. Weigl, Polyhedron, 2006, 25, 888–900 CrossRef CAS PubMed; (l) M. Steppert, C. Walther, A. Geist and T. Fanghänel, New J. Chem., 2009, 33, 2437–2442 RSC; (m) N. L. Banik, B. Schimmelpfennig, C. M. Marquardt, B. Brendebach, A. Geist and M. A. Denecke, Dalton Trans., 2010, 39, 5117–5122 RSC; (n) G. Benay, R. Schurhammer and G. Wipff, Phys. Chem. Chem. Phys., 2010, 12, 11089–11102 RSC; (o) S. Trumm, A. Geist, P. J. Panak and T. Fanghänel, Solvent Extr. Ion Exch., 2011, 29, 213–229 CrossRef CAS PubMed.
- For leading references, see: (a) M. G. B. Drew, M. R. S. J. Foreman, C. Hill, M. J. Hudson and C. Madic, Inorg. Chem. Commun., 2005, 8, 239–241 CrossRef CAS PubMed; (b) M. Nilsson, C. Ekberg, M. Foreman, M. Hudson, J.-O. Liljenzin, G. Modolo and G. Skarnemark, Solvent Extr. Ion Exch., 2006, 24, 823–843 CrossRef CAS PubMed; (c) V. Hubscher-Bruder, J. Haddaoui, S. Bouhroum and F. Arnaud-Neu, Inorg. Chem., 2010, 49, 1363–1371 CrossRef CAS PubMed; (d) C. Ekberg, E. Aneheim, A. Fermvik, M. Foreman, E. Löfström-Engdahl, T. Retegan and I. Spendlikova, J. Chem. Eng. Data, 2010, 55, 5133–5137 CrossRef CAS; (e) J.-C. Berthet, J. Maynadié, P. Thuéry and M. Ephritikhine, Dalton Trans., 2010, 39, 6801–6807 RSC; (f) L. M. Harwood, F. W. Lewis, M. J. Hudson, J. John and P. Distler, Solvent Extr. Ion Exch., 2011, 29, 551–576 CrossRef CAS PubMed; (g) F. W. Lewis, L. M. Harwood, M. J. Hudson, P. Distler, J. John, K. Stamberg, A. Núñez, H. Galán and A. G. Espartero, Eur. J. Org. Chem., 2012, 1509–1519 CrossRef CAS PubMed; (h) E. Aneheim, B. Grüner, C. Ekberg, M. R. S. J. Foreman, Z. Hájková, E. Löfström-Engdahl, M. G. B. Drew and M. J. Hudson, Polyhedron, 2013, 50, 154–163 CrossRef CAS PubMed.
- A. Geist, C. Hill, G. Modolo, M. R. S. J. Foreman, M. Weigl, K. Gompper, M. J. Hudson and C. Madic, Solvent Extr. Ion Exch., 2006, 24, 463–483 CrossRef CAS PubMed.
- (a) D. Magnusson, B. Christiansen, M. R. S. Foreman, A. Geist, J.-P. Glatz, R. Malmbeck, G. Modolo, D. Serrano-Purroy and C. Sorel, Solvent Extr. Ion Exch., 2009, 27, 97–106 CrossRef CAS PubMed; (b) E. Aneheim, C. Ekberg, A. Fermvik, M. R. S. J. Foreman, T. Retegan and G. Skarnemark, Solvent Extr. Ion Exch., 2010, 28, 437–458 CrossRef CAS PubMed; (c) E. Aneheim, C. Ekberg, A. Fermvik, M. R. S. J. Foreman, B. Grűner, Z. Hájková and M. Kvičalová, Solvent Extr. Ion Exch., 2011, 29, 157–175 CrossRef CAS PubMed; (d) A. Wilden, C. Schreinemachers, M. Sypula and G. Modolo, Solvent Extr. Ion Exch., 2011, 29, 190–212 CrossRef CAS PubMed.
- (a) F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, J. F. Desreux, G. Vidick, N. Bouslimani, G. Modolo, A. Wilden, M. Sypula, T.-H. Vu and J. P. Simonin, J. Am. Chem. Soc., 2011, 133, 13093–13102 CrossRef CAS PubMed; (b) F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, V. Hubscher-Bruder, V. Videva, F. Arnaud-Neu, K. Stamberg and S. Vyas, Inorg. Chem., 2013, 52, 4993–5005 CrossRef CAS PubMed.
- A. Afsar, L. M. Harwood, M. J. Hudson, P. Distler and J. John, Chem. Commun., 2014, 50, 15082–15085 RSC.
- (a) M. J. Hudson, M. G. B. Drew, M. R. S. J. Foreman, C. Hill, N. Huet, C. Madic and T. G. A. Youngs, Dalton Trans., 2003, 1675–1685 RSC; (b) F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, M. Sypula, G. Modolo, D. Whittaker, C. A. Sharrad, V. Videva, V. Hubscher-Bruder and F. Arnaud-Neu, Dalton Trans., 2012, 9209–9219 RSC.
- (a) B. Weaver and F. A. Kappelmann, TALSPEAK: A New Method of Separating Americum and Curium from the Lanthanides by Extraction from an Aqueous Solution of an Aminopolyacetic Acid Complex with a Monoacidic Organophosphate or Phosphonate, ORNL-3559, Oak Ridge National Laboratory, USA, 1964 Search PubMed; (b) J. Starý, Talanta, 1966, 13, 421–437 CrossRef; (c) B. Weaver and F. A. Kappelmann, J. Inorg. Nucl. Chem., 1968, 30, 263–272 CrossRef CAS; (d) G. Persson, I. Svantesson, S. Wingefors and J.-O. Liljenzin, Solvent Extr. Ion Exch., 1984, 2, 89–113 CrossRef CAS PubMed.
- (a) M. Nilsson and K. L. Nash, Solvent Extr. Ion Exch., 2007, 25, 665–701 CrossRef CAS PubMed; (b) M. Nilsson and K. L. Nash, Solvent Extr. Ion Exch., 2009, 27, 354–377 CrossRef CAS PubMed; (c) K. L. Nash, Solvent Extr. Ion Exch., 2015, 33, 1–55 CrossRef CAS PubMed.
- For other examples of N-donor An(III) selective aqueous complexing agents, see: (a) M. Heitzmann, F. Bravard, C. Gateau, N. Boubals, C. Berthon, J. Pecaut, M.-C. Charbonnel and P. Delangle, Inorg. Chem., 2009, 48, 246–256 CrossRef CAS PubMed; (b) M. Heitzmann, C. Gateau, L. Chareyre, M. Miguirditchian, M.-C. Charbonnel and P. Delangle, New J. Chem., 2010, 34, 108–116 RSC.
- (a) S. Trumm, P. J. Panak, A. Geist and T. Fanghänel, Eur. J. Inorg. Chem., 2010, 3022–3028 CrossRef CAS PubMed; (b) S. Trumm, G. Lieser, M. R. S. Foreman, P. J. Panak, A. Geist and T. Fanghänel, Dalton Trans., 2010, 39, 923–929 RSC.
- G. L. Traister and A. A. Schilt, Anal. Chem., 1976, 48, 1216–1220 CrossRef CAS.
- (a) A. Geist, U. Müllich, D. Magnusson, P. Kaden, G. Modolo, A. Wilden and T. Zevaco, Solvent Extr. Ion Exch., 2012, 30, 433–444 CrossRef CAS PubMed; (b) C. M. Ruff, U. Müllich, A. Geist and P. J. Panak, Dalton Trans., 2012, 41, 14594–14602 RSC; (c) M. Carrott, A. Geist, X. Hères, S. Lange, R. Malmbeck, M. Miguirditchian, G. Modolo, A. Wilden and R. Taylor, Hydrometallurgy, 2015, 152, 139–148 CrossRef CAS PubMed; (d) A. Wilden, G. Modolo, P. Kaufholz, F. Sadowski, S. Lange, M. Sypula, D. Magnusson, U. Müllich, A. Geist and D. Bosbach, Solvent Extr. Ion Exch., 2015, 33, 91–108 CrossRef CAS PubMed.
- For examples of this methodology, see: (a) E. N. Sidorenko and T. Y. Dutova, US pat., 6583284, 2003; (b) T. Y. Dutova, A. Y. Nokel, E. N. Sidorenko and S. V. Timofeev, US pat., 20050109986A1, 2005; (c) S. C. Vonwiller, D. D. Ridley, S. Indusegaram, S. M. Starling, G. Gonzaga and K. Silverbrook, WO2007002981A1, 2007; (d) A. Nokel, T. Nagatsuka and M. V. Paukshto, US20070248771A1, 2007; (e) F. W. Lewis, L. M. Harwood, M. J. Hudson, U. Müllich and A. Geist, Chem. Commun., 2015, 51, 9189–9192 RSC.
- See for example: (a) S. Yoshii, K. Miyamoto and T. Nishimura, Yakugaku Zasshi, 1988, 108, 50–57 CAS; (b) R. M. Abdel-Rahman, J. M. Morsy, H. A. Allimony and W. R. Abd El-Monem, Boll. Chim. Farm., 1999, 138, 176–185 CAS; (c) M. E. F. Braibante, H. T. S. Braibante, M. P. Uliana, C. C. Costa and M. Spenazzatto, J. Braz. Chem. Soc., 2008, 19, 909–913 CrossRef CAS PubMed.
- See for example: (a) M. O'Rourke, S. A. Lang Jr and E. Cohen, J. Med. Chem., 1977, 20, 723–726 CrossRef; (b) M. G. Mamolo, V. Falagiani, D. Zampieri, L. Vio and E. Banfi, Il Farmaco, 2000, 55, 590–595 CrossRef CAS.
- (a) R. J. Cremlyn, O. O. Shode and F. J. Swinbourne, J. Chem. Soc., Perkin Trans. 1, 1983, 1, 2181–2183 RSC; (b) R. J. Cremlyn, F. J. Swinbourne and O. Shode, J. Heterocycl. Chem., 1985, 22, 1211–1214 CrossRef CAS PubMed.
- V. N. Kozhevnikov, D. N. Kozhevnikov, O. V. Shabunina, V. L. Rusinov and O. N. Chupakhin, Tetrahedron Lett., 2005, 46, 1521–1523 CrossRef CAS PubMed.
- (a) Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91–103 CrossRef CAS; (b) G. Modolo, H. Asp, C. Schreinemachers and H. Vijgen, Solvent Extr. Ion Exch., 2007, 25, 703–721 CrossRef CAS PubMed; (c) G. Modolo, H. Asp, H. Vijgen, R. Malmbeck, D. Magnusson and C. Sorel, Solvent Extr. Ion Exch., 2008, 26, 62–76 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures and characterization data for all compounds. Tables and graphs of solvent extraction data. See DOI: 10.1039/c5sc01328c |
| This journal is © The Royal Society of Chemistry 2015 |