
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical selectivity of nucleobase adduction relative to in vivo mutation sites on exon 7 fragment of p53 tumor suppressor gene†
Spundana
Malla
a,
Karteek
Kadimisetty
a,
You-Jun
Fu
a,
Dharamainder
Choudhary
c,
Ingela
Jansson
b,
John B.
Schenkman
b and
James F.
Rusling
*abde
aDepartment of Chemistry, University of Connecticut, Storrs, CT 06269, USA. E-mail: james.rusling@uconn.edu
bDepartment of Cell Biology, University of Connecticut Health Center, Farmington, CT 06032, USA
cDepartment of Surgery, University of Connecticut Health Center, Farmington, CT 06032, USA
dInstitute of Material Science, University of Connecticut, Storrs, CT 06269, USA
eSchool of Chemistry, National University of Ireland at Galway, Ireland
First published on 24th June 2015
Abstract
Damage to p53 tumor suppressor gene is found in half of all human cancers. Databases integrating studies of large numbers of tumors and cancer cell cultures show that mutation sites of specific p53 codons are correlated with specific types of cancers. If the most frequently damaged p53 codons in vivo correlate with the most frequent chemical damage sites in vitro, predictions of organ-specific cancer risks might result. Herein, we describe LC-MS/MS methodology to reveal codons with metabolite-adducted nucleobases by LC-MS/MS for oligonucleotides longer than 20 base pairs. Specifically, we used a known carcinogen, benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE) to determine the most frequently adducted nucleobases within codons. We used a known sequence of 32 base pairs (bp) representing part of p53 exon 7 with 5 possible reactive hot spots. This is the first nucleobase reactivity study of a double stranded DNA p53 fragment featuring more than 20 base pairs with multiple reactive sites. We reacted the 32 bp fragment with benzo[a]pyrene metabolite BPDE that undergoes nucleophilic substitution by DNA bases. Liquid chromatography-mass spectrometry (LC-MS/MS) was used for sequencing of oligonucleotide products from the reacted 32 bp fragment after fragmentation by a restriction endonuclease. Analysis of the adducted p53 fragment compared with unreacted fragment revealed guanines of codons 248 and 244 as most frequently targeted, which are also mutated with high frequency in human tumors. Codon 248 is mutated in non-small cell and small cell lung, head and neck, colorectal and skin cancer, while codon 244 is mutated in small cell lung cancer, all of which involve possible BDPE exposure. Results suggest the utility of this approach for screening of adducted p53 gene by drugs and environmental chemicals to predict risks for organ specific cancers.
Introduction
Cancer is the second most common cause of death worldwide.1 Tumor suppressor genes in humans provide cancer protection pathways by coding for proteins that control cellular stress by inducing cell-cycle arrest, apoptosis and senescence.2 TP53 (or p53) was confirmed as a tumor suppressor gene in the 1980s,3 and other tumor suppressor genes such as retinoblastoma (RB), Wilms Tumor 1 (WT1), Adenomatosis Polyposis Coli (APC), and p16 have also been discovered. The p53 gene encodes p53 protein, which is involved in cell cycle regulation leading to cancer protection.4–7Tumor suppressor genes can be damaged by xenobiotic chemicals, by their metabolites and by radiation. Mutations in the p53 gene have been found in 50% of human cancers.8–12 Moreover, there are well-documented links between human exposure to various carcinogens and specific mutated codons in the p53 gene leading to the development of specific cancers.11–13 Most mutations in the p53 gene occur in exons 5 to 8.14,15
Thus, mutational spectra on the p53 gene are correlated with the incidence of tissue specific cancers. For example, data in the p53 database13 show that highly mutated reactive hot spots include codons 157, 158, 248, 249 in lung cancer, codon 273 in brain and prostate cancer, codons 175, 248 and 273 in breast cancer and codons 175, 282 and 248 in liver cancer.13,16 Codon hot spots for reactions of metabolites on the p53 gene have been linked to human exposure to particular carcinogens. Specifically, components of tobacco smoke are related to lung cancer, tobacco smoke and alcohol to head and neck cancers, aromatic amines to bladder cancer, aflatoxine-B1 and hepatitis B virus to liver cancer and ultraviolet light to skin cancer. Thus, exposure to specific carcinogens that lead to damage to the p53 gene can be correlated with organ-specific cancers.
These relationships between the mutational spectra of p53 to organ-specific cancers are clearly indicated in large databases integrating extensive p53 research.13,17 These data include genomic studies of patient tumors and cell lines, and show that the p53 gene has 7 reactive hot spots between bases 361 and 920 of the reading frame, one in exon 5, one in exon 6, five in exon 7 (Scheme 1), and two in exon 8. Screening of a wide range of carcinogens by assessing reactive hot spots on p53 in vitro could identify reactive nucleobases within codons that, if correlated with mutational spectra, could be used to predict tissue specific cancers. This sort of information is not available for large libraries of potentially reactive compounds or metabolites on p53, and is almost non-existent for other tumor suppressor genes.18 New methods for screening reactive metabolites for sequence specific tumor suppressor gene damage would be valuable tools to assess the potential of new drugs and environmental chemicals for organ-specific carcinogenicity.
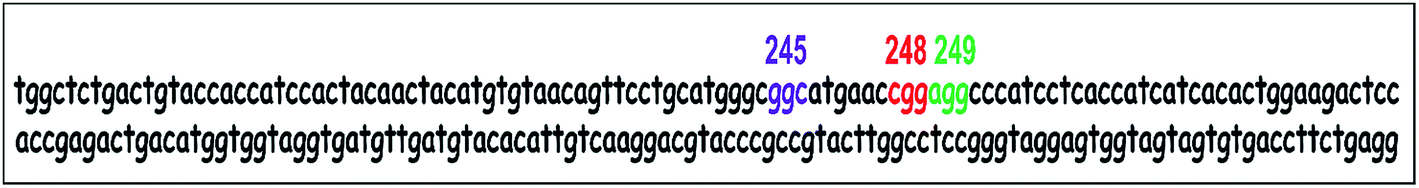 | ||
| Scheme 1 Exon 7 of the p53 gene. Known highly reactive hotspots 245, 248 and 249 are in purple, red and green respectively. Codons 244 and 247 are additional hot spots.13 | ||
Mass spectrometry (MS) is a valuable tool for structural analysis of DNA, and LC-MS/MS methodologies have been developed over the past decade for sizing and sequencing oligonucleotides of up to 20 base pairs (bp).19–23 Harsch, reacted a 10 base pair oligonucleotide derived from hypoxanthine-guanine phosphoribosyltransferase gene (HPRT gene) with benzo[c]phenanthrene and determined positional isomers in the product.24 Chowdhury and Guengerich reacted a 15 base pair oligonucleotide incorporating hot spot codon 157 on exon 5 of p53 gene with mutagenic molecules benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE) and N-hydroxy-4-aminobiphenyl (N-OH-4ABP) and used MS/MS to determine site reactivity.25 They also determined C-4 oxidized abasic sites on a 15-mer oligonucleotide.21 Sharma et al., reacted a 17-mer incorporating codon 135 of p53 with 2-acetylaminofluorene (AAF), and observed multiple adducts formed from reactions with guanosines.26 Satterwhite et al., reacted a 21-mer of p53 containing codon 273 with BPDE.27 Xiong et al., reacted a 14 mer ds DNA containing hot spot codons 157 and 158 with BPDE. Xiong et al., reacted a 14 mer ds DNA containing hot spot codons 157 and 158 with BPDE.19 Sharma et al., studied a 15 base pair DNA containing codon 135 with 2-AAF28 and also investigated the 14 mer ds DNA with codons 157 and 158 in reactions with BPDE, AAF and N-OH-4ABP.29
Previous studies have been restricted to ds-oligonucleotides smaller than 20 bp to enable direct LC-MS/MS sequencing. The short strands studied thus far contained only a single reaction site, and thus did not address relative reactivity between different sites or correlations with mutation frequencies in tumors. Further, short strands may exhibit altered structural specificity in nucleophilic addition compared to natural tumor suppressor genes that may show tertiary and quaternary structural influences on codon reactivity.30–34
In this paper, we report an LC-MS/MS study of ds fragment of 32 bp of p53 gene exon 7, from codon 242 to 253. This fragment exhibits up to 5 reactive hot spots,13–17 and our work represents metabolite reactivity of a p53 strand (>20 base pairs) with multiple mutation sites. The 32 bp fragment was chosen to be long enough to mimic higher order structure-reactivity of DNA.30 This fragment was reacted with BPDE, the major DNA-reactive metabolite of benzo[a]pyrene,35–37 and then a restriction enzyme was used to cleave the fragment into smaller fragments to enable LC-MS/MS sequencing.
Benzo[a]pyrene is a Group I carcinogen,38 and its reactive metabolite BDPE is implicated in lung, head and neck, colorectal and skin cancer. Mutational hot spots within this 32 bp fragment include 248, 245, and 249 associated with non-small cell lung cancer, 248, 249 and 244 for small cell lung cancer, 248 for head and neck cancer, 248 and 245 for colorectal cancer, 248 and 247 for skin cancer.13–15,18 Using the approaches described herein, we found that the most frequently BPDE-adducted guanines were within codons 244 and 248, which correlate with frequently mutated p53 sites in several cancers.
Experimental section
Caution: Benzo[a]pyrene-r-7,t-8-dihydrodiol-t-9,10-epoxide (±) (anti) (anti BPDE) is a chemical carcinogen. Protective measures including wearing gloves and protective eyewear and doing experiments in a closed hood were taken.Chemicals and Reagents
Benzo[a]pyrene-r-7,t-8-dihydrodiol-t-9,10-epoxide (±) (anti) (anti BPDE) was from National Cancer Institute Chemical Carcinogen Reference Standard Repository. Triethylammonium bicarbonate (TEAB, 1.0 M, pH 8.4–8.6) and 4-nitrobenzyl alcohol were from Sigma-Aldrich. Custom made single stranded DNA oligomers and the 32-base pair oligonucleotide were from Sigma-Aldrich. HPLC grade solvents, methanol, tetrahydrofuran and water were from Fischer Scientific. Restriction enzyme, NlaIII was from New England Biolabs.Reactions of oligonucleotides with BPDE
Reactions with BPDE were done with 4 single stranded (ss) oligonucleotides and the double stranded (ds) 32 bp portion of p53 gene representing codons 242 to 253. 0.1 nmol of each of the 4 ss oligonucleotides were treated with 100 nmol BPDE in a reaction volume of 150 μL at 37 °C for 48 hours in dark, 10 mM Tris buffer, pH 7.4. 2.5 μmol of ds 32 bp fragment was reacted with 0.1 μmole of BPDE in volume of 150 μL at 37 °C for 48 hours in dark, 10 mM Tris buffer, pH 7.4. A minimum of three replicates of DNA and BPDE reactions were done.Removal of excess BPDE
ss DNA fragments were extracted from the BPDE–ss DNA mixture by extracting with 150 μL of ethylacetate for three times. DNA was recovered from the aqueous phase. Samples were then dried and reconstituted in 100 μL of pure water to make a final concentration of 1 μM ss DNA.For the ds-DNA–BPDE mixtures, Millipore 3000 Dalton mass cutoff filter vials were used to remove unreacted BPDE (Scheme 2). Reaction mixture from above was put into a 3000 Da mass cutoff filter, (catalog # UFC500396) and centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. ds-DNA was retained on the filter, while the smaller BPDE molecules pass through. The filter was then removed, inverted and placed into a new centrifuge vial, and centrifuged for another 30 min. Approximately 50 μL of the DNA solution was recovered. Recovered solutions of ds oligonucleotides were subjected to restriction enzyme treatment (Scheme 2).
000 rpm for 30 min. ds-DNA was retained on the filter, while the smaller BPDE molecules pass through. The filter was then removed, inverted and placed into a new centrifuge vial, and centrifuged for another 30 min. Approximately 50 μL of the DNA solution was recovered. Recovered solutions of ds oligonucleotides were subjected to restriction enzyme treatment (Scheme 2).
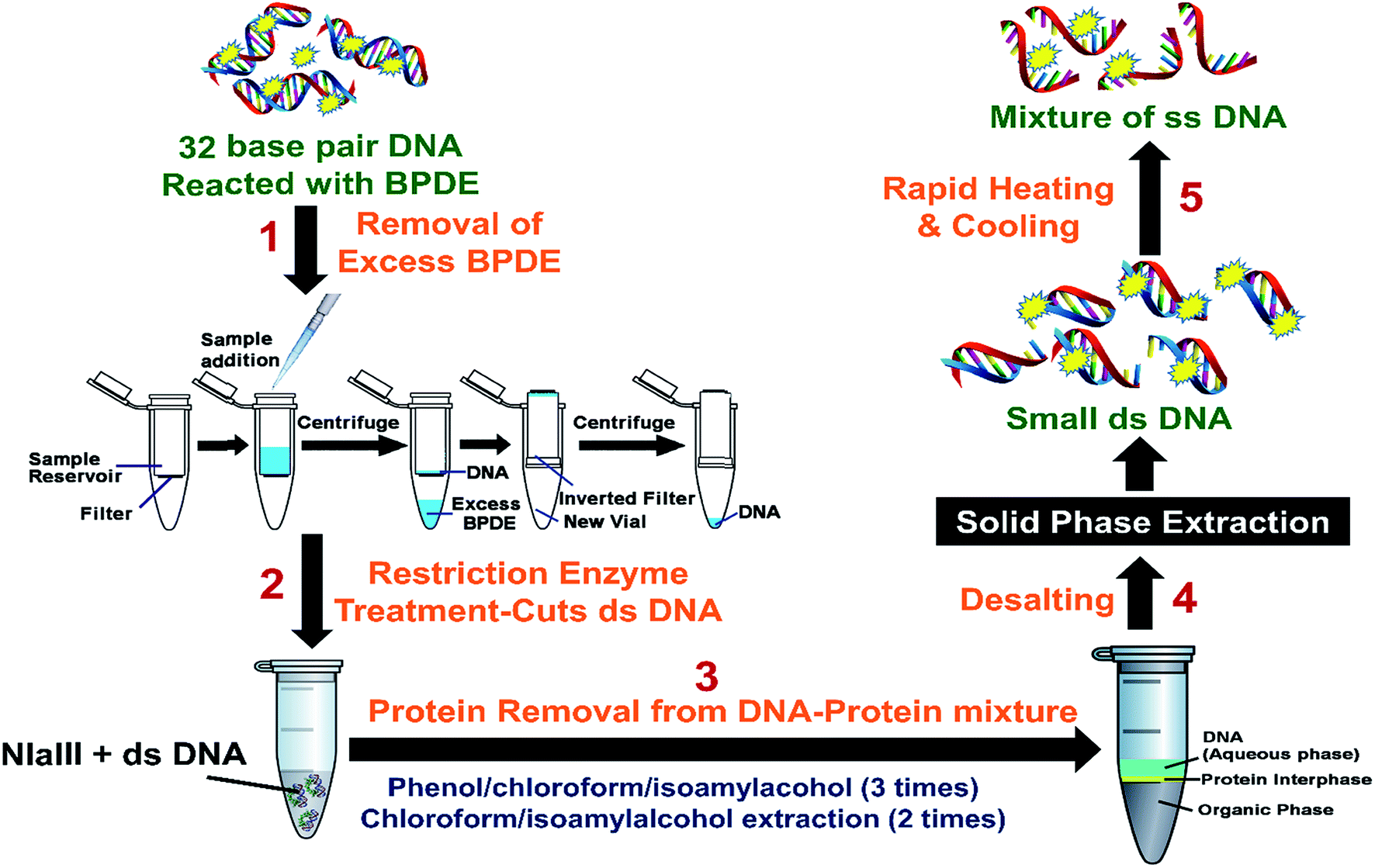 | ||
| Scheme 2 Protocol for sample preparation of 32-base pair p53 fragment reacted with BPDE involving steps: (1) removal of excess BPDE from DNA reaction mixture; (2) restriction enzyme treatment to cut DNA into smaller fragments; (3) protein removal; (4) desalting and (5) rapid heating and cooling to give ss DNA. | ||
Restriction enzyme treatment on ds DNA
Approximately 50 μg (2.5 μmol) of DNA was recovered from above step. 50 units (5 μL) of restriction enzyme, NlaIII was added to 50 μg of the ds 32 bp oligonucleotides to which 20 μL of 10× NE buffer supplied by New England Biolabs, was added and the reaction volume made up to 200 μL with pure water. This reaction mixture was incubated at 37 °C for 8 hours. The resulting solution is mixture of DNA fragments, protein and salt from the NE reaction buffer given by the manufacturer.Removal of proteins from DNA–protein–salt mixture
Proteins were removed from the DNA samples by extraction using 200 μL of water-saturated phenol/chloroform/isoamylalcohol, 25/24/1. Each extraction step involves vortexing on a rotor for 10 min followed by centrifugation for 10 min prior to aqueous phase collection. DNA in the aqueous phase was collected and the extraction procedure was repeated twice more. Further extraction of DNA contained in the aqueous phase was done twice with 200 μL of water-saturated chloroform/isoamylalcohol, 24/1. Finally the aqueous phases were combined and organic phase was discarded. The resulting sample containing DNA and salt was approximately 200 μL.Desalting
Solid phase extraction (SPE) was performed to remove salt from DNA–salt mixtures to obtain samples suitable for LC-MS/MS analysis. Waters Oasis HLB cartridges were used for desalting. The method involves initial equilibration of the cartridge with methanol and water. Sample was loaded and then washed with 5% methanol in water to remove salts followed by elution with 100% methanol. The resultant DNA sample was dried and reconstituted in 100 μL pure water. The ds DNA samples were heated and cooled rapidly to convert ds DNA to ss DNA and stored at −20 °C until LC-MS/MS analysis.LC-MS analysis
A Waters Capillary LC-XE (Milliford, MA) chromatograph with a Gemini C-18 column (0.3 mm × 150 mm, 5 μm for ss DNA fragments & 0.5 mm × 150 mm, 3 μm for ds DNA fragments) and photodiode array detector were used. Separation featured a binary solvent consisting of A – 25 mM TEAB and B – methanol. Separation was achieved with a gradient of 0–10% B for 10 min followed by increase from 10 to 50% B for 100 min and 50 to 100% B for the final 10 min at flow rate 10 μL min−1. m-Nitrobenzyl alcohol increases signal intensity and charge states of oligonucleotide fragments in negative mode, which enhances the fragmentation of oligonucleotides in tandem mass spectrometry.39 Thus, 0.1% m-nitrobenzyl alcohol was infused at a flow rate of 3 μL min−1 to mix with the LC flow post-column using a three way connector before entering the mass spectrometer to produce supercharged oligonucleotide fragments.ss DNA fragments were analyzed with an AB Sciex Qtrap 4000 mass spectrometer interfaced to the capillary LC. Enhanced multiple charge (EMC) mode for sizing (molecular weight measurement) and enhanced product ion mode (EPI) for sequencing of ss-DNA fragments in negative mode were employed. Potentials −4500 V (ion spray voltage), 130 V (declustering potential), −60 eV (collision energy), and 20 eV (collision energy spread) were used while GS1, GS2, and temperature were kept at 35, 20, and 350 °C, respectively.
For analysis of ds-DNA samples, an AB Sciex QSTAR mass spectrometer was interfaced with the capillary LC. Negative ion mode with a −4500 V ion spray voltage, −130 V declustering potential and 300 °C temperature was used. DNA fragments were subjected to time of flight mass scan (TOF-MS) mode for sizing of unreacted and adducted DNA fragments and product ion scan mode was used at −45 eV collision energy for MS/MS sequencing. Only singly adducted fragments were discussed in this study.
Results
Initial studies centered on reacting single-strand fragments GGCGGCATG (ss-DNA fragment 1), including hot spot codon 244 and 245, AACCGGAGGCCCATCCTCA (ss-DNA fragment 2), including hot spot codon 248 and 249, TGAGGATGGGCCTCCGGTTCATG (ss-DNA fragment 3, complementary to fragment 2) and CCGCCCATG (ss-DNA fragment 4, complementary to fragment 1) with BPDE to optimize the LC-MS/MS methodology without using the restriction enzyme.Our protocol for the 32 base pair exon 7 fragment involves reacting p53 oligonucleotide sequences with BPDE, then using restriction enzymes to cut the reacted fragment into smaller fragments suitable for LC-MS/MS (Scheme 3). Our 32 bp exon 7 fragment extends from codon 242 to 253 and includes possible reactive hot spots at codons 244, 245, 247, 248 and 249 related to the various cancers described above. All codons contain guanine except 247 (ACC), which contains a possible reactive adenine. Within this sequence, codon 248 is the most frequently mutated in all cancers.13 Codons 245, 248 and 249 are adducted for most cancers related to BPDE, which adds to DNA bases by nucleophilic substitution. Our 32 base pair fragment is a combination of ss-DNA fragment 1 and ss-DNA fragment 2 with four additional bases CATG added before the ss-DNA fragment 1. This was done in order to eliminate the possibility of having terminal guanines with enhanced reactivity.
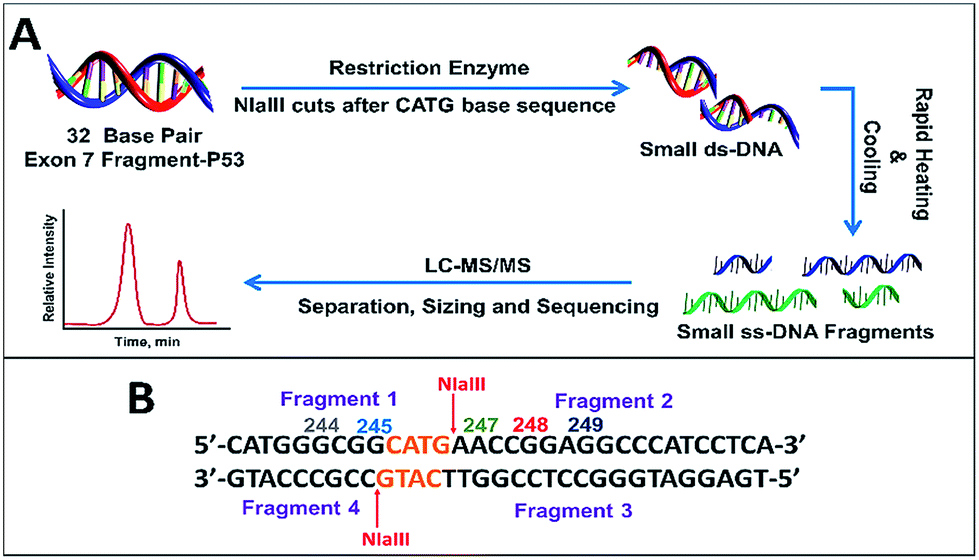 | ||
| Scheme 3 (A) Sample preparation for LC-MS/MS sizing and sequencing of fragment. (B) 32 bp exon 7 fragment showing cut points for restriction enzyme NlaIII along with resulting fragments obtained. | ||
In order to sequence products of 32 base pair oligonucleotide reactions with BPDE by LC-MS/MS, we used the restriction enzyme NlaIII, which cuts DNA after the sequence CATG (Scheme 3). For our 32 bp fragment, this results in two fragments of 13 and 19 bases that are double stranded except for 4 bases at their ends (Scheme 3B). 13 mer was obtained instead of the expected 9 mer since the efficiency of restriction enzymes to cut the ds-DNA decreases drastically when the recognition site in closer to the 5′ end. A minimum of 3–4 extra bases flanking the recognition site on the 5′ end was required for restriction enzyme to act on ds-DNA.40 These two ds-fragments were rapidly heated and cooled to give in 4 ss-oligonucleotide fragments (Scheme 3A).
We used enhanced multiple charge scanning to size the ss-oligonucleotides, followed by collision-induced dissociation (CID) MS/MS for sequencing. CID of oligonucleotides shows a characteristic fragmentation pattern (Scheme 4) featuring an-bn and wn ions.41–44 Adduction of BPDE to specific nucleobases within codons was monitored by comparing un-adducted oligonucleotides to those reacted with BPDE, and detecting the difference in m/z of the an-bn and wn ions.
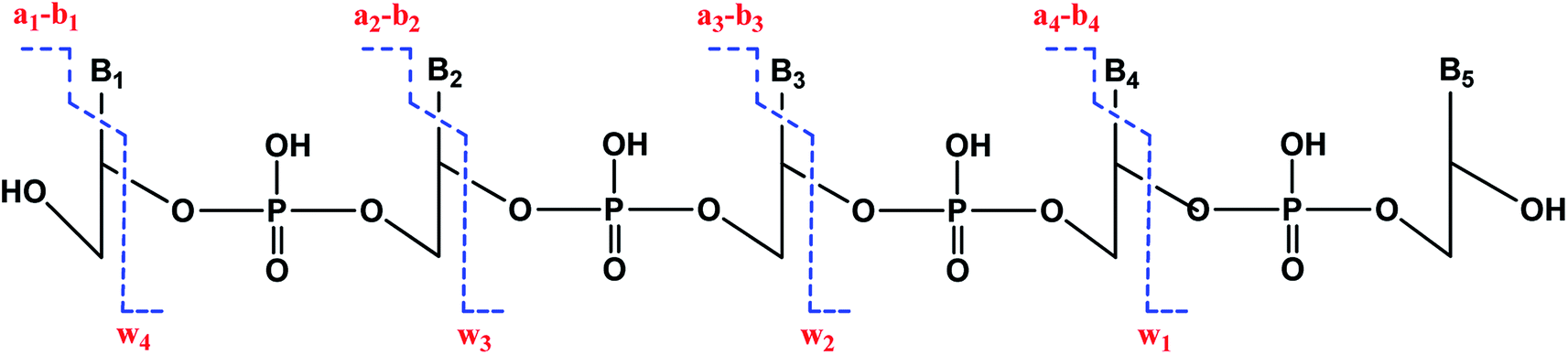 | ||
| Scheme 4 Collision induced dissociation (MS/MS or tandem MS) of DNA fragments resulting in the generation of wn and an-bn ions. | ||
Enhanced multiple charge scans (EMC) were used to determine the m/z of standard and BPDE-adducted DNA fragments that have an additional mass of 302.323. Expected multiple charged species possible were calculated using the online database Mongo oligomass calculator.45
We first present results for the ss-fragments. Possible m/z for standard and singly adducted ss-DNA fragments are as shown in Table S1† for ss-fragment 1. With the ionization conditions used, we observed an m/z of 1388.9 for unreacted ss-fragment 1 with a charge of −2 and m/z of 1540.5 for the reacted fragment indicating a singly adducted strand also with charge −2. Multiple peaks for a single fragment ion were observed due to isotopic distribution (see Fig. S1,† fragment ion 1540.5).
Extracted ion chromatogram of a selected ion as a function of retention time of singly adducted ss-DNA fragment 1 of m/z 1540.5 shows (Fig. 1A) that there is a possibility of 2 positional isomers for the singly adducted ss-fragment 1 at retention times 38.67 and 43.75 min. CID of fragment ion 1540.5 for peak I is as shown in Fig. 1B and for peak II in Fig. 1C.
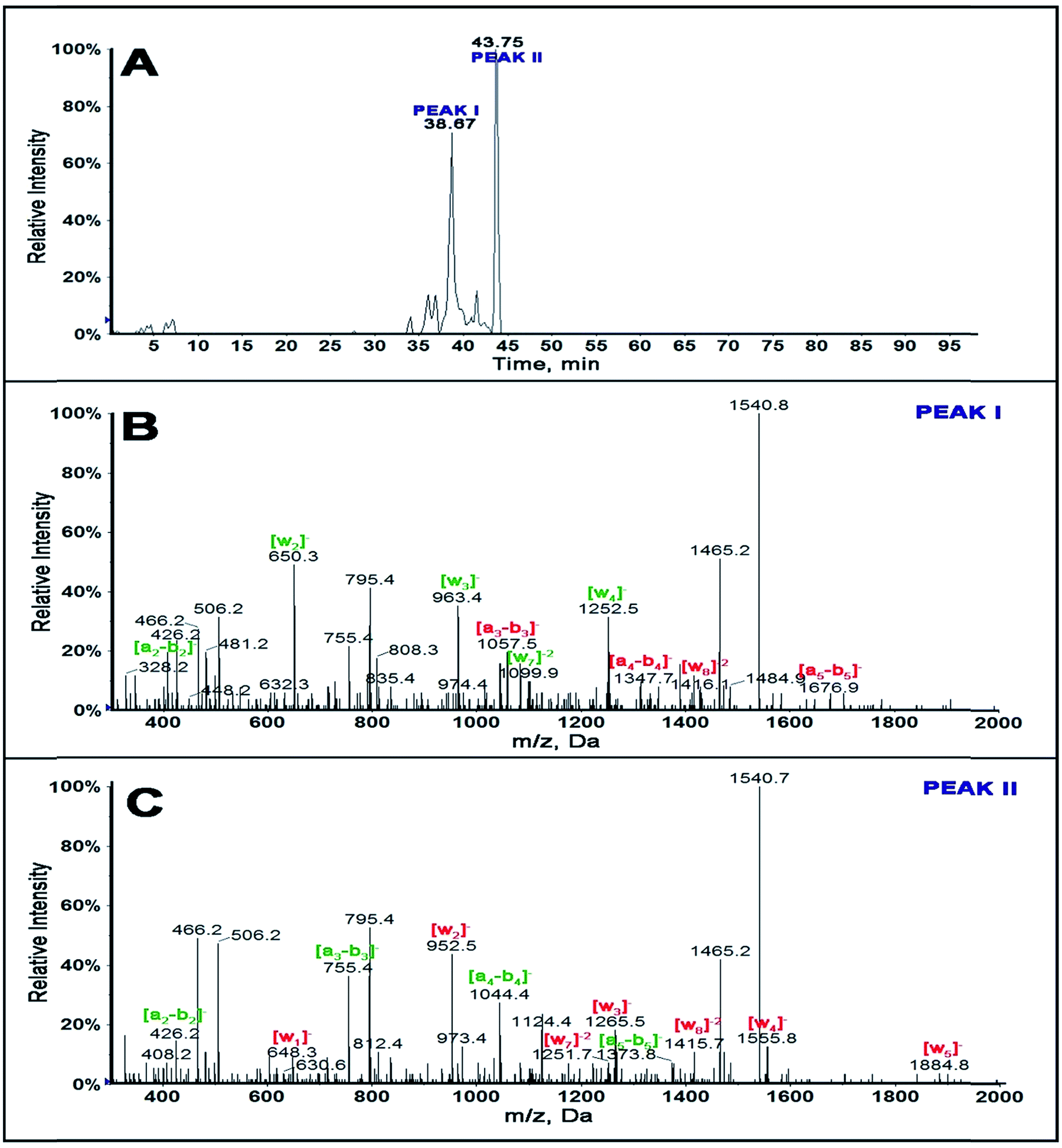 | ||
| Fig. 1 LC-MS of ss-DNA fragment 1 (A) extracted ion chromatogram for fragment m/z 1540.5 representing z = −2 singly adducted fragment 1. (B) MS/MS spectrum of singly adducted BPDE ss-DNA fragment 1, m/z 1540.5 for peak I eluting at 38.67 min and (C) MS/MS of peak II eluting at 43.75 min (1540.7 or 1540.8 m/z was observed instead of 1540.5 due to isotopic distribution). | ||
Differences in m/z of a-b and w ions between the standard ss-DNA fragment 1 and the singly adducted DNA fragment help locate the exact reacted position on a given oligonucleotide.46–48Table 1 summarizes fragment ions obtained for standard ss-DNA fragment 1, singly adducted ss-fragment 1 for peaks I and II. Red numbers indicate those ions increased in mass by 302.32 from adducting with BPDE compared to the unreacted fragment.
| Fragment ion | m/z | ||
|---|---|---|---|
| Standard | Peak I | Peak II | |
| a2-b2 | [426.2]− | [426.2]− | [426.2]− |
| a3-b3 | [755.5]− | [1057.5]− | [755.4]− |
| a4-b4 | [1044.7]− | [1347.7]− | [1044.3]− |
| a5-b5 | [1373.5]− | [1676.9]− | [1373.8]− |
| w1 | [346.2]− | [346.2]− | [648.3]− |
| w2 | [650.3]− | [650.3]− | [952.5]− |
| w3 | [963.4]− | [963.4]− | [1265.5]− |
| w4 | [1252.8]− | [1252.5]− | [1555.8]− |
| w5 | [1582.0]− | n/d | [1884.7]− |
| w6 | n/d | n/d | n/d |
| w7 | [1099.7]2− | [1099.9]2− | [1251.6]2− |
| w8 | [1263.3]2− | [1416.1]2− | [1415.7]2− |
MS/MS spectrum for peak I of singly adducted ss-fragment 1 (Fig. 1B) reveals increase in mass of all the ions from a3-b3 to a6-b6 and w8 compared with that of unreacted ss DNA standard (Table 1). This indicates covalent binding of BPDE on the second G base, GG*CGGCATG. This is additionally confirmed by the presence of ion a2-b2 with an m/z of 426.2− and all ions from w1 to w7 with m/z the same as that of the standard.
MS/MS spectrum for peak II of singly adducted ss-fragment 1 (Fig. 1C) reflects increases in m/z for all the w ions compared to unreacted standard (Table 1). This indicates covalent binding of BPDE to the last G base, GGCGGCATG*, which was supported by all a-b ions having m/z similar to unreacted standard. Similar experiments were done for other ss-DNA fragments 2, 3 and 4 (Table 2, and ESI-Fig. S2–S4 and Tables S2 and S3†).
| Standard ss-DNA | Sequence | LC-MS/MS DATA | Common Hot Spots from Database13–15,18 | |
|---|---|---|---|---|
| Base adducted | Codon | |||
| a Possible cancers due to BPDE. b Non small cell lung cancer. c Small cell lung cancer. d Head and neck cancer. e Colorectal cancer. f Skin cancer. | ||||
| Fragment 1 | GGCGGCATG | GG*CGGCATG | 244c | 244c, 245be |
| GGCGGCATG* | 246 | 244c, 245be | ||
| Fragment 2 | AACCGGAGGCCCATCCTCA | AACCGGAGG*CCCATCCTCA | 249bc | 248bcdef, 249bc |
| Complementary strands | ||||
| Fragment 3 | GTGAGGATGGGCCTCCGGTTCATG | GTGAG*GATGGGCCTCCGGTTCATG | ||
| Fragment 4 | CCGCCCATG | CCG*CCCATG | ||
| CCGCCCA*TG | ||||
| CCGCCCATG* | ||||
After establishing and optimizing reproducible methodology on the standard single stranded DNA fragments, the 32 bp ds-p53 exon 7 fragment, which is a combination of fragments 1 and 2 along with the complementary strand, was reacted with BPDE. Four bases, CATG were added to the front of fragment 1 to avoid terminal guanines. The sequence added is consistent with the exon 7 sequence. Excess BPDE was removed using mass cutoff filters, then the aqueous extract was treated with restriction enzyme NlaIII to cut the fragment into smaller fragments suitable for LC-MS (Schemes 3 and S1†). Subsequently, heating and rapid cooling gave the four single stranded fragments. The m/z values possible for the standard four single stranded fragments and the single adducted fragments are shown in Table S4 of ESI.†
In order to determine sequence specificity on the entire 32 bp fragment, collision induced dissociation (CID or MS/MS) was performed on all possible single adducted oligonucleotide fragments. Singly adducted fragment 2 is described here as the fragment containing hot spot 248 and 249 active for a number of cancers.13–15,18 Singly adducted fragment 2 upon MS ionization produced a ion of m/z 1530.3 with z = −4. The extracted ion chromatogram (XIC) of m/z 1530.3 (Fig. 2A) gave only one major peak indicating that there is one major adduct on the singly adducted fragment 2 of the reacted 32 bp fragment.
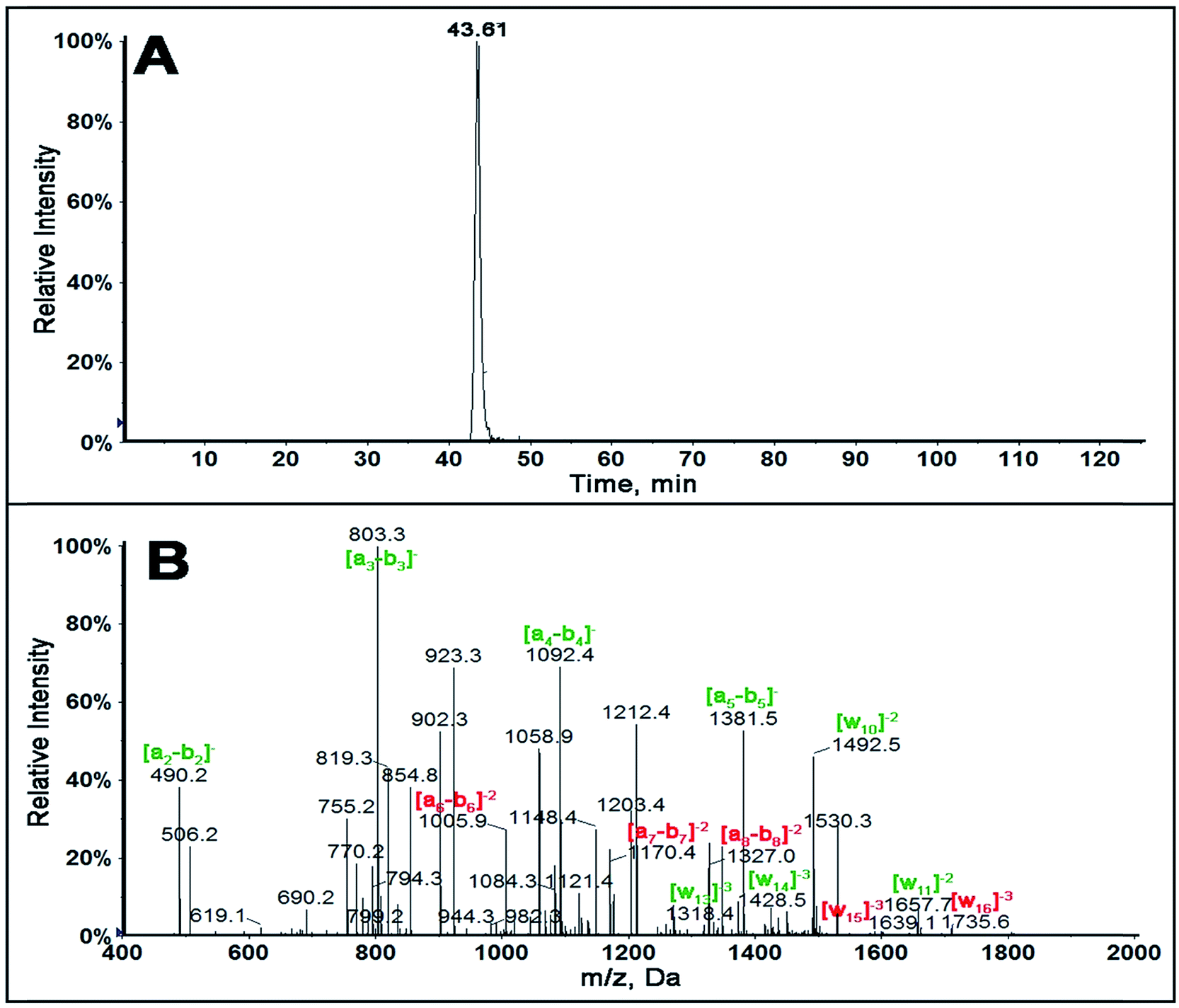 | ||
| Fig. 2 LC-MS of ds-32 bp exon 7 fragment: (A) extracted ion chromatogram of singly adducted fragment 2, m/z 1530.3, z = −4. (B) MS/MS spectrum of 1530.3 showing an-bn and wn ions. Ions with m/z similar to standard labeled in green and ions with increased m/z in red. | ||
MS/MS spectra for singly adducted fragment 2, m/z 1530.3, (Fig. 2B) shows all an-bn ions up to a5-b5 have m/z similar to that of unreacted standard. An increase in m/z of 302.323 (z = −1) was observed for a6-b6, a7-b7, a8-b8. This suggests that the possible modification is on base 5, (AACCG*GAGGCCCATCCTCA). This was confirmed by increases in m/z for w15 and w16 ions, while all w ions below w14 have m/z similar to that of unreacted standard (Table 3).
| Fragment ion | m/z | |
|---|---|---|
| Unadducted | Singly adducted | |
| a2-b2 | [490.2]− | [490.2]− |
| a3-b3 | [803.3]− | [803.3]− |
| a4-b4 | [1092.4]− | [1092.4]− |
| a5-b5 | [1381.5]− | [1381.5]− |
| a6-b6 | [1710.6]− | [1005.8]2− |
| a7-b7 | [1019.4]2− | [1170.4]2− |
| a8-b8 | [1176.2]2− | [1327.0]2− |
| w9 | [1348.0]2− | [1348.0]2− |
| w10 | [1492.7]2− | [1492.5]2− |
| w11 | [1657.7]2− | [1657.7]2− |
| w12 | n/d | n/d |
| w13 | [1318.4]3− | [1318.4]3− |
| w14 | [1428.3]3− | [1428.3]3− |
| w15 | [1538.0]3− | [1639.2]3− |
| w16 | [1634.5]3− | [1735.8]3− |
Similar experiments were done on the other singly adducted fragments obtained from the reacted ds-32 bp fragment, giving fragment 1 (m/z 1437.8, z = −3), fragment 3 (m/z 1012.7, z = −3) and fragment 4 (m/z 1234.9, z = −6). MS/MS spectra are shown in ESI (Fig. S5–S7 & Table S5–S7†), and results summarized in Table 4. These results were identical when the chemical reaction was done in the same buffer containing 10 mM NaCl, to see if reaction conditions would affect the specificity (Fig. S8†).
| Fragment name | Sequence | LC-MS/MS data | Common hot spot database13–15,18 | |
|---|---|---|---|---|
| Base adducted | Codon | |||
| a Possible cancers due to BPDE. b Non small cell lung cancer. c Small cell lung cancer. d Head and neck cancer. e Colorectal cancer. f Skin cancer. | ||||
| Fragment 1 | CATGGGCGGCATG | CATG*GGCGGCATG | 243 | 244c, 245be |
| CATGG*GCGGCATG | 244c | 244c, 245be | ||
| Fragment 2 | AACCGGAGGCCCATCCTCA | AACCG*GAGGCCCATCCTCA | 249bc | 248bcdef, 249bc |
| Complementary strands | ||||
| Fragment 3 | TGAGGATGGGCCTCCGGTTCATG | TGAGG*ATGGGCCTCCGGTTCATG | ||
| Fragment 4 | CCGCCCATG | CCG*CCCATG | ||
Since the modification is relatively small in the adducted DNA fragments, we assume the adducted and unadducted fragments have only minor changes in electrospray ionization behavior and MS response. Relative abundance of specific adducted nucleobases within codons were estimated by comparing areas of the extracted ion chromatograms of adducted fragments to that of unadducted fragments, and these ratios were further compared with other adducted fragments ratios. Ratios of relative amounts of adduction in the 32 bp fragment for codons 248/244 was 1.5 and for codons 248/243 was 1.3 with a relative standard deviation less than 6.5% (n = 3). According to p53 handbook database,13 the mutation frequency ratio of codons 248/243 is 24, 248/244 is 7, 248/249 is 2.7 and 248/245 is 2.1. Increased reactivity of codon 243 compared to p53 mutation frequency ratio may be related to guanine in codon 243 being the first guanine in our 32 bp fragment which may be more available for reaction due to its position near the end of the strand.
Discussion
The 32 bp p53 exon 7 fragment reacted with BPDE had all reactive bases as Gs, and the most frequently reacted guanine was in codon 248. Guanines within codons 243 and 244 were adducted to a lesser extent. Codon 248 is the major mutation hot spot for most tissue specific cancers such as lung, and head and neck. Thus, there is qualitative correlation of the high reactive frequency of codon 248 in our fragment with its high rate of mutation in many cancers, as well as the observed reactivity of codons 244 and 243. However, the relative mutation frequencies of codons 244 and 243 are lower that the relative reactivities we find in the 32 bp p53 fragment.Differences in reactivity vs. mutation frequency in tumors at codons 244 and 245 is undoubtedly related to the complexity of living tumors and cell cultures as compared to our in vitro reaction system. In addition to complications from organ-based metabolism, which is absent in our present reactions, differences may be due to DNA being bound to histones and other proteins in vivo as well as to in vivo DNA repair. Also, sequences in both 244 and 245 codons are GGC. Codon 243 is close to the end of the fragment, adjacent to an AT sequence, and may be partially unwound exposing the guanine adjacent to it due to fewer hydrogen bonds.49 Codon 243 may thus be more reactive in the fragment than in the full p53 gene. Also, under in vivo conditions, a considerable fraction of cytosines are methylated which can mediate codon reactivity.50,51 5-Methyl cytosines increase the nucleophilicity of exocyclic amine of adjacent guanine as result of inductive electronic effects.52 We are currently exploring this issue by additional experiments.
Covalent binding of BPDE preferentially targets guanines in DNA,53,54 because guanine is the best nucleophile of all the DNA bases. The N7 position of guanine is the most nucleophilic site but the exocyclic amine group, also a good nucleophile, is the most favorable target for polyaromatic hydrocarbon diol epoxides like BPDE. This is because proximity of the N7 group to the hydrophilic sugar chain of the DNA makes it less susceptible to attack of hydrophobic pyrenyl moieties (Scheme S2†).55–57
All exocyclic amines of guanines in single stranded DNA are available for covalent reaction with BPDE. In double stranded DNA the exocyclic amines are involved in hydrogen bonding with the carbonyl oxygen atom of cytosine hence are not freely available for reactions with BPDE. Guanines specifically in the minor groove of duplex DNA can form covalent adducts with BPDE, thereby disrupting the hydrogen bonding between the GC base pair as shown in ESI, Scheme S3.†58–60
Codon 247 is a hot spot for melanoma and skin squamous cell carcinoma, admittedly with a small number of mutations.13 Although guanine is not in this codon, the potential of BPDE to react with adenine exists. Since we did not find any adduction within codon 247, selectivity of BPDE to guanines was further affirmed.
Our results are consistent with the fact that most damaged codons in tumors and cancer cell cultures involve guanines as the major site of attack by BPDE.49,50,61 Guanines were also exclusive targets in the experiments with ss-oligonucleotides (Table 2). Structural differences of ss-DNA and ds-DNA may play a role in sequence specific DNA damage. Short single stranded oligonucleotides used in our study have a more flexible structure unprotected with a complementary strand and featuring a free axis of rotation along the phosphodiester backbone.62,63 Thus, while specificity would be expected to be much lower for ss-DNA compared with that of ds-DNA we still find that high mutation frequency codons 249 and 244 are highly reactive with BDPE in the short ss-fragments. These results suggest that the relative reactivity of the ds-DNA may be a combination of inherent chemistry of the codon sites and the ds-and higher structure of the DNA.
Double stranded DNAs are relatively inflexible due to GC and AT base pairing and double helical structure resulting in their complicated secondary and tertiary structures.30 The double helical structure also features major and minor groves in duplex DNA. BPDE specifically attacks the electron donating exocyclic amine in the minor groove containing the nucleophilic GC base pair of DNA. Therefore reactivity of ds-DNA will be different to that of ss-DNA.55,56 However in our experiments, we see that the ss-fragments and ds-32 bp fragment both have selective reactivity that includes codons with high frequency of mutation in p53. Thus, selectivity is different in the ss- and ds-fragments, but still exhibited by the single strands.
Very few studies thus far have focused on differential selectivity of specific base sequences.64 Jernstroem, et al. suggest that guanines adjacent to or flanked by guanines are more reactive62–66 In contradiction, other studies predict that guanines flanked by pyrimidine bases are more reactive due to less steric effects as compared to guanines flanked by purine bases.56,67–69 The reactive site of codon 248 in our 32 bp fragment is a guanine flanked by guanine, while codons 244 and 243 are guanines flanked by purine on one side and pyrimidine on the other side and show intermediate reactivity. Codon adduction sites in ss-DNA oligonucleotides are guanines sandwiched between one purine and one pyrimidine. Hence, our results are in qualitative agreement with the predictions quoted above. However, relating full guanine sequence specificity to adjacent bases is undoubtedly an oversimplification, and in addition to base sequence, specificity most likely involves complex structural and environmental factors in vivo including bound histones and proteins.
In summary, we have described methodology to screen chemicals, drugs and metabolites for reactions with oligonucleotides to determine the most frequently adducted nucleobase within codons. Results show that LC-MS/MS and restriction enzymes can be used to prepare carcinogen modified oligonucleotides so as to identify the site of adduction on oligonucleotide fragments longer than 20 base pairs. Our 32 bp fragment reactions represent the first study of a p53 gene fragment representing more than 20 base pairs with multiple mutation hot spots. The highest reaction frequency was at codon 248, consistent with the highest mutation frequency of the p53 gene in many cancers. Also, BPDE reactivity found at codons 243 and 244 is consistent with these codons being mutated in tissue cancers. Future work will focus on the use of metabolic enzymes to generate metabolites in the presence of longer, more representative p53 gene.
Acknowledgements
The authors thank the National Institute of Environmental Health Sciences (NIEHS), NIH, USA, Grant No. ES03154 for financial support.References
- American Cancer Society, Cancer Facts & Figures 2015, Atlanta, Ga, American Cancer Society, 2014, http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2015/, accessed April 16, 2015 Search PubMed.
- D. Haber and E. Harlow, Nat. Genet., 1997, 16, 320–322 CrossRef CAS PubMed.
- P. May and E. May, Oncogene, 1999, 18, 7621–7636 CrossRef CAS PubMed.
- M. Isobe, B. S. Emanuel, D. Givol, M. Oren and C. M. Croce, Nature, 1986, 320, 84–85 CrossRef CAS PubMed.
- S. E. Kern, K. W. Kinzler, A. Bruskin, D. Jarosz, P. Friedman, C. Prives and B. Vogelstein, Science, 1991, 252, 1708–1711 CrossRef CAS.
- G. Matlashewski, P. Lamb, D. Pim, J. Peacock, L. Crawford and S. Benchimol, EMBO J., 1984, 3, 3257–3262 CAS.
- M. C. Maiuri, E. Tasdemir, A. Criollo, E. Morselli, J. M. Vicencio, R. Carnuccio and G. Kroemer, Cell Death Differ., 2009, 16, 87–93 CrossRef CAS PubMed.
- T. Soussi and K. G. Wiman, Cancer Cell, 2007, 12, 303–312 CrossRef CAS PubMed.
- T. Soussi, Oncogene, 2007, 26, 2145–2156 CrossRef CAS PubMed.
- C. F. Cheok, C. S. Verma, J. Baselga and D. P. Lane, Nat. Rev. Clin. Oncol, 2011, 8, 25–37 CrossRef CAS PubMed.
- G. P. Pfeifer and A. Besaratinia, Hum. Genet., 2009, 125, 493–506 CrossRef CAS PubMed.
- T. Ozaki and A. Nakagawara, J. Biomed. Biotechnol., 2011, 603925, 1–13 CrossRef PubMed.
- L. Hjortsberg, J. M. Rubio-Nevado, D. Hamroun, C. Béroud, M. Claustre and T. Soussi, The p53 Mutation handbook 2.0, available online, http://p53.free.fr/Database/p53_cancer_db.html, accessed April 16, 2015 Search PubMed.
- T. Soussi, Adv. Cancer Res, 2011, 110, 107–139 CrossRef CAS PubMed.
- B. Leroy, M. Anderson and T. Soussi, Hum. Mutat., 2014, 35, 672–688 CrossRef CAS PubMed.
- M. Olivier, S. P. Hussain, d. F. C. Caron, P. Hainaut and C. C. Harris, IARC Sci. Publ., 2004, 157, 247–270 Search PubMed.
- http://www.binfo.ncku.edu.tw/TAG/GeneFinder.php, accessed April 16, 2014.
- J. C. States, M. Ouyang and C. W. Helm, Cancer Epidemiol., 2014, 38, 321–327 CrossRef PubMed.
- W. Xiong, J. Glick, Y. Lin and P. Vouros, Anal. Chem., 2007, 79, 5312–5321 CrossRef CAS PubMed.
- Q. Liao, C. Shen and P. Vouros, J. Mass Spectrom., 2009, 44, 549–556 CrossRef CAS PubMed.
- G. Chowdhury and F. P. Guengerich, Chem. Res. Toxicol., 2009, 22, 1310–1319 CrossRef CAS PubMed.
- G. Chowdhury and F. P. Guengerich, Curr Protoc Nucleic Acid Chem, 2011, ch. 7, Unit 7.16.pp. 1–7.16.11 Search PubMed.
- N. Tretyakova, P. W. Villalta and S. Kotapati, Chem. Rev., 2013, 113, 2395–2436 CrossRef CAS PubMed.
- A. Harsch, J. M. Sayer, D. M. Jerina and P. Vouros, Chem. Res. Toxicol., 2000, 13, 1342–1348 CrossRef CAS PubMed.
- G. Chowdhury and F. P. Guengerich, Angew. Chem., Int. Ed., 2008, 47, 381–384 CrossRef CAS PubMed.
- V. K. Sharma, J. Glick, Q. Liao, C. Shen and P. Vouros, J. Mass Spectrom., 2012, 47, 490–501 CrossRef CAS PubMed.
- J. E. Satterwhite, A. M. Pugh, A. S. Danell and E. G. Hvastkovs, Anal. Chem., 2011, 83, 3327–3335 CrossRef CAS PubMed.
- V. K. Sharma, J. Glick and P. Vouros, J. Chromatogr. A, 2012, 1245, 65–74 CrossRef CAS PubMed.
- V. K. Sharma, W. Xiong, J. Glick and P. Vouros, Eur. J. Mass Spectrom., 2014, 20, 63–72 CrossRef CAS.
- C. Bergonzo, R. Galindo-Murillo and T. E. Cheatham, Current Protocols in Nucleic Acid Chemistry, 2014, 55, 7.9.1–7.9.27 Search PubMed.
- A. A. Kornyshev, Phys. Chem. Chem. Phys., 2010, 12, 12352–12378 RSC.
- C. R. Cantor and P. R. Schimmel, Biophysical Chemistry, Pt. 1: The Conformation of Biological Macromolecules, 1st edn, Freeman, 1980, pp. 155–201 Search PubMed.
- Z. Chen, G. Zhang, X. Chen, J. Chen and J. Liu, J. Antibiot., 2012, 65, 517–522 CrossRef CAS PubMed.
- E. L. Loechler, Carcinogenesis, 1996, 17, 895–902 CrossRef CAS PubMed.
- A. N. Neilson, The Handbook of Environmental Chemistry, Anthropogenic Compounds, Part I: PAHs and Related Compounds: Chemistry, vol. 3, Springer, 1998, pp. 41–45 Search PubMed.
- H. V. Gelboin, Physiol. Rev., 1980, 60, 1107–1166 CAS.
- R. G. Harvey, Metabolic activation, DNA binding, and mechanisms of carcinogenesis. Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity, Cambridge University Press, New York, 1991, pp. 50–78 Search PubMed.
- http://monographs.iarc.fr/ENG/Classification/, (accessed April 6, 2015).
- B. Brahim, S. Alves, R. B. Cole and J. Tabet, J. Am. Soc. Mass Spectrom., 2013, 24, 1988–1996 CrossRef CAS PubMed.
- V. Jung, S. B. Pestka and S. Pestka, Nucleic Acids Res., 1990, 18, 6156 CrossRef CAS PubMed.
- K. K. Murray, J. Mass Spectrom., 1996, 31, 1203–1215 CrossRef CAS.
- D. P. Little, R. A. Chorush, J. P. Speir, M. W. Senko, N. L. Kelleher and F. W. McLafferty, J. Am. Chem. Soc., 1994, 116, 4893–4897 CrossRef CAS.
- N. Potier, D. Van Alain, Y. Cordier, O. Roch and R. Bischoff, Nucleic Acids Res., 1994, 22, 3895–3903 CrossRef CAS PubMed.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- http://mods.rna.albany.edu/masspec/Mongo-Oligo, accessed April 16, 2015.
- S. A. McLuckey, B. G. Van and G. L. Glish, J. Am. Soc. Mass Spectrom., 1992, 3, 60–70 CrossRef CAS.
- S. A. McLuckey and S. Habibi-Goudarzi, J. Am. Soc. Mass Spectrom., 1994, 5, 740–747 CrossRef CAS.
- J. Rozenski and J. A. McCloskey, J. Am. Soc. Mass Spectrom., 2002, 13, 200–203 CrossRef CAS.
- C. R. Calladine, H. R. Drew, B. F. Luisi and A. A. Travers, Understanding DNA: The Molecule & how it Works, 3rd edn, Elsevier Ltd, 2004, pp. 70–72 Search PubMed.
- M. F. Denissenko, J. X. Chen, M. Tang and G. P. Pfeifer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3893–3898 CrossRef CAS.
- K. E. Varley, J. Gertz, K. M. Bowling, S. L. Parker, T. E. Reddy, F. Pauli-Behn, M. K. Cross, B. A. Williams, J. A. Stamatoyannopoulos, G. E. Crawford, D. M. Absher, B. J. Wold and R. M. Myers, Genome Res., 2013, 23, 555–567 CrossRef CAS PubMed.
- R. Guza, D. Kotandeniya, K. Murphy, T. Dissanayake, C. Lin, G. M. Giambasu, R. R. Lad, F. Wojciechowski, S. Amin, S. J. Sturla, R. H. E. Hudson, D. M. York, R. Jankowiak, R. Jones and N. Y. Tretyakova, Nucleic Acids Res., 2011, 39, 3988–4006 CrossRef CAS PubMed.
- G. Hermanson, Nucleic acid and oligonucleotide conjugation and modification, Bioconjugate Techniques, ed. J. Audet and M. Preap, 3rd edn, Academic Press, 2013, pp. 959–987 Search PubMed.
- K. S. Gates, Chem. Res. Toxicol., 2009, 22, 1747–1760 CrossRef CAS PubMed.
- F. A. Beland and M. C. Poirier, Methods to Assess DNA Damage and Repair: lnterspecies Comparisons, ed. R. G. Tardiff, P. H. Lohman and G. N. Wogan, SCOPE, John Wiley & Sons, New York, 1994, pp. 29–55 Search PubMed.
- W. M. Baird, L. A. Hooven and B. Mahadevan, Environ. Mol. Mutagen., 2005, 45, 106–114 CrossRef CAS PubMed.
- G. Lenglet and M. David-Cordonnier, J. Nucleic Acids, 2010, 290935, 1–17 CrossRef PubMed.
- N. E. Geacintov, M. Cosman, B. E. Hingerty, S. Amin, S. Broyde and D. J. Patel, Chem. Res. Toxicol., 1997, 10, 111–146 CrossRef CAS PubMed.
- M. Penning, Poly Aromatic Hydrocarbons: Multiple Metabolic Pathways and the DNA Lesions formed, The Chemical Biology of DNA Damage, ed. N. E. Geacintov and S. Broyde, Wiley-VCH Verlag GmbH & Co, KGaA, 2010, pp. 131–155 Search PubMed.
- C. Deligkaris and J. H. Rodriguez, Phys. Chem. Chem. Phys., 2014, 16, 6199–6210 RSC.
- S. Pan, D. Li, L. Zhao, J. B. Schenkman and J. F. Rusling, Chem. Res. Toxicol., 2013, 26, 1229–1239 CrossRef CAS PubMed.
- M. Levitt, Folding of nucleic acids, Int. Assoc. Hydrol. Sci. Publ., 1972, 7, 147–171 CAS.
- H. Lodish, A. Berk, P. Matsudaira, C. A. Kaiser and M. Krieger, Structure of Nucleic Acids. Molecular Cell Biology, 5th ed., W. H. Freeman, New York, 2003; pp. 102–107 Search PubMed.
- B. Jernstroem and A. Graeslund, Biophys. Chem., 1994, 49, 185–199 CrossRef CAS.
- B. Said and R. C. Shank, Nucleic Acids Res., 1991, 19, 1311–1316 CrossRef CAS PubMed.
- F. A. Rodriguez, Y. Cai, C. Lin, Y. Tang, A. Kolbanovskiy, S. Amin, D. J. Patel, S. Broyde and N. E. Geacintov, Nucleic Acids Res., 2007, 35, 1555–1568 CrossRef CAS PubMed.
- T. Musafija-Jeknic, A. Luch, A. Seidel, C. Johns, C. Pereira and W. M. Baird, Polycyclic Aromat. Compd., 2005, 25, 103–111 CrossRef CAS PubMed.
- J. J. Schwartz, H. H. S. Lau and W. M. Baird, Chem. Res. Toxicol., 1994, 7, 29–40 CrossRef CAS.
- L. A. Margulis, V. Ibanez and N. E. Geacintov, Chem. Res. Toxicol., 1993, 6, 59–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI file contains 14 additional figures and 7 tables describing LC-MS/MS results and 3 schemes discussing the mechanism of chemical damage. See DOI: 10.1039/c5sc01403d |
| This journal is © The Royal Society of Chemistry 2015 |