
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction of a diaryldigermyne with ethylene†
Takahiro
Sasamori
*a,
Tomohiro
Sugahara
a,
Tomohiro
Agou
a,
Koh
Sugamata
a,
Jing-Dong
Guo
ab,
Shigeru
Nagase
b and
Norihiro
Tokitoh
*a
aInstitute for Chemical Research, Kyoto University, Gokasho Uji, Kyoto 611-0011, Japan. E-mail: sasamori@boc.kuicr.kyoto-u.ac.jp
bFukui Institute for Fundamental Chemistry, Kyoto University, Kyoto 606-8103, Japan
First published on 24th June 2015
Abstract
Reaction of the stable digermyne BbtGe![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) GeBbt (Bbt = 2,6-[CH(SiMe3)2]2-4-[C(SiMe3)3]-C6H2) with ethylene initially afforded the corresponding 1,2-digermacyclobutene. Depending on the reaction conditions applied, further reaction of this 1,2-digermacyclobutene with ethylene furnished two different reaction products: a 1,4-digermabicyclo[2.2.0]hexane or a bis(germiranyl)ethane. Combined experimental and theoretical results suggested that the 1,4-digermabicyclo[2.2.0]hexane and the bis(germiranyl)ethane are the thermodynamic and kinetic reaction products, respectively. A reaction mechanism in agreement with these results was proposed.
GeBbt (Bbt = 2,6-[CH(SiMe3)2]2-4-[C(SiMe3)3]-C6H2) with ethylene initially afforded the corresponding 1,2-digermacyclobutene. Depending on the reaction conditions applied, further reaction of this 1,2-digermacyclobutene with ethylene furnished two different reaction products: a 1,4-digermabicyclo[2.2.0]hexane or a bis(germiranyl)ethane. Combined experimental and theoretical results suggested that the 1,4-digermabicyclo[2.2.0]hexane and the bis(germiranyl)ethane are the thermodynamic and kinetic reaction products, respectively. A reaction mechanism in agreement with these results was proposed.
Control over the modification of olefin groups is important in organic synthesis, as a variety of preparative methods for the introduction of functional groups start from C–C multiple bonds. Even though several olefin addition reactions, such as hydrosilylation,1 hydroboration2 and olefin polymerisation,3 are well established, the use of transition metal catalysts is required in many cases. However, divalent or multiple-bonded compounds of heavier group 14 elements have recently received much attention as potential transition metal-free catalysts.4 These compounds generally react with olefins or other compounds that have carbon-containing multiple bonds to form the corresponding cycloadducts, tantamount to a strong propensity to activate small inert molecules. Unfortunately, low-coordinate species of heavier main group elements are usually difficult to isolate, mostly due to their inherently high reactivity towards addition reactions involving atmospheric moisture and/or aerobic oxygen and self-oligomerisation. Nevertheless, these compounds can be isolated while retaining their characteristic reactivity when sterically demanding substituents are used to provide kinetic stabilisation.5 Power and co-workers have, for example, reported the isolation of the heavier acetylene analogues ArDipGe
GeArDip (1)6 and ArDipSnSnArDip (2)7 as stable compounds. The reactions of 1 and 2 with ethylene proceed smoothly in the absence of any transition metal catalyst at room temperature to afford the corresponding 4-membered cycloadducts 3 and 4 (Type I; Scheme 1).8,9 Subsequently, 4 is able to undergo a thermal retro-cycloaddition to generate 2, concomitant with the release of two molecules of ethylene. Accordingly, distannyne 2 can, in contrast to digermyne 1, be considered as an ethylene-storage molecule. In this context, the reaction of a comparable disilyne with ethylene should also be of great interest. Independently, the groups of Wiberg and Sekiguchi have reported the stereoselective [2+2] cycloaddition of stable disilynes 5a,b (RSiSiSiRSi; 5a: RSi = Si(Me)[Si(t-Bu)3]2,105b: RSi = Si[CH(SiMe3)2](i-Pr)11) with alkenes (RHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR) to afford disilenes 6a,b (6a: R = H, 6b: R = Me).10,12 However, neither the further reaction of 6a,b with ethylene, nor any possible retro-reaction were reported.
CHR) to afford disilenes 6a,b (6a: R = H, 6b: R = Me).10,12 However, neither the further reaction of 6a,b with ethylene, nor any possible retro-reaction were reported.
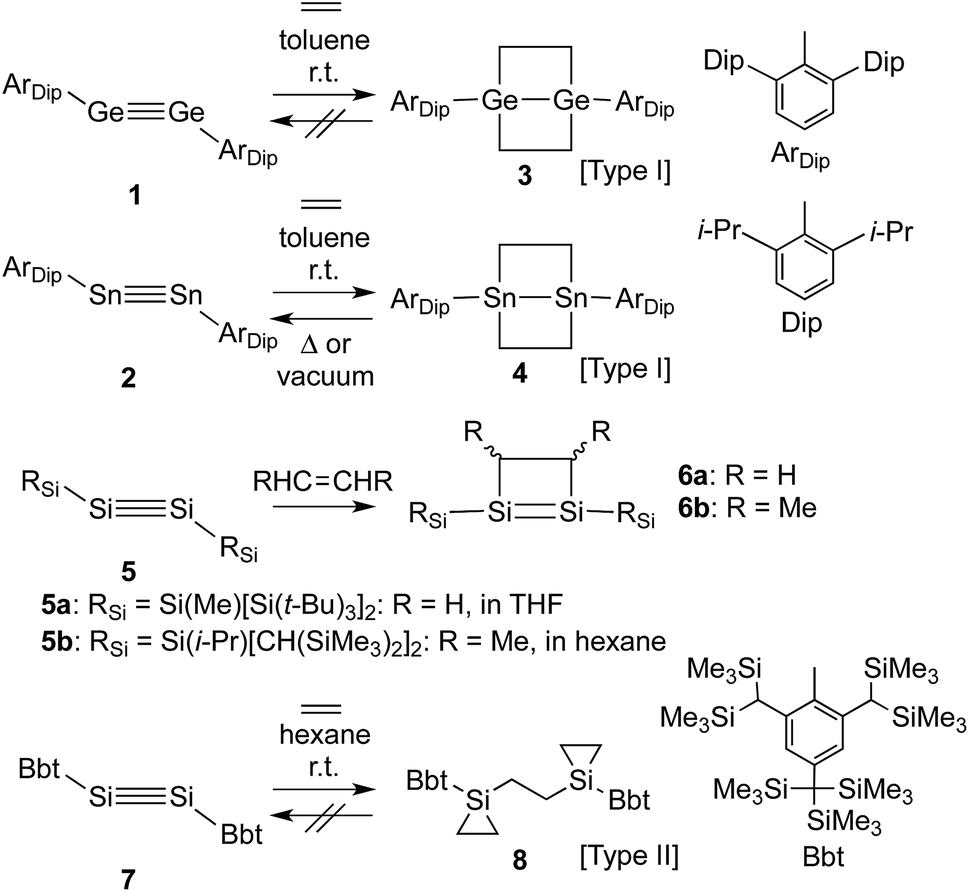 | ||
| Scheme 1 Reactions of dimetallynes 1, 2, 5 and 7 with ethylene. | ||
Previously, we have reported the synthesis of the stable diaryldisilyne BbtSiSiBbt (7, Bbt = 2,6-[CH(SiMe3)2]-4-[C(SiMe3)3]-C6H2).13 The reaction of 7 with ethylene resulted in the unexpected formation of 8 (Type II; Scheme 1), containing two silacyclopropane moieties. Compound 8 was found to be remarkably stable, as decomposition of these silacyclopropane moieties was not observed, even upon heating.14 Subsequently, we began to investigate the reactivity difference between diaryldisilynes and diaryldigermynes. Herein, we report the reaction of the stable diaryldigermyne BbtGeGeBbt (9)15 with ethylene to afford the corresponding 1,2-digermacyclobutene (10), which is the formal [2+2] cycloadduct of 9. Depending on the reaction conditions, further treatment of 10 with ethylene resulted in the formation of two products, specifically a four-membered cycloadduct (12, Type I; Scheme 2) and a three-membered cycloadduct (11, Type II; Scheme 2).
 | ||
| Scheme 2 Reaction of diaryldigermyne 9 with ethylene. | ||
A hexane solution of digermyne 9 was frozen (−196 °C) and degassed in a J-Young tube, before being charged with ethylene.16 The colour of the solution changed from dark red to purple. Removal of the solvent from the reaction mixture afforded 1,2-digermacyclobutene 10. The formation of 10 from the reaction of digermyne 9 with ethylene can be explained by the same mechanism used to describe the reaction of disilynes with olefins:12 initially, interaction between ethylene and one of the Ge atoms in the GeGe bond generates germirane-substituted germylene 13 as an intermediate,17 which subsequently inserts intramolecularly into the Ge–C bond of the germirane moiety (Scheme 3). X-ray crystallographic analysis of 10 revealed a non-planar structure for the four-membered GeGe–C–C ring (Fig. 1).18 The two Bbt groups were found to be oriented in opposite directions, resulting in a trans-bent geometry for the GeGe moiety with trans-bent angles of 39.5° (Ge1) and 39.7° (Ge2). A GeGe bond length of 2.4132(5) Å was observed, which is slightly shorter than a typical Ge–Ge single bond (ca. 2.44 Å),19 but consistent with previously reported GeGe double bonds in digermenes (ca. 2.2–2.5 Å).19 These structural features suggested that the GeGe double bond in 10 should be weakened by the severe intrinsic strain of the four-membered GeGe–C–C ring and the highly trans-bent geometry. The 1H NMR spectrum of 10 exhibited signals commensurate with two identical Bbt groups, as well as signals consistent with two equivalent SiMe3 groups at the ortho-positions of the Bbt groups, thus confirming a fast inversion of the trans-bent geometry of the GeGe bond in 10 in solution.
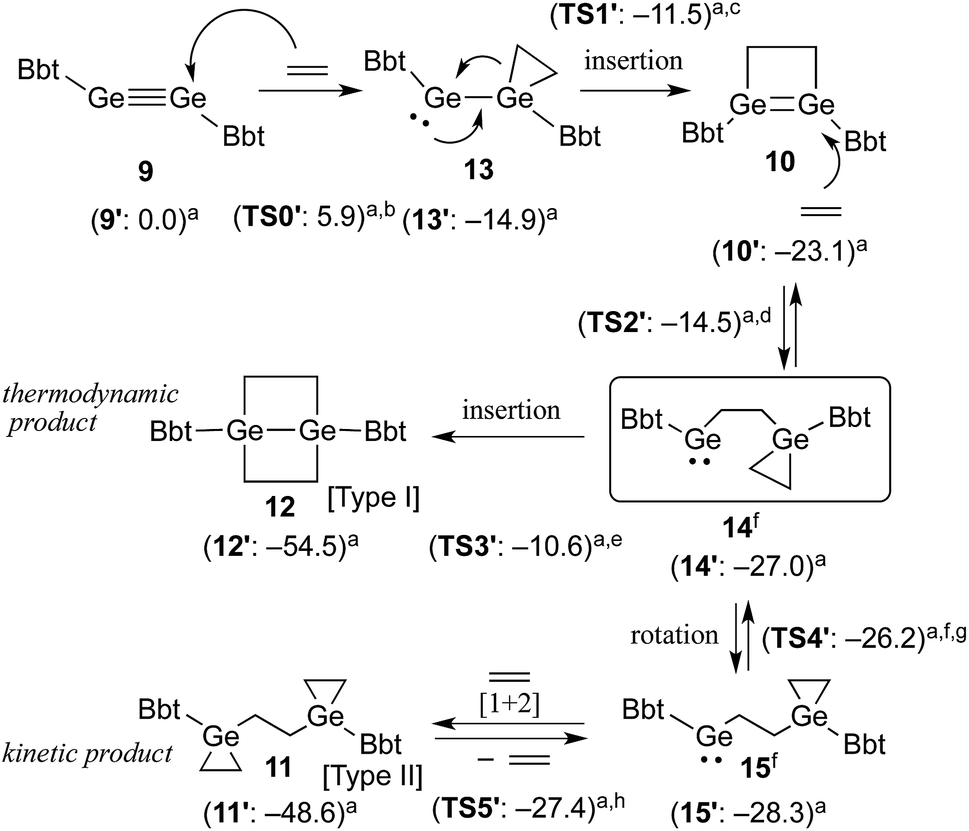 | ||
| Scheme 3 Proposed mechanism for the reaction of 9 with ethylene. (a) Calculated relative energies (kcal mol−1) for model compounds bearing 2,6-[CH(SiMe3)2]2-C6H3 (Bbp) groups instead of Bbt groups. (b) ΔE‡ (9′–13′) = 5.9 kcal mol−1. (c) ΔE‡ (13′–10′) = 3.4 kcal mol−1. (d) ΔE‡ (10′–14′) = 8.6 kcal mol−1. (e) ΔE‡ (14′–12′) = 16.4 kcal mol−1. (f) 14 and 15 are rotational isomers with respect to a rotation around the central GeH2C–CH2Ge bond. (g) ΔE‡ (14′–15′) = 0.8 kcal mol−1. (h) ΔE‡ (15′–11′) = 0.9 kcal mol−1. | ||
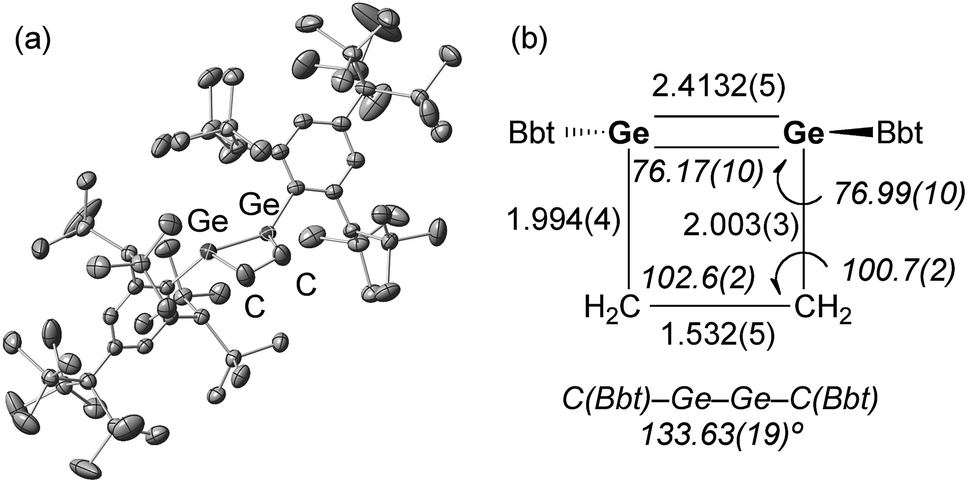 | ||
| Fig. 1 (a) Molecular structure of 10 (thermal ellipsoids at 50% probability; hydrogen atoms omitted for clarity), and (b) selected metric parameters for the digermacyclobutene core in 10. | ||
In order to induce a further reaction of 10 with a second molecule of the alkene, ethylene was condensed into a sealed vessel, which contained a frozen and degassed C6D6 solution of 10 at −196 °C. Subsequently, the reaction mixture was allowed to warm to r.t. in this sealed tube, and based on the volume of the tube, 10 was treated with an excess of ethylene (ca. 5 atm). The purple colour of 10 disappeared immediately,20 and 11 (Type II; Scheme 2) was obtained as a colourless precipitate.21 Upon opening the sealed tube in an argon-filled glove box, the colourless powder turned purple again, and on the basis of its 1H NMR spectrum it could be established that 11 retroconverted quantitatively to afford 10 within a few minutes at r.t. Accordingly, the reaction of 10 with ethylene to furnish 11 is, depending on the ethylene pressure, reversible. On the other hand, exposure of a degassed THF solution of 10 to ethylene at ambient pressure (ca. 1 atm; −78 °C to r.t.; 1 d) afforded stable colourless crystals of 12 (Type I; Scheme 2) in quantitative yield. Depending on the reaction conditions, the reaction of 10 with ethylene thus delivers different reaction products. The molecular structures of 11 and 12 were determined unambiguously by spectroscopic and X-ray crystallographic analyses.22
In order to elucidate the underlying reaction mechanism (Scheme 3), the reaction between digermyne 9 and ethylene was monitored by 1H NMR spectroscopy in THF-d8. After exposing a degassed THF-d8 solution of 9 to ethylene (ca. 1 atm) at −78 °C and then allowing it to warm to r.t., the colour of 9 disappeared and only signals associated with 10 were observed. After 10 min, the intensity of these signals decreased, and additional signals consistent with the formation of 11 were observed (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 = ca. 1:1). After 20 min, signals in agreement with the formation of 12 appeared, and after 5 hours, the quantitative formation of 12 was observed. These experimental results suggested that the reaction of 10 with ethylene furnishes 11 and 12 as the kinetic and thermodynamic products, respectively.
:11 = ca. 1:1). After 20 min, signals in agreement with the formation of 12 appeared, and after 5 hours, the quantitative formation of 12 was observed. These experimental results suggested that the reaction of 10 with ethylene furnishes 11 and 12 as the kinetic and thermodynamic products, respectively.
Taking all the previously discussed results into consideration, the reaction mechanism for the reaction between digermyne 9 and ethylene can most likely be interpreted as follows: the reaction is initiated by a nucleophilic attack of ethylene towards the LUMO of 9 to afford 13, which readily undergoes an intramolecular ring-expansion, affording 10via a germylgermylene–digermene rearrangement.23 Subsequently, nucleophilic attack of another molecule of ethylene towards the LUMO of 10 affords germylene 14, which is expected to easily undergo a [1+2] cycloaddition reaction between a further molecule of ethylene and the second germylene moiety. While this [1+2] cycloaddition reaction should be reversible,24 considering the results of the NMR monitoring reactions, the intramolecular C–Ge insertion of 14 is expected to proceed irreversibly to provide the thermodynamically stable product 1,4-digerma-bicyclo[2.2.0]hexane (12). The solubility of 11 in benzene was found to be limited, and the precipitation of 11 in the form of a colourless solid was observed when the reaction was conducted in this solvent. When the same reaction was carried out in THF, the kinetic product 11 was generated at an early stage in the reaction, and subsequently both 10 and 11 were converted to the thermodynamic product 12. It can thus be concluded that the reactions of such digermynes are mostly initiated by nucleophilic attack of π-electrons towards the in-plane π* orbital (LUMO) of the Ge–Ge triple bond, which is consistent with the previously reported reactivity of π-bond compounds containing heavier group 14 elements.
The proposed reaction pathways were also examined by density functional theory (DFT) calculations (see Fig. S11†), using appropriate model compounds (9′–15′) bearing Bbp (Bbp = 2,6-[CH(SiMe3)2]2-C6H3) instead of Bbt groups (Scheme 3).25 The results suggested that intermediate 13′ is formed with a small reaction barrier of 5.9 kcal mol−1, and is thermodynamically more stable than 9′ + ethylene by 14.9 kcal mol−1. Subsequently, 13′ can afford 10′ (8.2 kcal mol−1 more stable) with a small reaction barrier of 3.4 kcal mol−1. Following that, the reaction of 10′ with ethylene can provide key intermediate 14′ (3.9 kcal mol−1 more stable) with a reaction barrier of 8.6 kcal mol−1. The second molecule of ethylene can then react smoothly with 14′ to give product 11′via intermediate 15′, which is a rotational isomer with a very low reaction barrier (<1.0 kcal mol−1), while product 12′ is produced with a large barrier of 16.4 kcal mol−1, which is 5.9 kcal mol−1 more stable than product 11′. The results of these DFT calculations corroborated the hypothesis that the reaction of 10 with ethylene should furnish 11 and 12 as the kinetic and thermodynamic products, respectively.26
Finally, the reactivity difference between the reaction of ethylene with digermynes (1 and 9) and that with disilyne (7) can be explained as follows: for the reaction with 1 (ArDipGeGeArDip), the calculations draw the conclusion that the corresponding Type II product with three-membered rings should be the kinetic product, while the Type I product 3, i.e. 1,4-digermabicyclo[2.2.0]hexane, should be the thermodynamic product, indicating that the observation of the kinetic product under these reaction conditions is unlikely.27 These conclusions are in agreement with our experimental observations. For the reaction of 7 with ethylene, theoretical calculations indicated that Type II product 8 should be both the kinetically and the thermodynamically favoured product.28 These results could be interpreted in terms of the relative stability of the Ge- or Si-containing three-membered rings.
Conclusions
In summary, we found that the reaction of digermyne 9 with ethylene affords two different reaction products (11, 12), depending on the reaction conditions applied. The stable digermacyclobutene 10, which is an intermediate in this reaction, could be isolated and subsequently treated under controlled reaction conditions with a second molecule of ethylene. A combined theoretical and experimental investigation of these reactions allowed the assignment of 11 and 12 as the kinetic and thermodynamic reaction products, respectively.Acknowledgements
This work was partially supported by the following grants: Grants-in-Aid for Scientific Research (B) (no. 2235001722350017), Scientific Research on Innovative Areas, “New Polymeric Materials Based on Element-Blocks” [#2401] (no. 2510251925102519), “Stimuli-Responsive Chemical Species for the Creation of Functional Molecules” [#2408] (no. 2410901324109013), and the MEXT Project of Integrated Research on Chemical Synthesis from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, as well as the “Molecular Systems Research” project of the RIKEN Advanced Science Institute and the Collaborative Research Program of Institute for Chemical Research, Kyoto University. We would like to thank Mr Toshiaki Noda and Ms Hideko Natsume at Nagoya University for the expert manufacturing of custom-tailored glassware. The manuscript for this publication was written at the Rheinische Friedrich-Wilhelms-Universität Bonn during the tenure of an Alexander von Humboldt Research Award (NT) and a Friedrich Wilhelm Bessel Research Award (TS). NT and TS would like to express their gratitude to Prof. Rainer Streubel and his co-workers for their kind hospitality, as well as to the Alexander von Humboldt Stiftung for their generosity.Notes and references
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440 CrossRef CAS.
- H. C. Brown and B. Singaram, Acc. Chem. Res., 1988, 21, 287 CrossRef CAS.
- V. Busico and R. Cipullo, Prog. Polym. Sci., 2001, 26, 443 CrossRef CAS.
- (a) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed; (b) P. P. Power, Acc. Chem. Res., 2011, 44, 627 CrossRef CAS PubMed; (c) C. M. Cui, M. M. Olmstead, J. C. Fettinger, G. H. Spikes and P. P. Power, J. Am. Chem. Soc., 2005, 127, 17530 CrossRef CAS PubMed.
- (a) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877 CrossRef CAS PubMed; (b) T. Sasamori and N. Tokitoh, Bull. Chem. Soc. Jpn., 2013, 86, 1005 CrossRef CAS; (c) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479 CrossRef CAS PubMed; (d) S. Nagase, Bull. Chem. Soc. Jpn., 2014, 87, 167 CrossRef CAS; (e) M. Asay and A. Sekiguchi, Bull. Chem. Soc. Jpn., 2012, 85, 1245 CrossRef CAS.
- M. Stender, A. D. Phillips, R. J. Wright and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 1785 CrossRef CAS.
- A. D. Phillips, R. J. Wright, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2002, 124, 5930 CrossRef CAS PubMed.
- Y. Peng, B. D. Ellis, X. P. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668 CrossRef CAS PubMed.
- The reduction of Tsi(Cl)Ge: using Mg/MgBr2 in the presence of ethylene in THF was reported to afford the corresponding 1,4-digerma-bicyclo[2.2.0]hexane, which is the Type I ethylene-adduct of a transient TsiGeGeTsi species; see: T. Ohtaki and W. Ando, Organometallics, 1996, 15, 3103 CrossRef CAS.
- N. Wiberg, S. K. Vasisht, G. Fischer and P. Mayer, Z. Anorg. Allg. Chem., 2004, 630, 1823 CrossRef CAS.
- A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755 CrossRef CAS PubMed.
- R. Kinjo, M. Ichinohe, A. Sekiguchi, N. Takagi, M. Sumimoto and S. Nagase, J. Am. Chem. Soc., 2007, 129, 7766 CrossRef CAS PubMed.
- (a) T. Sasamori, K. Hironaka, Y. Sugiyama, N. Takagi, S. Nagase, Y. Hosoi, Y. Furukawa and N. Tokitoh, J. Am. Chem. Soc., 2008, 130, 13856 CrossRef CAS PubMed; (b) T. Sasamori, J. S. Han, K. Hironaka, N. Takagi, S. Nagase and N. Tokitoh, Pure Appl. Chem., 2010, 82, 603 CrossRef CAS.
- J. S. Han, T. Sasamori, Y. Mizuhata and N. Tokitoh, J. Am. Chem. Soc., 2010, 132, 2546 CrossRef CAS PubMed.
- Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023 CrossRef CAS PubMed.
- Experimental details and chemical data for previously unreported compounds are given in the ESI.†.
- Given the structure and NBO charge (C2H4 part, +0.045) of transition state TS0′, the initial interaction between ethylene and digermyne 9 can be feasibly explained by an interaction between the HOMO of ethylene and the in-plane π* orbital (LUMO) of 9. Conversely, the geometry of intermediate 13′ should, considering its geometry and NBO charge (C2H4 part, −0.586), be interpreted in terms of an orbital interaction between the π* orbital of ethylene and the out-of-plane π orbital of 9. For further details, see ref. 15 and the ESI.†.
- Torsion angle: 11.06(18)°.
- For example, the Ge–Ge and GeGe bond lengths in ArDipH2Ge–GeH2ArDip and ArDipHGeGeHArDip are 2.4019(10) Å and 2.3026(3) Å, respectively; see: G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232 CrossRef CAS PubMed.
-
10: λmax = 494 nm (ε 10000).
- Diaryldigermyne ArTipGeGeArTip [ArTip = 2,6-bis(2,4,6-triisopropylphenyl)phenyl] was reported to undergo a similar reaction with three molecules of 2,3-dimethyl-1,3-butadiene; see: M. Stender, A. D. Phillips and P. P. Power, Chem. Commun., 2002, 1312 RSC.
- Crystallographic data and selected structural parameters of 11 and 12 are given in the ESI.†.
- Interconversion between germylgermylenes and digermenes via 1,2-substituent-migration has been reported previously; see: (a) K. M. Baines, J. A. Cooke and J. J. Vittal, J. Chem. Soc., Chem. Commun., 1992, 1484 RSC; (b) K. M. Baines, J. A. Cooke, C. E. Dixon, H. W. Liu and M. R. Netherton, Organometallics, 1994, 13, 631 CrossRef CAS.
- The mechanism of formal [1+2] and [1+4] cycloaddition reactions of germylenes has been well studied, see: L. A. Huch and W. J. Leigh, Organometallics, 2009, 28, 6777 CrossRef.
- Level of theory: B3PW91/6-311+G(2d)[Ge,Si] and 6-31G(d,p)[C,H]//B3PW91/Lanl2dz+d[Ge], 3-21G*[Si], and 3-21G[C,H]. M. J. Frisch, et al., Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- In general, [2+2] cycloaddition reactions between a CC π-bond and a GeGe π-bond (with out-of-plane π/π* orbital) are symmetry forbidden according to the Woodward–Hoffmann rules. Thus, the concerted [2+2] cycloaddition pathway from 9 to 10 as well as from 10 to 12 can be excluded. In fact, a reaction barrier of ΔE‡ = 25.4 kcal mol−1 was calculated for the concerted [2+2] cycloaddition from 10′ to 12′, which is substantially larger than the barrier in the pathway shown in Scheme 3 (ΔE‡ = 16.4 kcal mol−1). Another possibility of a [2+2] cycloaddition via a biradical pathway was suggested in a previous paper (K. L. Hurni and K. M. Baines, Chem. Commun., 2011, 47, 8382), but could be excluded on the basis of the results of attempted calculations on these reaction pathways, which showed substantially larger barriers.
- The Type I product 3, i.e. 1,4-digerma-bicyclo[2.2.0]hexane, was calculated to be 1.3 kcal mol−1 more stable than the Type II product, i.e. the bis(germiranyl)ethane. A reaction barrier of ca. 19 kcal mol−1 was calculated for the retro-reaction from the Type II product to the ArDip analogue of intermediate 14′, indicating a reversible reaction at r.t. Conversely, a barrier of ca. 40 kcal mol−1 was obtained for the conversion of Type I product 3 to the intermediate, suggesting an irreversible reaction. Accordingly, 3 should be the final product under thermodynamically controlled conditions. In our case, the low solubility in non-polar solvents such as benzene or hexane most probably enabled us to isolate the kinetic product 11.
- In the case of Si, the Type II product, i.e.8, was calculated to be 23.1 kcal mol−1 more stable than the Type I product, i.e. the 1,4-disila-bicyclo[2.2.0]hexane analogue. Levels of theory: 6-311+G(2df)[Si] and 6-31G(d,p)[C,H]//B3PW91/3-21G*.
Footnote |
| † Electronic supplementary information (ESI) available: experimental and computational details, as well as X-ray crystallographic data for 10–12 are available. CCDC 1054594–1054596. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01266j |
| This journal is © The Royal Society of Chemistry 2015 |