
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExceptional CO2 working capacity in a heterodiamine-grafted metal–organic framework†
Woo Ram
Lee
a,
Hyuna
Jo
a,
Li-Ming
Yang
b,
Hanyeong
Lee
c,
Dae Won
Ryu
a,
Kwang Soo
Lim
a,
Jeong Hwa
Song
a,
Da Young
Min
d,
Sang Soo
Han
b,
Jeong Gil
Seo
c,
Yong Ki
Park
d,
Dohyun
Moon
e and
Chang Seop
Hong
*a
aDepartment of Chemistry, Korea University, Seoul 136-713, Korea. E-mail: cshong@korea.ac.kr
bCenter for Computational Science, Korea Institute of Science and Technology (KIST), Hwarangno 14-gil 5, Seongbuk-gu, Seoul 136-791, Korea
cDepartment of Energy and Biotechnology, Myongji University, Myongji-ro 116, Cheoin-gu, Yongin, Gyeonggi-do 449-728, Korea
dCenter for Carbon Resources Conversion, Korea Research Institute of Chemical Technology, Daejeon, 305-600, Korea
eBeamline Division, Pohang Accelerator Laboratory, Pohang, Kyungbuk 790-784, Korea
First published on 22nd April 2015
Abstract
An amine-functionalized metal–organic framework (MOF), dmen-Mg2(dobpdc) (dmen = N,N-dimethylethylenediamine), which contains a heterodiamine with both primary and tertiary amines, was prepared via a post-synthetic method. This material exhibits a significant selectivity factor for CO2 over N2 that is commensurate with top-performing MOFs. It is remarkable that the solid is fully regenerated under vacuum or flowing Ar at low desorption temperatures, and following this can take up CO2 at more than 13 wt%. An exceptionally high working capacity is achieved at low regeneration temperatures and after exposure to humid conditions, which are important parameters for a real post-combustion CO2 capture process.
Introduction
Carbon capture and sequestration (CCS) has been investigated to mitigate atmospheric CO2 concentrations and to address global environmental concerns.1–5 To capture CO2 selectively from power plant flue gas, aqueous alkanolamine solutions have been intensively investigated. However, these amine solutions require an enormous energy input (∼30% of the produced power) to regenerate the solvents.6 This energy penalty that arises from heating the water of the amine solutions makes solid adsorbents with low heat capacities, and other prospective alternatives, advantageous.7 Thus, one of the current research topics in the field of developing innovative materials for post-combustion CO2 capture is the reduction of the regeneration energy of the capture media.To evaluate the actual amount of CO2 captured during adsorption and desorption cycles in a practical post-combustion process, it is critically important to determine the working, rather than the absolute, CO2 capacity. The estimated working capacity of porous materials such as porous polymer networks (PPNs)8–11 and monoethanolamine (MEA)1,12 gradually decreases with lower desorption temperatures, based on a simplified model using CO2 isotherms (the difference between the adsorbed quantities at 0.15 bar CO2 and 40 °C, and at 1 bar CO2 and different desorption temperatures).9,13 The working capacities of the aforementioned materials become negligible at lower desorption temperatures; however, the regeneration temperature of the capture materials should be minimized to improve upon the high energy penalty of the current amine scrubbers.
Metal–organic frameworks (MOFs) with tunable pore shapes and other properties have been investigated for CO2 capture.14–23 Of these MOFs, the MOF-74 series exhibits a high density of exposed Lewis acidic metal sites aligned along the pore walls.24–27 The coordinatively unsaturated sites cause strong interactions with CO2 due to their large quadrupole moments and their polarizability.28 For instance, an exceptionally high CO2 capacity of 20.6 wt% under relevant post-combustion flue gas conditions was observed for Mg–MOF-74.13 However, in a breakthrough experiment, after exposure to 70% relative humidity (RH) and subsequent regeneration at high temperature, Mg–MOF-74 showed only about 16% recovery of its initial amount of CO2.29
Improving CO2 capacities at lower CO2 partial pressures has also been achieved by decorating the open metal sites of MOFs with diamines.1,30–35 In this case, homodiamines were selected to modify the interior of the pores of the MOF such that one amine was grafted onto an open metal site while the other remained uncoordinated. Amine-functionalized MOFs reveal outstanding CO2 capacities at low CO2 concentrations because the free amine groups located toward the pore centers can strongly couple to CO2.36–40 In particular, Mg2(dobpdc) (H4dobpdc = 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylic acid) grafted with primary en (= ethylenediamine) and secondary mmen (= N,N′-dimethylethylenediamine) ligands, an extended version of Mg–MOF-74, showed a better CO2 capacity, even upon exposure to water vapor when reactivated under a flow of pure Ar.41 In general, amine functionalization allows for the chemisorption of CO2 onto the free amine groups when primary or secondary diamines are introduced. Therefore, the working capacity of amine-grafted MOFs with chemisorbed CO2 is expected to be low if the regeneration temperature is not sufficiently high. This presents a substantial challenge to identify a promising adsorbent that can maintain a high working capacity even at low desorption temperatures.
Herein, we report the synthesis and sorption properties of N,N-dimethylethylenediamine (dmen)-functionalized Mg2(dobpdc) (1-dmen), constructed by grafting the open metal sites of Mg2(dobpdc) (1) with dmen, composed of primary and tertiary amines. In addition to having an exceptionally high selectivity for CO2 over N2, this framework exhibits a record high working capacity at low regeneration temperatures among MOFs; this is a critical metric in determining the practical application in a post-combustion CO2 capture process. This working capacity is retained after several temperature-swing adsorption (TSA) cycles, and even after exposure to humidity.
Results and discussion
Synthesis and structure
Mg2(dobpdc)–DMF was prepared by reacting MgBr2·6H2O and H4dobpdc in N,N′-dimethylformamide (DMF)–EtOH under microwave irradiation, and then evacuation at 390 °C to produce the activated sample (1). 1 was immersed in anhydrous hexane in the presence of 20 equiv. of dmen to yield the diamine-grafted sample. The amine-functionalized sample was treated at 130 °C under vacuum for 30 min to remove trace hexane from the pores. The dried sample (1-dmen) was exposed to a stream of CO2 for 30 min to give the CO2-captured material (1-dmen-CO2). The CO2-adsorbed sample was evacuated at 130 °C for 30 min to give the regenerated sample (1-dmen-re). The structural variations of these samples were examined by collecting synchrotron powder X-ray diffraction (PXRD) data (Fig. 1 and S1–S3†). The PXRD profiles were analyzed using Pawley refinement. The cell volume of V = 2789.8 Å3 for 1-dmen is reduced to V = 2650.2 Å3 for 1-dmen-CO2, a 5% reduction, indicating that CO2 adsorption on 1-dmen causes a shrinkage of the framework. The cell reduction (5%) of this system is substantially smaller than that of en-Mg2(dobpdc) with the primary diamine en (which gave a 13.5% cell reduction), and may be a result of the bulkier dmen in the pores.41 The remaining CO2 is completely desorbed by heating 1-dmen-CO2 under vacuum at 130 °C, as confirmed by the synchrotron PXRD data in which the cell volume (V = 2805.7 Å3) of 1-dmen-re is close to the original volume of 1-dmen.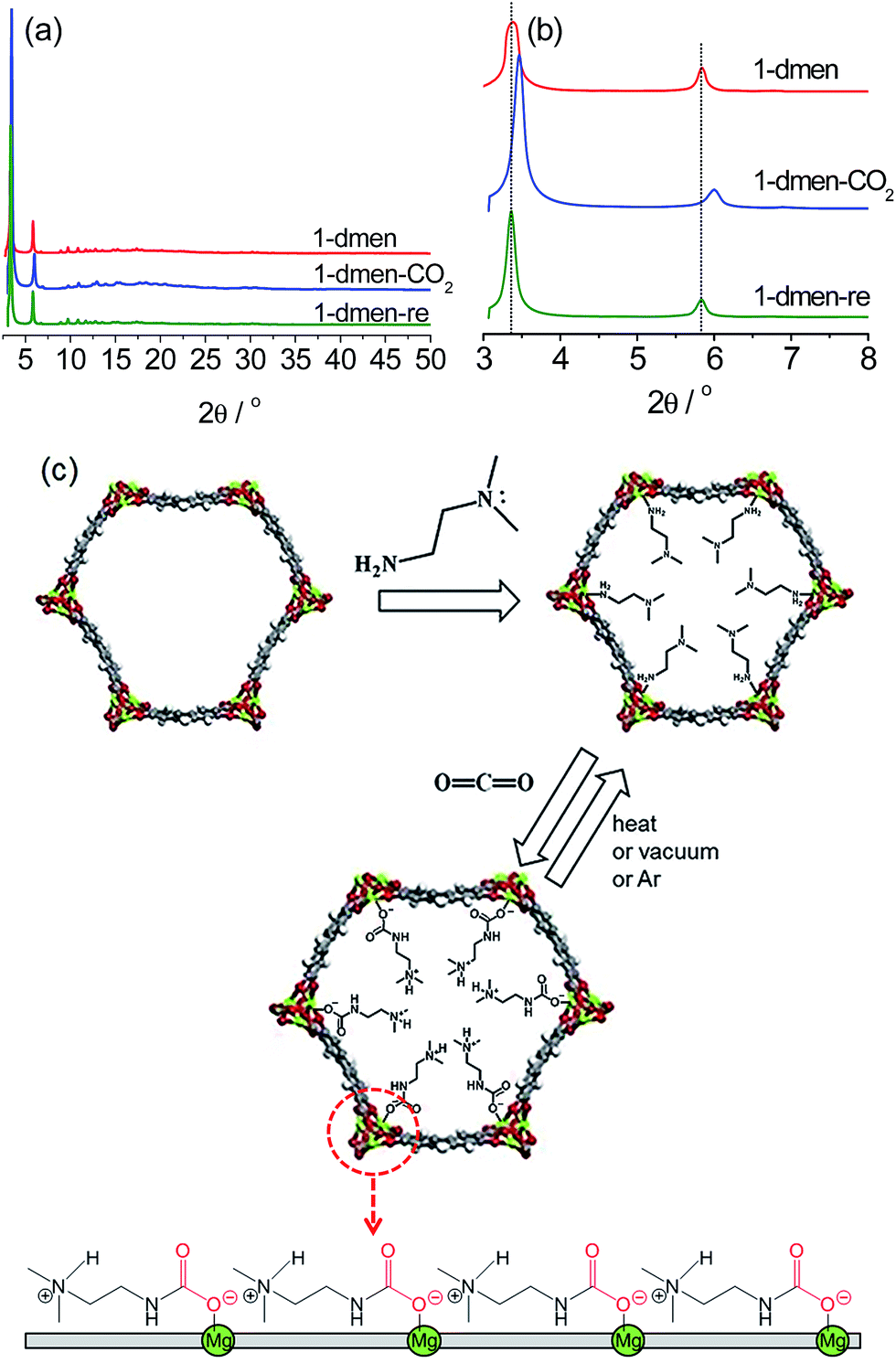 | ||
| Fig. 1 (a) Synchrotron PXRD profiles of 1-dmen, 1-dmen-CO2, and 1-dmen-re. (b) Enlargement of the low-angle region of the PXRD data. (c) Framework structure of 1 with open metal sites, grafting modes of dmen onto the open metal sites, and subsequent CO2 adsorption. The schematic diagram (bottom) indicates the arrangement of ammonium carbamates running along the c-axis. | ||
IR spectroscopy
We utilized in situ infrared (IR) spectroscopy to probe the CO2 adsorption mechanism. The IR cell and the interior of the instrument were thoroughly purged with N2 to remove CO2 prior to the IR measurements. We used an airtight IR cell (with KBr windows) and an oil bubbler to isolate the cell atmosphere from air. 1-dmen was put into the IR cell to record the IR data under an Ar atmosphere. The distinct peaks centered at 3330 and 3246 cm−1 for 1-dmen can be assigned to the N–H stretching vibrations of the primary amine of dmen (Fig. S4†).41–43 When 15% CO2 flowed into the cell for 3 min, the primary amine peaks weakened but still remained due to the presence of anchoring primary amines, while a new peak appeared at 3397 cm−1. This new peak in 1-dmen is associated with the N–H stretching frequency of a carbamate, as can also be observed in the IR spectra of amine-functionalized silica44–47 and the secondary diamine-functionalized mmen-Mg2(dobpdc).38In addition, there are other strong peaks associated with the combination bands (ν3 + ν1 at 3728 and 3704 cm−1; ν3 + 2ν2 at 3625 and 3599 cm−1) and the asymmetric stretching mode (ν3 at around 2377 cm−1) of CO2.48,49 To remove free CO2 from the cell, it was purged with a N2 flow for 1 min. The characteristic N–H stretching bands are retained even after purging with N2 for 10 min. For the CO2-captured sample, a broad peak centered at 2200 cm−1 is visible together with multiple peaks in the range of 1700–1630 cm−1 (Fig. S5†). The broad peak is assignable to a combination band from the ammonium cations, and the multiple peaks are related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the amide group and the asymmetric deformation of the ammonium cation.45,47 These peaks simultaneously disappear as the N–H stretching peak at 3397 cm−1 vanishes, suggesting the formation of a carbamate group during CO2 adsorption, although there is also the possibility of concomitant carbamic acid formation, as observed in amine-modified silica and as supported by DFT calculations on mmen-Mg2(dobpdc).35,45 The presence of the N–H stretching band of carbamate indicates that the tertiary amines of dmen are grafted onto the open metal sites, while the primary amines that protrude in the pore direction react with CO2 to produce the carbamate species with the assistance of neighboring amine groups.41,45–47 To understand the CO2 adsorption behavior of 1-dmen as a function of temperature, we measured the in situ IR data at several temperatures (Fig. S6†). At 25 °C, the N–H stretch (3397 cm−1) for the chemisorbed species is present together with the primary N–H stretching bands (3330 and 3246 cm−1). In addition, there is a characteristic band around 2200 cm−1, assigned to ammonium cations. As the temperature increases, the N–H and ammonium bands weaken while the primary N–H stretching peaks increase. From this result, it appears that the chemisorbed species is disrupted and the free primary amine groups reform. At 130 °C, almost no CO2 is adsorbed on 1-dmen.
O stretching of the amide group and the asymmetric deformation of the ammonium cation.45,47 These peaks simultaneously disappear as the N–H stretching peak at 3397 cm−1 vanishes, suggesting the formation of a carbamate group during CO2 adsorption, although there is also the possibility of concomitant carbamic acid formation, as observed in amine-modified silica and as supported by DFT calculations on mmen-Mg2(dobpdc).35,45 The presence of the N–H stretching band of carbamate indicates that the tertiary amines of dmen are grafted onto the open metal sites, while the primary amines that protrude in the pore direction react with CO2 to produce the carbamate species with the assistance of neighboring amine groups.41,45–47 To understand the CO2 adsorption behavior of 1-dmen as a function of temperature, we measured the in situ IR data at several temperatures (Fig. S6†). At 25 °C, the N–H stretch (3397 cm−1) for the chemisorbed species is present together with the primary N–H stretching bands (3330 and 3246 cm−1). In addition, there is a characteristic band around 2200 cm−1, assigned to ammonium cations. As the temperature increases, the N–H and ammonium bands weaken while the primary N–H stretching peaks increase. From this result, it appears that the chemisorbed species is disrupted and the free primary amine groups reform. At 130 °C, almost no CO2 is adsorbed on 1-dmen.
Adsorption isotherm and mechanism
From the N2 uptake for 1-dmen at 77 K (Fig. S7†), it is observed that the BET surface area of 675 m2 g−1 is between those of en-Mg2(dobpdc) (1253 m2 g−1) and mmen-Mg2(dobpdc) (70 m2 g−1),38,41 which is related to the bulkiness of the appended heterodiamines on the unsaturated metal sites. From the analysis of the pore size distributions, the average pore size of 1-dmen is smaller than those of both en-Mg2(dobpdc) and mmen-Mg2(dobpdc) (Fig. S8†). The CO2 adsorption isotherms for 1-dmen were collected at several temperatures after the samples had been activated at 75 °C for 4 h prior to each measurement (Fig. 2a). The isotherm curves show steps at certain pressures, depending on the adsorption temperature. The shift of the step pressure may be associated with the greater thermal motion of the grafted dmen and CO2 molecules at elevated temperatures. Similar behavior was also observed in Mg2(dobpdc) functionalized with primary and secondary diamines.38,41 At 1 bar, the adsorbed quantity is 4.34 mmol g−1 at 40 °C, comparable to those at 50 and 60 °C. Remarkably, however, almost no CO2 is adsorbed on 1-dmen at 75 °C and 1 bar, which is comparable with the adsorption behaviors of en-Mg2(dobpdc) and mmen-Mg2(dobpdc), which both show non-adsorption at temperatures above 120 °C.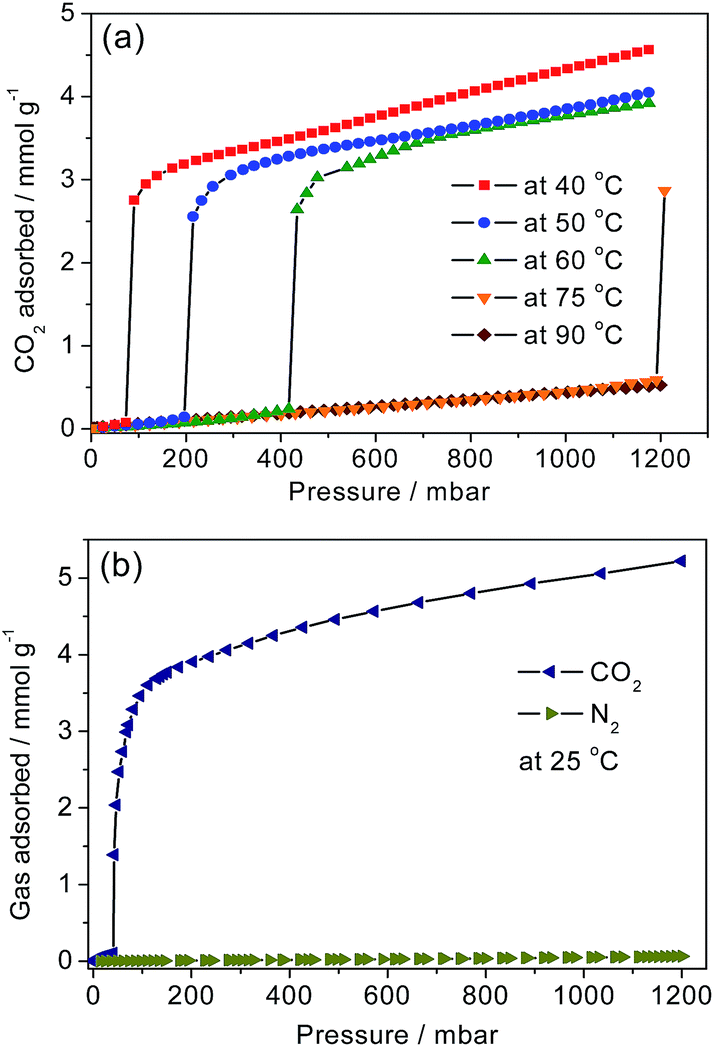 | ||
| Fig. 2 (a) Adsorption isotherms of CO2 for 1-dmen at the indicated temperatures. The solid lines are eye-guides. (b) CO2 and N2 isotherms at 25 °C. | ||
This notable feature indicates a weaker interaction between CO2 and the amine groups in 1-dmen than that between CO2 and the amines in en-Mg2(dobpdc) and mmen-Mg2(dobpdc).
The isosteric heat of adsorption (Qst), which represents the mean binding energy of a gas molecule at a specific site of an adsorbent, was estimated by employing a dual-site Langmuir–Freundlich model to fit the CO2 isotherms. The precise pressures corresponding to the adsorbed CO2 amounts were introduced as input parameters to the Clausius–Clapeyron equation to calculate the adsorption enthalpy (Fig. S9†). Using this calculation, the estimated heat of adsorption (−Qst) increases to 75 kJ mol−1 at a loading of 0.25 mmol g−1, and remains almost invariant in the range of 71–75 kJ mol−1 below loadings of 2.6 mmol g−1. This overall feature of the adsorption enthalpy for 1-dmen resembles that observed for the other Mg2(dobpdc) moieties functionalized with diamines.38,41 The initial low Qst values at ultradilute CO2 concentrations are unusual for chemisorbents. To investigate the adsorption behavior at the CO2 pressures before and after the step in the isotherm, we measured the IR in situ at 40 °C by flowing simulated air (0.39 ppm CO2 balanced with N2) or pure CO2 into the cell (Fig. S10†). There is no evidence of chemisorption at the CO2 pressure before the step, while the peak at 3397 cm−1 appears after the step pressure, indicating the formation of the chemisorbed species. To understand the CO2 adsorption mechanism, we performed DFT calculations on the binding energy of each possible Mg–amine pair. From the results, the primary amine–Mg pair has a binding energy of −174.3 kJ mol−1, which is thermodynamically more stable than that of the tertiary amine–Mg pair (−135.6 kJ mol−1) (Fig. 1 and S11†). This result suggests that the open metal site is primarily occupied by the primary amine end of 1-dmen, although some tertiary amine ends are probably grafted onto the exposed metal site as well. On the basis of the in situ IR data and DFT calculations, one possible CO2 adsorption mechanism is as follows: the low-affinity adsorption at low pressures is highly uncommon for chemisorbents, and cannot be rationalized in the absence of sorbent restructuring under CO2 dosing conditions. At a certain partial pressure, the amine is reorganized so that it is now available to bind CO2via chemisorption causing a jump in the binding energy. Therefore, we speculate that under CO2 conditions, deprotonation of the coordinate amine group takes place with the assistance of the basic tertiary amine group of the dangling dmen molecule. Concomitant nucleophilic attack of CO2 forms an alkylammonium carbamate. The generated ion-pair interacts to weaken the Mg–N bond strength of the neighboring dmen and in turn accelerates cooperative CO2 insertion, which can explain the abrupt rise in the CO2 isotherm (Fig. 2a). The carbamate species is rearranged at a critical temperature and pressure (Fig. 1c), as recently reported for mmen-Mg2(dobpdc).50 In this scenario, the carbamates are aligned along the c-direction, which may account for the structural shrinkage upon CO2 adsorption (Fig. 1c).50
CO2 selectivity
The isotherms of CO2 and N2 at 25 °C were measured to evaluate the adsorption selectivity for CO2 over N2 (Fig. 2b). This selectivity is required for post-combustion CO2 capture to effectively separate CO2 from the flue gas where CO2 and N2 coexist at partial pressures of 0.15 and 0.75 bar, respectively. At 25 °C and 0.15 bar, 1-dmen takes up 3.77 mmol g−1 of CO2, an adsorption capacity larger than those of en-Mg2(dobpdc) (3.62 mmol g−1) and mmen-Mg2(dobpdc) (3.13 mmol g−1).38,41 Using single-component isotherms, the selectivity factor, which is normalized to the composition of the gases, is defined as S = (qCO2/qother)/(pCO2/pother), where qn and pn are the adsorption capacity and the pressure of the component n, respectively. The purity of the adsorbed gas, a practical and meaningful value for the transport and sequestration of captured CO2, is calculated using 100qCO2/(qCO2 + qother). For the practical purity of the adsorbed gas, it is not only the adsorbed species that must be considered, but also the composition of the residual gas in the column that is not adsorbed; the bulk density of the adsorbent is very important. The calculations done here assume that all non-adsorbed gas is removed by vacuum from the column before desorption, which is not feasible for flue gas capture. The selectivity of 1-dmen for CO2 over N2 and the purity of CO2 are estimated to be 554 and 99%, respectively. The selectivity of 1-dmen is comparable to those of the top-performing MOFs, mmen-Mg2(dobpdc) (S = 200), en-Mg2(dobpdc) (S = 230), mmen-Cu-BTTri (S = 327), and PEI-MIL-101 (S = 600), at pressures and temperatures relevant to practical post-combustion CO2 capture.1,13,36,51To further explore the relative selectivity of 1-dmen, we examined the competitive sorption kinetics of different gas compositions, including pure CO2, CH4/CO2 = 50/50, H2/CO2 = 70/30, and N2/CO2 = 85/15, at 40 °C. The gas uptake is equivalent to 18.8 wt% for pure CO2, 16.6 wt% for CH4/CO2 = 50/50, 15.0 wt% for H2/CO2 = 70/30, and 14.1 wt% for N2/CO2 = 85/15, while no obvious adsorption was observed for pure CH4, H2, or N2 (Fig. S12†). Each CO2 adsorption amount is similar to that at the corresponding CO2 partial pressure in the isotherm at 40 °C (Fig. 2a). These results suggest that CO2 adsorbs more strongly than CH4, H2, and N2, thus favorably taking up the available adsorption sites and pores while the other gases are excluded. The observed sorption kinetics of the gas mixtures indicate a relatively high CO2 selectivity over the other gases.
Facile regeneration and recyclability
Adsorbed CO2 can be removed from the solid at 30 °C, lower than the uptake temperature of 40 °C, followed by a degree of CO2 adsorption with a capacity identical to the first cycle's (Fig. 3a). Fig. 3b presents the cycling behavior for the adsorption (40 °C)-activation (40 °C) protocols, showing a significant adsorption capacity as well as a facile desorption of the CO2 adsorbed at low temperature (Fig. S13†); this is supported by in situ IR spectroscopy (Fig. S14†). A recyclability test using a TSA procedure was also run (Fig. 3c). CO2 was adsorbed at 40 °C for 1 h and desorbed at 75 °C for 1 h under Ar. After 24 cycles, no capacity loss was observed, suggesting that the material is thermally stable under these experimental conditions. Thus, 1-dmen could be potentially applicable for capturing CO2 from coal-fired power plant emissions, because its regeneration requires very low desorption temperatures, and it has a high adsorption capability and no capacity decay even after many TSA cycles. This finding is valuable in that our material requires a lower energy input to regenerate the capture material, which is one of the most essential objectives for current CCS technologies.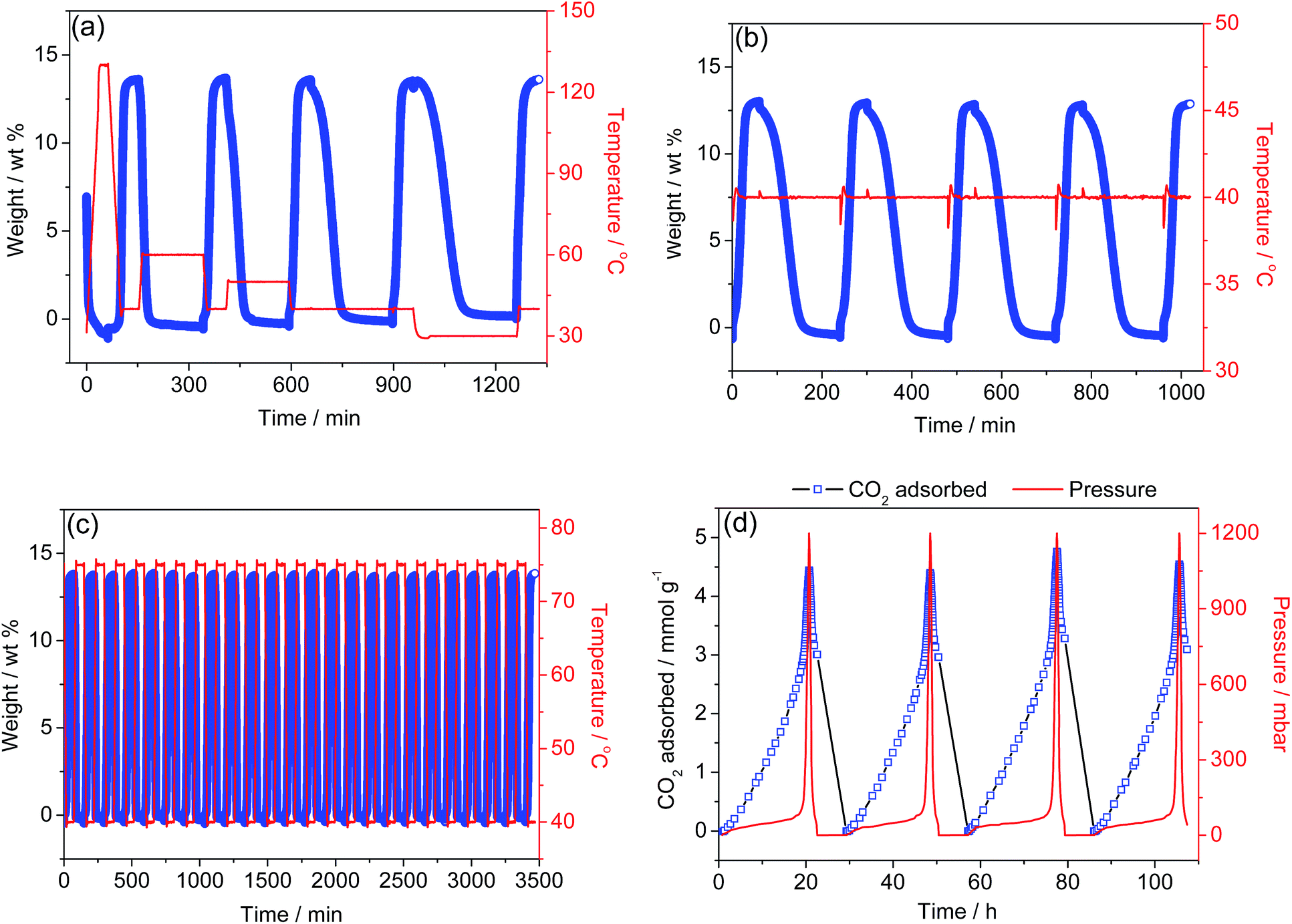 | ||
| Fig. 3 (a) Adsorption–desorption cycling of CO2 for 1-dmen, showing reversible uptake from simulated flue gas (0.15 bar CO2 balanced with N2). Adsorption temperature was 40 °C and desorption temperatures were 130 °C, 60 °C, 50 °C, 40 °C, and 30 °C under Ar. (b) Adsorption–desorption cycling of CO2 at 40 °C by switching atmospheres between simulated flue gas and Ar. (c) Adsorption–desorption cycling of CO2 at adsorption (40 °C) and desorption (75 °C) under Ar. (d) Vacuum-swing adsorption at 25 °C using a Micromeritics ASAP2020 instrument. | ||
In another experiment, the desorption behavior of 1-dmen was further investigated via the vacuum-swing adsorption (VSA) method52,53 using an ASAP2020 analyzer (Fig. 3d). At 25 °C, 1-dmen was saturated with CO2 at 1.2 bar and then placed under high vacuum. The removal of adsorbed CO2 from the solid was performed repeatedly by applying a vacuum to the adsorbent; this CO2 desorption under vacuum was confirmed by IR data in which the peaks relating to CO2 adsorption progressively vanish upon the application of a high vacuum (Fig. S15†). Thus, notably, even such a large amount of CO2 as is adsorbed onto 1-dmen at 1.2 bar (∼4.5 mmol g−1) can be completely desorbed only under vacuum, without heating. Such facile desorption of captured CO2 under evacuation supports the hypothesis that the alkylammonium carbamate species is weakly bound, as was also found in amine-grafted SBA-15.47
CO2 working capacity
The working capacity that corresponds to the true amount of CO2 adsorbed during an adsorption–desorption process is a decisive metric to evaluate the material performance of CO2 capture in real-world post-combustion applications, and is a far more important parameter than the absolute CO2 uptake under flue gas conditions. As previously shown for sorbents,9,13 an estimation of the working capacity can be made on the basis of the difference between the quantities adsorbed at the adsorption and desorption temperatures (WCq = qads − qdes) when the operation temperature is fixed at Tads = 40 °C.In this case, the adsorption amounts were obtained from CO2 isotherms at disparate loading conditions (qads at Pads = 0.15 bar CO2, Tads = 40 °C; qdes at Pdes = 1 bar CO2, Tdes = corresponding desorption state temperature). The obtained working capacity is significant: 11.6 wt%, even at Tdes = 75 °C (Fig. S16†). Notably, the working capacity of this material, with its significantly lower desorption temperature, can be compared to those of MOFs such as Mg–MOF-74 and MOF-177, functionalized PPN-6, zeolite NaX, and MEA, in which almost zero or negative values of working capacities were recorded at the identical temperature.9,13 In particular, the increased temperature does not improve the working capacity of 1-dmen because CO2 is hardly adsorbed onto the sample at temperatures higher than 75 °C. On the other hand, we performed thermogravimetric (TG) experiments to monitor the mass change upon regeneration and CO2 adsorption using TSA processes, which can give valuable information about the working capacity for practical CO2 capture applications. 1-dmen was activated at 130 °C under a pure CO2 atmosphere for 90 min, and then 15% CO2 in N2 was flowed over the sample at 40 °C for 60 min (Fig. 4a and S17†). A CO2 adsorption capacity of 13.3 wt% was observed, and was maintained after 7 cycles. We examined the working capacities (WCTGA) of 1-dmen at different desorption temperatures and compared them to those of other solid adsorbents such as zeolite 13X and MOFs with open metal sites or grafted amines (Fig. 4b). The working capacities of 1-dmen were exceptionally high, mostly in the range of 11.7–13.5 wt%, even though the desorption temperature changed from 130 °C to 90 °C; these mark the highest values among MOFs at relatively low temperatures. In other words, the obtained capacity of 1-dmen at a desorption temperature of 130 °C is substantially superior to those of some top performing MOFs such as Mg–MOF-74 (3.7 wt%), Mg2(dobpdc) (4.5 wt%), en-Mg2(dobpdc) (2.9 wt%), mmen-Mg2(dobpdc) (2.1 wt%), and tmen-Mg2(dobpdc) (3.9 wt%) (tmen = N,N,N′,N′-tetramethylethylenediamine), and to that of zeolite 13X (7.0 wt%). As the desorption temperature decreased below 90 °C, the working capacity of 1-dmen sharply reduced to almost zero at 70 °C. The primary en-Mg2(dobpdc) and secondary mmen-Mg2(dobpdc) show lower working capacities than the tertiary diamine-grafted solid tmen-Mg2(dobpdc), even though the actual CO2 adsorptions of en-Mg2(dobpdc) and mmen-Mg2(dobpdc) are larger than that of tmen-Mg2(dobpdc) upon activation in pure Ar. This implies that successful regeneration under pure CO2 contributes to the enhanced working capacity of tmen-Mg2(dobpdc). On the other hand, the working capacity of 1-dmen exceeds that of tmen-Mg2(dobpdc) because it exhibits high CO2 uptake as well as facile regeneration. Therefore, both a large CO2 uptake and easy regeneration under pure CO2 are responsible for a high working capacity.
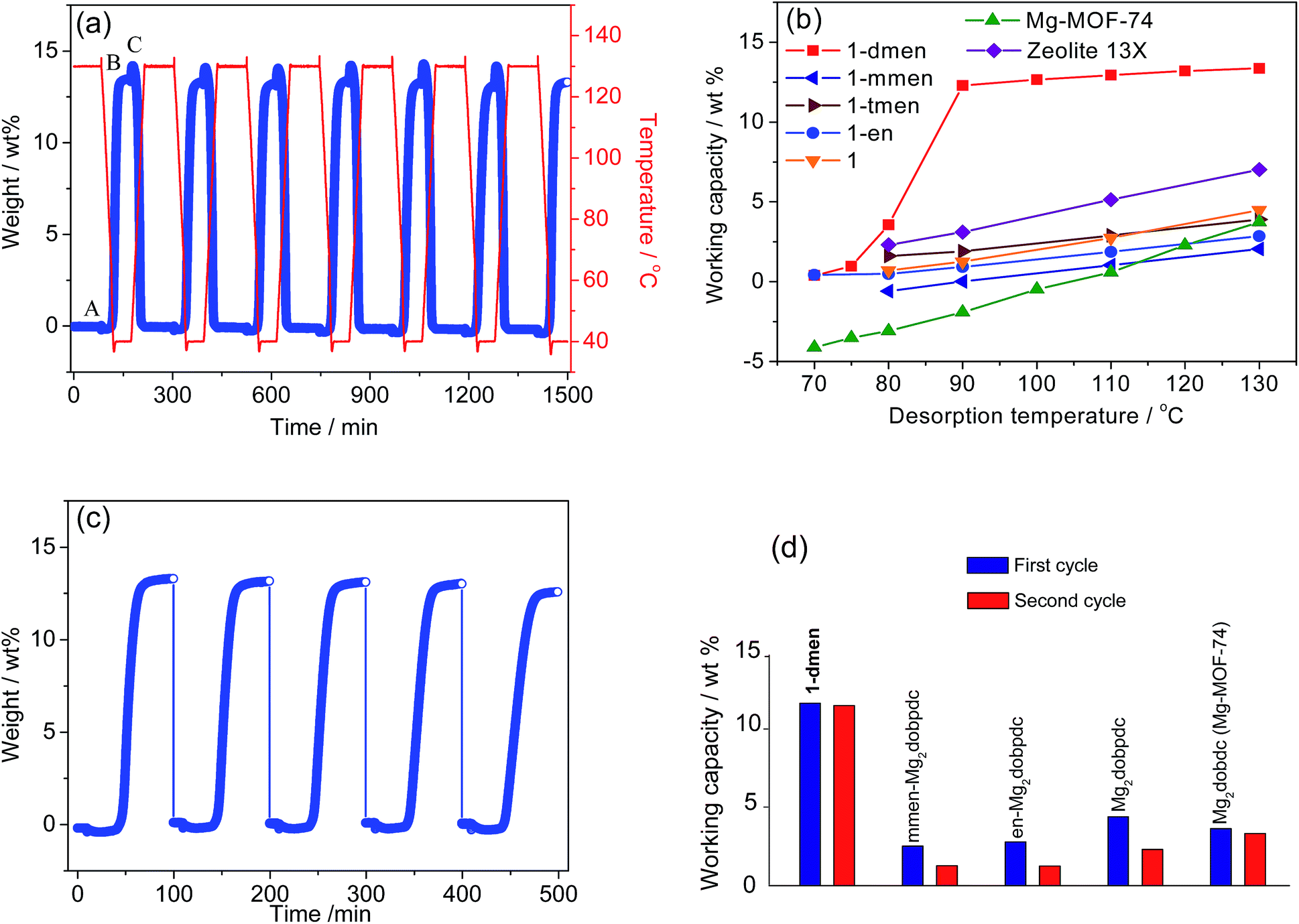 | ||
| Fig. 4 (a) Adsorption–desorption cycling of CO2 for 1-dmen. The adsorption of simulated flue gas (0.15 bar CO2 balanced with N2) was recorded at 40 °C and desorption was performed at 130 °C under flowing pure CO2. The symbol C represents the influx of pure CO2 to regenerate the sample. The working capacity is determined as the difference between A and B. (b) The working capacities of 1-dmen and the other porous solids obtained under the same conditions. (c) CO2 adsorption of 1-dmen in flue gas using the sequence (adsorption at 40 °C – desorption at 130 °C under pure CO2 – 10 min exposure to 100% RH). (d) Working capacities of the CO2 uptake in the first and second cycles for 1-dmen, mmen-Mg2(dobpdc), en-Mg2(dobpdc), Mg2(dobpdc), and Mg–MOF-74 using TG experiments. The first cycle was measured with fully activated samples in flue gas, and the second cycle was measured by following the sequence. | ||
CO2 uptake capacity under humid conditions
The reusability of a solid adsorbent in the presence of water vapor is also essential to be relevant for CO2 capture applications.54 We assessed the material's performance by repeated exposure to humid environments. The solid sample can be fully saturated with CO2 within the exposure time (10 min), because its initial rate of adsorption is estimated to be roughly 1.7 wt% min−1 (Fig. S18†). In this experiment, 1-dmen was exposed to 100% RH for 10 min. The humidified sample was then reactivated under a pure CO2 flow at 130 °C for 4 h, followed by CO2 adsorption at 40 °C. After 5 cycles run in this sequence (adsorption at 15% CO2 and 40 °C, humidification at 100% RH, activation at 130 °C under pure CO2), a capacity loss of 5% is observed (Fig. 4c). To compare the CO2 working capacity of 1-dmen upon exposure to humidity with those of other adsorbents, we applied the same experimental protocols to the amine-grafted MOFs, en-Mg2(dobpdc) and mmen-Mg2(dobpdc), and the MOFs with open metal sites, Mg–MOF-74 and Mg2(dobpdc) (Fig. 4d). The capacity of 1-dmen retains significantly high values in subsequent cycles, while in comparison, the working capacities of the other MOFs during the first cycle are below 4.5 wt% and are reduced even further in the second cycle. From these results, the CO2 working capacity of 1-dmen outperforms those of the other tested materials that have primary and secondary diamines or open metal sites.To examine the CO2 adsorption capability under humid conditions, we performed time-dependent in situ IR spectroscopy at 40 °C upon simultaneous exposure to 15% CO2 and 100% RH (Fig. S19†). The N–H stretching band (3397 cm−1) of the chemisorbed species appears under humid CO2 flowing conditions. The characteristic peak still survives after purging the cell with pure N2 for 1 min, while the free CO2 peaks disappear. This observation definitely supports the conclusion that CO2 is preferably adsorbed onto the grafted amine groups even under humid conditions.
Moreover, to probe the true effect of water vapor on CO2 uptake, we performed humid adsorption–desorption cycles, exposing 1-dmen to 15% CO2 and 3.75% H2O balanced with He. The sample was placed in an automated chemisorption analyzer (Autochem II 2920) to test its adsorption of CO2 at 40 °C for 30 min, followed by CO2 desorption at 130 °C for 1 h. The reversible adsorption–desorption events occurred while a CO2 uptake of around 14.6 wt% was maintained (Fig. 5a), similar to the CO2 adsorption amount under dry conditions. The framework structure of 1-dmen remains intact after the cycling experiment in humid conditions, as shown by the PXRD data (Fig. S20†). To explore the dynamic separation capability, we ran breakthrough tests for a gas mixture containing 15% CO2 in an N2 gas stream. The sample bed was preheated at 130 °C for 4 h in a He atmosphere. The gas mixture was introduced to the solid packed in a column, and the effluent was detected by a gas chromatograph (Agilent 7890A). Under dry conditions, the solid held CO2 for up to 14.7 min g−1 relative to the N2 breakthrough time (Fig. 5b). To identify dynamic CO2 separation in the presence of moisture, we carried out humid breakthrough experiments. While 5% water vapor was flowed into the adsorbent bed prior to the breakthrough runs, a gas mixture of 15% CO2 and 80% N2 was infused into the fixed bed. The breakthrough time for CO2 was equivalent to 21.1 min g−1, which is greater than that of the dry CO2 capacity. The shorter retention time of CO2 in dry conditions can be understood by the facile desorption of CO2 from 1-dmen under flowing dry inert gas, as shown in Fig. 3b. From the breakthrough data, it is evident that this material exhibits effective CO2 separation from the gas mixture, due to the more enhanced interaction of CO2 with the amine groups than N2, which will be beneficial for practical CO2 capture applications.
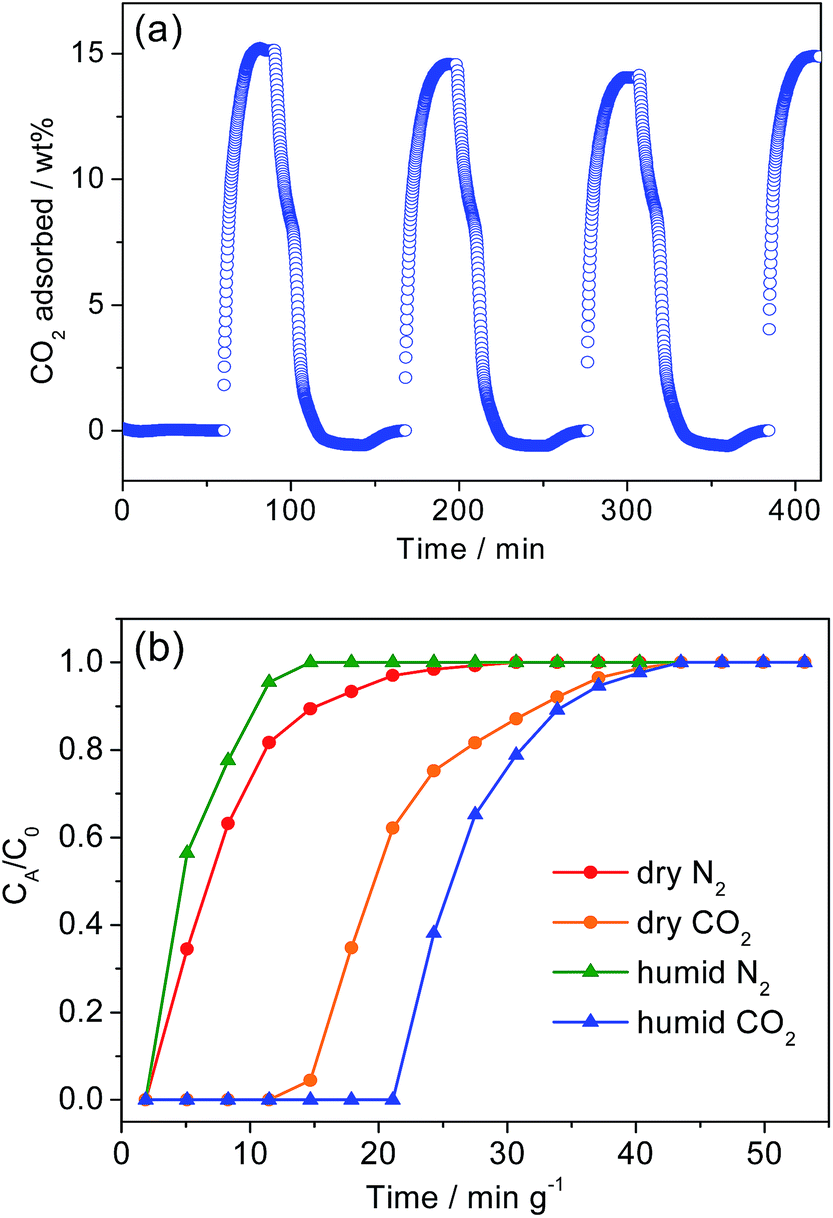 | ||
| Fig. 5 (a) CO2 adsorption–desorption cycling curve for 1-dmen in humid conditions. (b) Breakthrough curves for 1-dmen under dry (N2, red circle; CO2, orange circle) and humid conditions (N2, green triangle; CO2, blue triangle). | ||
Conclusions
The presented results show that the structure of 1-dmen undergoes shrinkage upon CO2 adsorption, and that the original structure is recovered when CO2 is released from the solid. The selectivity factor for CO2 over N2 for this solid is on a par with those of the foremost CO2-capturing MOFs. Remarkably, the regeneration of the adsorbent is completed under vacuum or flowing Ar even at very low desorption temperatures, while the material maintains a high CO2 adsorption capacity (above 13 wt%). Notably, the working capacity of 1-dmen, relevant to post-combustion CO2 capture applications, is exceptionally high at low desorption temperatures and upon exposure to humidity; its capacity exceeds those of the tested MOFs with amine grafting and with open metal sites in identical conditions. Overall, this material, with a record high working capacity at low regeneration temperatures, will be a promising adsorbent for a real post-combustion CO2 capture process. To establish its use for other practical applications, it will be necessary to determine how this material will perform in the presence of not only humidity but also NOx and SO2.Experimental
All chemicals and solvents in the synthesis were reagent grade and used as received. H4dobpdc and 1-DMF were prepared according to the literature.38[Mg2(dobpdc)(dmen)1.8(H2O)0.2] (1-dmen)
A sample of fully activated 1 (100 mg, 0.31 mmol) was loaded in a Schlenk flask in a glove box. A solution of dry hexane (100 mL) with 20 equiv. of N,N-dimethylethylenediamine (dmen, 0.68 mL, 6.27 mmol) was transferred to the flask using a cannula. The suspension was stirred for 18 h at room temperature. The solid was separated by filtration and washed with dry hexane several times. The resulting residue was immersed in dry hexane for 72 h and then evacuated at 130 °C for 4 h to obtain an off-white powder. Yield: 150 mg (99.5%). Anal. calcd for C21.2H28.6Mg2N3.6O6.4 [1-dmen-(H2O)0.3]: C, 52.34; H, 5.87; N, 10.24. Found: C, 52.34; H, 5.93; N 10.37.Acknowledgements
This work was supported by the Korea CCS R&D Center (KCRC) grant funded by the Korea government (The Ministry of Science, ICT & Future Planning (MSIP)) (NRF-2014M1A8A1049253) and by PAL. WRL was partly supported by a Korea University Grant. LMY and SSH are grateful for the financial support from the Korea Institute of Science and Technology (Grant no. 2E24630).Notes and references
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- R. S. Haszeldine, Science, 2009, 325, 1644–1645 CrossRef PubMed.
- R. Quadrelli and S. Peterson, Energy Policy, 2007, 35, 5938–5952 CrossRef PubMed.
- M. Pera-Titus, Chem. Rev., 2014, 114, 1413–1492 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, G. K. Surya Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9–20 CrossRef CAS.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- J. P. Sculley, W. M. Verdegaal, W. Lu, M. Wriedt and H. C. Zhou, Adv. Mater., 2013, 25, 3957–3961 CrossRef CAS PubMed.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna and H.-C. Zhou, J. Phys. Chem. C, 2013, 117, 4057–4061 CAS.
- M. Zhang, Z. Perry, J. Park and H.-C. Zhou, Polymer, 2014, 55, 335–339 CrossRef CAS PubMed.
- R. H. Weiland, J. C. Dingman and B. Cronin, J. Chem. Eng. Data, 1997, 42, 1004–1006 CrossRef CAS.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 CAS.
- Y. Liu, Z. U. Wang and H.-C. Zhou, Greenhouse Gases: Sci. Technol., 2012, 2, 239–259 CrossRef CAS PubMed.
- A.-H. Lu and G.-P. Hao, Inorg. Chem., 2013, 109, 484–503 CAS.
- M. Zhang, M. Bosch, T. Gentle III and H.-C. Zhou, CrystEngComm, 2014, 16, 4069–4083 RSC.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 RSC.
- W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- R. B. Lin, D. Chen, Y. Y. Lin, J. P. Zhang and X. M. Chen, Inorg. Chem., 2012, 51, 9950–9955 CrossRef CAS PubMed.
- W. J. Phang, W. R. Lee, K. Yoo, B. Kim and C. S. Hong, Dalton Trans., 2013, 42, 7850–7853 RSC.
- X. Si, C. Jiao, F. Li, J. Zhang, S. Wang, S. Liu, Z. Li, L. Sun, F. Xu, Z. Gabelica and C. Schick, Energy Environ. Sci., 2011, 4, 4522–4527 CAS.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed.
- S. R. Caskey, A. G. Wong-F and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- L. C. Lin, J. Kim, X. Kong, E. Scott, T. M. McDonald, J. R. Long, J. A. Reimer and B. Smit, Angew. Chem., Int. Ed., 2013, 52, 4410–4413 CrossRef CAS PubMed.
- W. Lou, J. Yang, L. Li and J. Li, J. Solid State Chem., 2014, 213, 224–228 CrossRef CAS PubMed.
- A. L. Dzubak, L.-C. Lin, J. Kim, J. A. Swisher, R. Poloni, S. N. Maximoff, B. Smit and L. Gagliardi, Nat. Chem., 2012, 4, 810–816 CrossRef CAS PubMed.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Fe'rey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS PubMed.
- S. Choi, T. Watanabe, T.-H. Bae, D. S. Sholl and C. W. Jones, J. Phys. Chem. Lett., 2012, 3, 1136–1141 CrossRef CAS.
- J. M. Gu, T. H. Kwon, J. H. Park and S. Huh, Dalton Trans., 2010, 39, 5608–5610 RSC.
- A. Das, M. Choucair, P. D. Southon, J. A. Mason, M. Zhao, C. J. Kepert, A. T. Harris and D. M. D'Alessandro, Microporous Mesoporous Mater., 2013, 174, 74–80 CrossRef CAS PubMed.
- Y. Hu, W. M. Verdegaal, S. H. Yu and H. L. Jiang, ChemSusChem, 2014, 7, 734–737 CrossRef CAS PubMed.
- N. Planas, A. L. Dzubak, R. Poloni, L. C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS PubMed.
- T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Chem. Sci., 2011, 2, 2022–2028 RSC.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Remy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gandara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed.
- W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 CAS.
- Z. Bacsik, R. Atluri, A. E. Garcia-Bennett and N. Hedin, Langmuir, 2010, 26, 10013–10024 CrossRef CAS PubMed.
- X. Wang, V. Schwartz, J. C. Clark, X. Ma, S. H. Overbury, X. Xu and C. Song, J. Phys. Chem. C, 2009, 113, 7260–7268 CAS.
- M. H. Jamroz and J. C. Dobrowolski, Vib. Spectrosc., 2002, 29, 217–221 CrossRef CAS.
- Z. Bacsik, N. Ahlsten, A. Ziadi, G. Zhao, A. E. Garcia-Bennett, B. Martin-Matute and N. Hedin, Langmuir, 2011, 27, 11118–11128 CrossRef CAS PubMed.
- C. Knöfel, C. Martin, V. Hornebecq and P. L. Llewellyn, J. Phys. Chem. C, 2009, 113, 21726–21734 Search PubMed.
- A. Danon, P. C. Stair and E. Weitz, J. Phys. Chem. C, 2011, 115, 11540–11549 CAS.
- P. Galhotra, J. G. Navea, S. C. Larsen and V. H. Grassian, Energy Environ. Sci., 2009, 2, 401–409 CAS.
- Q. Liu, A. Mace, Z. Bacsik, J. Sun, A. Laaksonen and N. Hedin, Chem. Commun., 2010, 46, 4502–4504 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- Y. Lin, Q. Yan, C. Kong and L. Chen, Sci. Rep., 2013, 3, 1859–1865 Search PubMed.
- C. Gebald, J. A. Wurzbacher, P. Tingaut and A. Steinfeld, Environ. Sci. Technol., 2013, 47, 10063–10070 CrossRef CAS PubMed.
- N. Hedin, L. Andersson, L. Bergström and J. Yan, Appl. Energy, 2013, 104, 418–433 CrossRef CAS PubMed.
- S. Keskin, T. M. van Heest and D. S. Sholl, ChemSusChem, 2010, 3, 879–891 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural, sorption, TGA, and in situ IR data. See DOI: 10.1039/c5sc01191d |
| This journal is © The Royal Society of Chemistry 2015 |