DOI:
10.1039/C5SC01027F
(Edge Article)
Chem. Sci., 2015,
6, 3706-3711
Carbene catalyzed umpolung of α,β-enals: a reactivity study of diamino dienols vs. azolium enolates, and the characterization of advanced reaction intermediates†
Received
21st March 2015
, Accepted 30th April 2015
First published on 30th April 2015
Abstract
Since their discovery by Bode and Glorius in 2004, N-heterocyclic carbene catalyzed conjugate umpolung reactions of α,β-enals have been postulated to involve the formation of diamino dienols (“homoenolates”) and/or azolium enolates (“enolates”), typically followed by addition to electrophiles, e.g. Michael-acceptors. In this article, we provide evidence, for the first time, for the postulated individual and specific reactivity patterns of diamino dienols (γ-C–C-bond formation) vs. azolium enolates (β-C–C-bond formation). Our study is based on the pre-formation of well defined diamino dienols and azolium enolates, and the in situ NMR monitoring of their reactivities towards enone electrophiles. Additionally, reaction intermediates were isolated and characterized, inter alia by X-ray crystallography.
Introduction
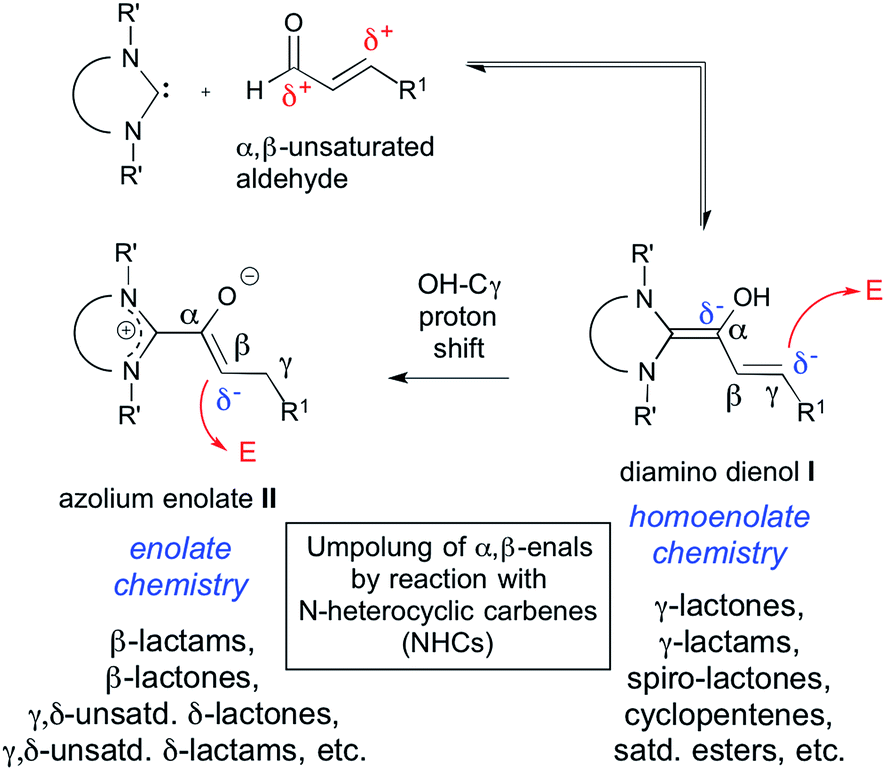
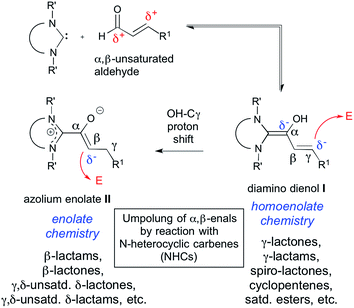
In N-heterocyclic carbene (NHC) organocatalysis,1 the “conjugate umpolung” of α,β-unsaturated aldehydes is a most thriving and proliferative field. As schematically shown in Scheme 1, interaction of an α,β-enal (a3) with an NHC first generates a Breslow-type2 intermediate, the diamino dienol I. A subsequent proton shift from the diamino dienol's –OH to Cγ leads to the azolium enolate II. The diamino dienol I carries a partial negative charge on Cγ, and therefore represents a homoenolate equivalent (d3). On the other hand, the azolium enolate II is nucleophilic at Cβ, and therefore behaves as an enolate equivalent (d2). Numerous experimental studies have revealed that the homoenolate vs. enolate behaviour of α,β-enals, when exposed to NHCs, can be influenced by the type of catalyst employed, and by the reaction conditions.3,4 For example, homoenolate chemistry is favoured by imidazolium precatalysts, in combination with strong bases.3,4 Reactions proceeding via the homoenolate pathway have been used to provide γ-lactones,5 spiro-lactones,6 spiro-bis-lactones,7 bicyclic lactones,8 γ-lactams,9 bicyclic β-lactams,10 cyclopentenes,5c,11 and saturated esters.12 Enolate chemistry, on the other hand, is favoured by triazolium precatalysts in combination with weak bases.3,4 Azolium enolates have been generated by the combination of NHCs with ketenes,13 aldehydes,3a,14 and esters.15 Reactions proceeding via the azolium enolate pathway have been used to provide β-lactams,13b,c β-lactones,13d,e unsaturated δ-lactams,14b,f,15b,c and unsaturated δ-lactones.14a,14e–g,15c
 |
| | Scheme 1 Early intermediates in the NHC-catalyzed umpolung of α,β-unsaturated aldehydes. | |
As outlined in Scheme 1, it is generally believed that diamino dienols I and the tautomeric azolium enolates II are the starting points of divergent reaction pathways, leading to different (isomeric) products when exposed to one and the same electrophilic reaction partner. This divergent reactivity is interpreted in the sense that diamino dienols I add electrophiles at Cγ, whereas the tautomeric azolium enolates II react at Cβ. In stark contrast to their pivotal importance in α,β-enal umpolung, no investigations of the reaction modes of pre-formed diamino dienols I and azolium enolates II (i.e. C–C bond formation with C-electrophiles at Cβ vs. Cγ) appear to have been reported to date.16 Several azolium enolates II are described in the literature. However, they were accessed by addition of carbenes to ketenes,16,17 and not by reaction of α,β-unsaturated aldehydes with N-heterocyclic carbenes (NHCs). With this in mind, we set out to investigate the reactivity patterns of pre-formed diamino dienols I and azolium enolates II with enone Michael acceptors. The first successful generation of both diamino dienols I and azolium enolates II from α,β-unsaturated aldehydes and carbenes, and their characterization by NMR and X-ray, was recently reported by our group.18
Results and discussion
Reactivity studies of diamino dienols
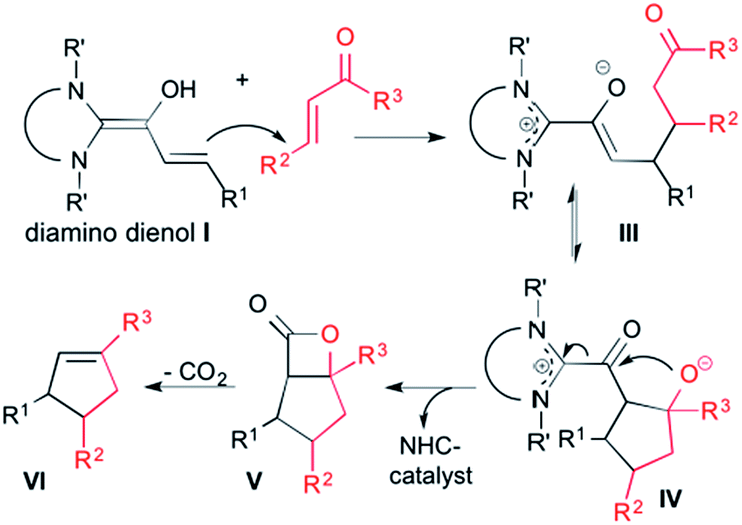
Cyclopentene formation with enones.
In 2006, Nair et al. reported that the NHC-catalyzed reaction of cinnamic aldehydes with enones affords 1,3,4-trisubstituted cyclopentenes.11a As schematically shown in Scheme 2, this transformation was interpreted by homoenolate addition to the Michael acceptor, giving rise to the intermediate III.19 Aldol ring closure leads to intermediate IV. From there, the β-lactone V is formed, with concomitant regeneration of the NHC catalyst. Decarboxylation of the β-lactone V finally gives the cyclopentene product VI.
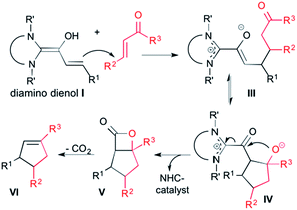 |
| | Scheme 2 Proposed mechanism for cyclopentene (VI) formation from diamino dienol I and an enone Michael acceptor. | |
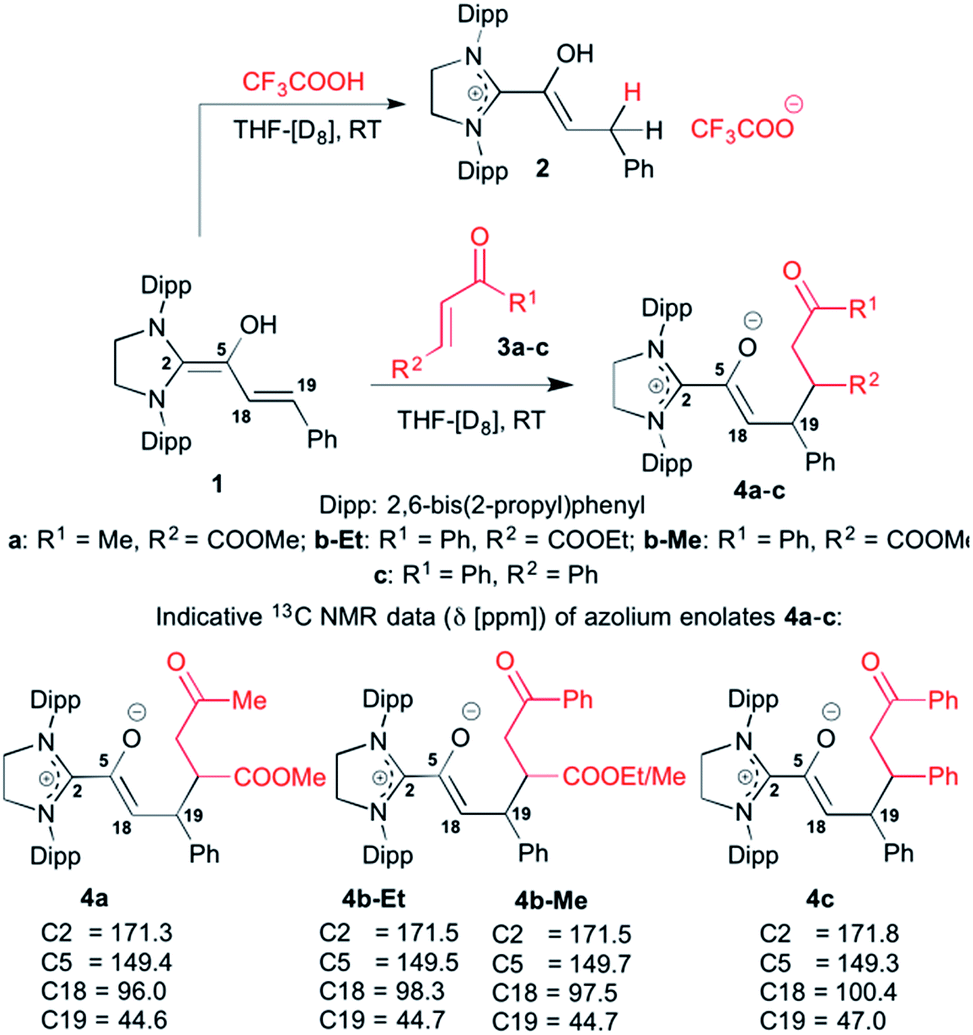
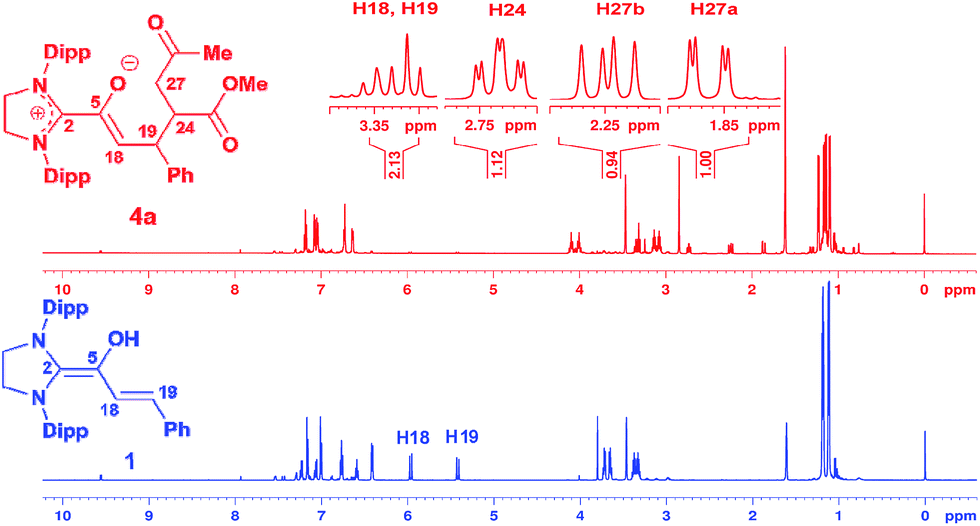
We had reported earlier18 that under strictly oxygen-free conditions, the saturated imidazolidinylidene SIPr (1,3-bis[2,6-di-(2-propylphenyl)]imidazolidin-2-ylidene) reacts smoothly with E-cinnamic aldehyde in THF at room temperature to the diamino dienol 1 (Scheme 3). Protonation of the latter exclusively gives the Cγ-protonation product 2 (an azolium enol), and thus nicely proves Cγ-nucleophilicity (Scheme 3, top). When the pre-formed and stable diamino dienol 1 was exposed to an equimolar amount of methyl-E-4-oxo-2-pentenoate 3a (Scheme 3, middle) under 1H NMR monitoring at room temperature, we observed the instantaneous disappearance of the signals characteristic of the diamino dienol 1 (Fig. 1, bottom: doublets at δ = 5.96 ppm, 3JHH = 15.2 Hz, 1H, H18, and δ = 5.42 ppm, 3JHH = 15.2 Hz, 1H, H19), with concomitant formation of a new species (Fig. 1, top). The newly formed sets of signals are consistent with the formation of the Michael addition product, the azolium enolate 4a that results from C–C bond formation at Cγ of the diamino dienol 1. For example, characteristic 1H NMR signals of 4a are a multiplet at δ = 3.36–3.30 ppm (2H, H18, H19), a triplet of doublets at δ = 2.73 ppm (3JH24–H27a = 2.9 Hz, 3JH24–H27b = 11.4 Hz, 3JH24–H19 = 11.4 Hz, 1H, H24), a doublet of doublets at δ = 2.25 ppm (3JH27b–H24 = 11.4 Hz, 2JH27b–H27a = 17.4 Hz, 1H, H27b), and a doublet of doublets at δ 1.86 ppm (2JH27a–H27b = 17.4 Hz, 3JH27a–H24 = 2.9 Hz, 1H, H27a). Similarly indicative, in the 13C NMR spectrum, the signals of C2, C5, C18 and C19 shifted from 145.0 to 171.3 ppm, 114.0 to 149.4 ppm, 125.3 to 96.0 ppm, and 110.0 to 44.6 ppm, respectively (see ESI† for 1D and 2D NMR characterization of 4a).
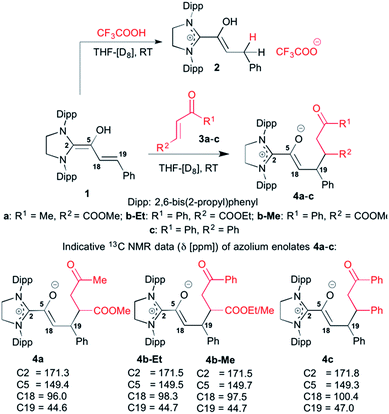 |
| | Scheme 3 Top: diamino dienol 1 reacts with TFA to the azolium enol 2, and (middle) with the enone electrophiles 3a–c to afford the Michael addition adducts 4a–c; bottom: characteristic 13C NMR shifts [ppm] of C2, C5, C18 and C19 of the Michael addition products 4a–c ([D8]THF, 25 °C); Dipp = 2,6-bis(2-propyl)phenyl. | |
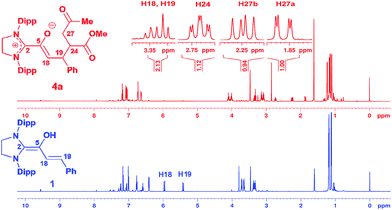 |
| | Fig. 1 Top: 1H NMR spectrum ([D8]THF, 600 MHz) obtained upon addition of methyl E-4-oxo-2-pentenoate (3a) to the diamino dienol 1, indicating the formation of Michael addition product 4a; bottom: 1H NMR of the starting diamino dienol 1; Dipp = 2,6-bis(2-propyl)phenyl. | |
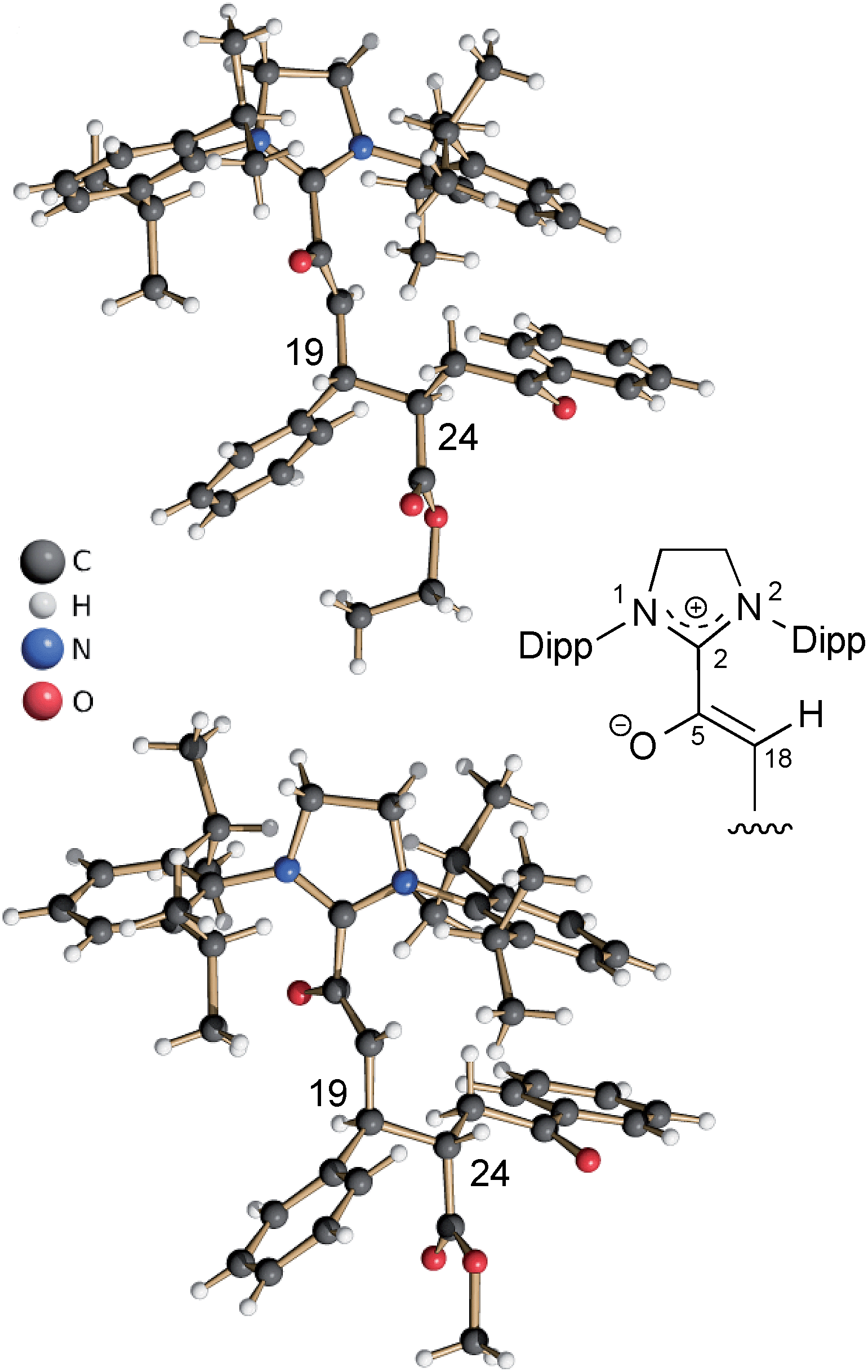
In the same manner, we exposed the diamino dienol 1 to an equimolar amount of ethyl E-3-benzoylacrylate (3b-Et). Again, NMR monitoring revealed the instantaneous disappearance of diamino dienol 1, with concomitant formation of the corresponding Michael product 4b-Et (Scheme 3, middle; see ESI† for the full 1D and 2D NMR characterization of 4b-Et). In addition, crystallization of this Michael product 4b-Et and of its methyl analogue, 4b-Me [obtained from methyl 3-benzoylacrylate (3b-Me)], was achieved from benzene and THF solution, respectively, by slow addition of n-hexane at room temperature, and under strictly anaerobic conditions. The X-ray crystal structures of the azolium enolates 4b-Et and 4b-Me are shown in Fig. 2. First of all, the X-ray structures provide unambiguous proof for the formation and the constitution of the Michael addition products 4b-Et/Me. Furthermore, they nicely reveal the almost orthogonal arrangement of the imidazolium ring and the enolate moiety, as evidenced by the dihedral angles O–C5–C2–N1 = 44.5(4)° and O–C5–C2–N2 = −132.3(3)° for 4b-Et, and [O–C5–C2–N1 = −128.7(4)° and O–C5–C2–N2 = 47.8(5)° for 4b-Me. Along the 5-oxy-4-pentenoate chain of the Michael products 4b-Et/Me, the substituents at C19 (phenyl) and at C24 (phenacetyl) occupy anti-positions.
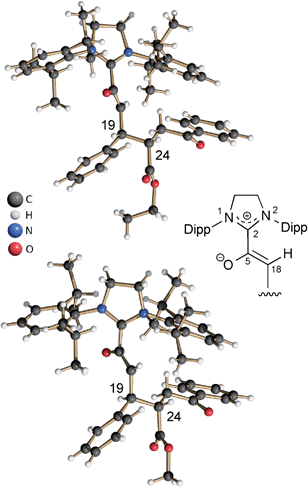 |
| | Fig. 2 Top: X-ray crystal structure of the Michael product 4b-Et, obtained from the addition of ethyl E-3-benzoylacrylate (3b-Et) to the diamino dienol 1; bottom: X-ray crystal structure of the Michael product 4b-Me obtained from diamino dienol 1 and methyl E-3-benzoylacrylate (3b-Me). | |
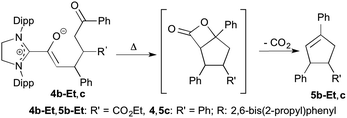 |
| | Scheme 4 Heating-induced conversion of the Michael addition products 4b-Et and 4c to the cyclopentenes 5b-Et and 5c, respectively; Dipp = 2,6-bis(2-propyl)phenyl. | |
When the diamino dienol 1 was exposed to E-chalcone (3c) in an analogous manner, the slow formation of the Michael addition product 4c was observed (Scheme 3, middle; ca. 80% conversion at room temperature after ca. 12 h; see ESI† for full NMR characterization of 4c). In summary, in all four cases studied (diamino dienol 1 + enones 3a, 3b-Et/Me, 3c), C–C bond formation had indeed occured at C-γ, of the diamino dienol and gave the azolium enolate intermediates 4a, 4b-Et/Me and 4c postulated for cyclopentene formation.11 The further conversion of the azolium enolate intermediates such as 4a, 4b-Et/Me, and 4c is typically formulated as an aldol addition of the enolate to the ketone moiety, followed by β-lactone formation and decarboxylation (vide supra, Scheme 2). Note that intermediate azolium enolates such as 4a, 4b-Et/Me and 4cen route to β-lactones/cyclopentenes had not been observed before. By employing the saturated NHC SIPr, we achieved sufficient stabilization of these intermediates such that the subsequent intramolecular aldol addition to the 5-membered carbocycles does not occur spontaneously at room temperature. However, as studied exemplarily with the Michael addition adducts 4b-Et and 4c, heating to 80 °C for 12 h in THF or toluene indeed resulted in the formation of the expected cyclopentene derivatives 5b-Et and 5c, along with the disappearance of the starting azolium enolates 4b-Et,c (Scheme 4; see ESI† for NMR spectra).
γ-Butyrolactone formation with aldehydes.
Diamino dienols I have been postulated as intermediates in γ-butyrolactone (VII) formation from enals and aldehydes (Scheme 5, top).5 Exposition of the diamino dienol 1 to benzaldehyde (6) in THF at 70 °C indeed resulted in a slow conversion (ca. 50% after 24 h) to the saturated lactone 7 (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis 3.3:1; Scheme 5, bottom). The most characteristic 1H NMR signals of 7 are a doublet at δ = 5.44 ppm [3JHH = 9.0 Hz, 1H, H4 (trans)] and a doublet at δ = 5.85 ppm [3JHH = 6.6 Hz, 1H, H4 (cis)]. In line with our earlier experience,18 the liberated NHC SIPr reacted with benzaldehyde to cleanly afford the diamino enol 8 (see ESI† for the NMR identification of lactone 7 and diamino enol 8).
:cis 3.3:1; Scheme 5, bottom). The most characteristic 1H NMR signals of 7 are a doublet at δ = 5.44 ppm [3JHH = 9.0 Hz, 1H, H4 (trans)] and a doublet at δ = 5.85 ppm [3JHH = 6.6 Hz, 1H, H4 (cis)]. In line with our earlier experience,18 the liberated NHC SIPr reacted with benzaldehyde to cleanly afford the diamino enol 8 (see ESI† for the NMR identification of lactone 7 and diamino enol 8).
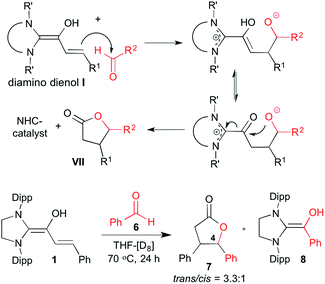 |
| | Scheme 5 Top: general reaction scheme for the NHC-catalyzed formation of γ-butyrolactones VII from enals and aldehydes: bottom: diamino dienol 1 reacts with benzaldehyde (6) to afford the saturated lactone 7 and the diamino enol 8; Dipp = 2,6-bis(2-propyl)phenyl. | |
Reactivity studies of azolium enolates
Formation of γ,δ-unsaturated δ-lactones with enones.
As discussed above, the conversion of α,β-unsaturated aldehydes to cyclopentenes VI proceeds via initial diamino dienol formation and subsequent reaction of the latter with an enone electrophile (Scheme 2). In contrast, the conversion of α,β-unsaturated aldehydes with enones to γ,δ-unsaturated δ-lactones VIII (i.e. same starting materials, but different products) is assumed to involve additional tautomerization of the diamino dienol I to an azolium enolate II (see Scheme 1). The latter then reacts with the enone Michael acceptor, ultimately affording the γ,δ-unsaturated δ-lactone VIII (Scheme 6).
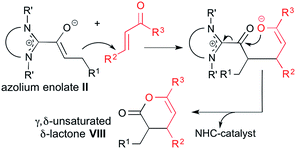 |
| | Scheme 6 Reaction scheme for the NHC-catalyzed conversion of α,β-enals, via azolium enolates II, to γ,δ-unsaturated δ-lactones VIII. | |
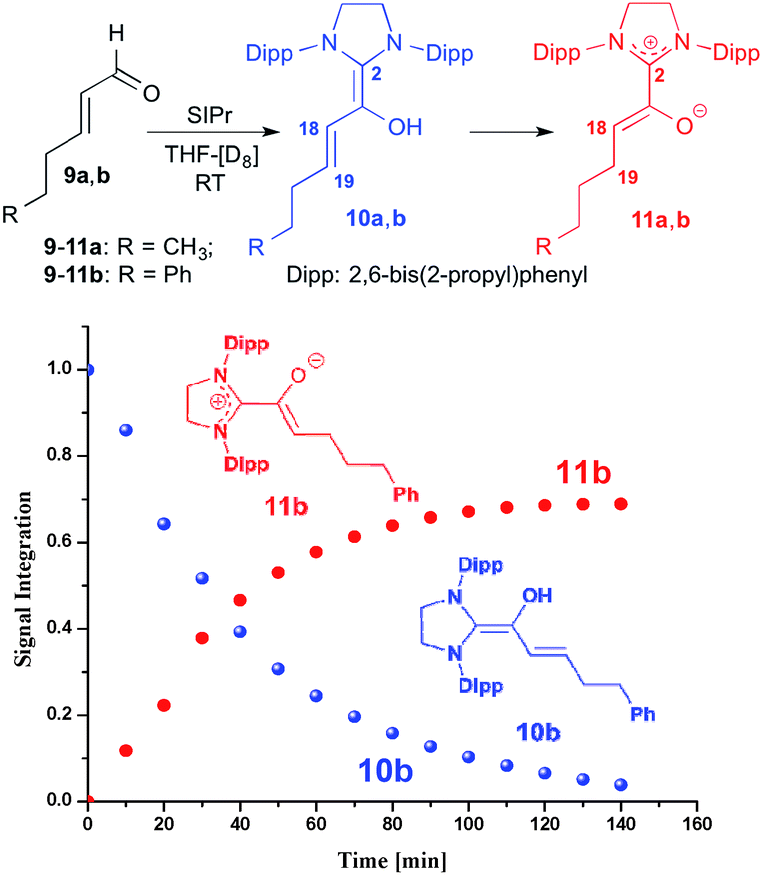
For studying the reactivity of preformed azolium enolates, we chose the two stable representatives 11a and 11b shown in Fig. 3 (top). Upon addition of n-hexenal (9a) to SIPr in THF-[D8] at room temperature, we observed the instantaneous disappearance of the aldehyde signal characteristic of 9a, and the appearance of diamino dienol 10a, as evidenced by a doublet at δ = 5.32 ppm (3JHH = 12.0 Hz, 1H, H18), a multiplet at δ = 4.71–4.66 ppm (1H, H19) and singlet at δ = 3.40 ppm (OH). At room temperature, the diamino dienol 10a tautomerized to the azolium enolate 11a within ca. 20 min.20 The latter shows a characteristic 1H NMR triplet at δ = 3.46 ppm (3JHH = 7.0 Hz, 1H, H18), and a multiplet at δ = 1.82–1.78 ppm, (2H, H19). Indicative 13C NMR resonances are those of C2 and C18, appearing at δ = 172.5 ppm and 100.5 ppm, respectively (see ESI† for further NMR data of 11a).
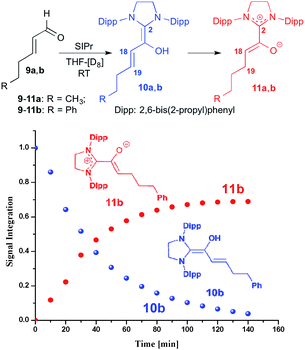 |
| | Fig. 3 Top: generation of the azolium enolates 11a,b from the enals 9a,bvia diamino dienols 10a,b: bottom: time course of the tautomerization of 10b to the azolium enolate 11b. | |
In a similar manner, when we exposed E-5-phenylpent-2-enal (9b) to SIPr, 1H NMR monitoring first revealed the instantaneous formation of the diamino dienol 10b, characterized by a doublet at δ = 5.39 ppm (3JHH = 14.9 Hz, H18), a multiplet at δ = 4.80–4.75 ppm (H19), and a singlet at δ = 3.42 ppm (OH) (see ESI† for further NMR data of 11b). After 10 min, the formation of the azolium enolate 11b was noticeable, and its concentration increased over time (Fig. 3, bottom). The azolium enolate 11b is characterized by a 1H NMR triplet at δ = 3.55 ppm (3JHH = 7.1 Hz, 1H, H18), and a multiplet at δ = 1.89–1.86 ppm (2H, H19). In the 13C NMR spectrum, the formation of 11b is evidenced by the characteristic signals of C2 and C18, appearing at δ = 172.4, 99.4 ppm respectively (see ESI† for further NMR data of 11b). Note that in an earlier report from our laboratory, we had observed that diamino dienols derived from enals with additional conjugation (e.g. E-cinnamic aldehyde, sorbic aldehyde) do not undergo tautomerization to azolium enolates.18b Tautomerization occurs only in the absence of this additional conjugative stabilization of the diamino dienol state, for example with E-hexenal (9a) and E-5-phenylpent-2-enal (9b) as reported here, or with E-crotonic aldehyde as substrate aldehyde.18b
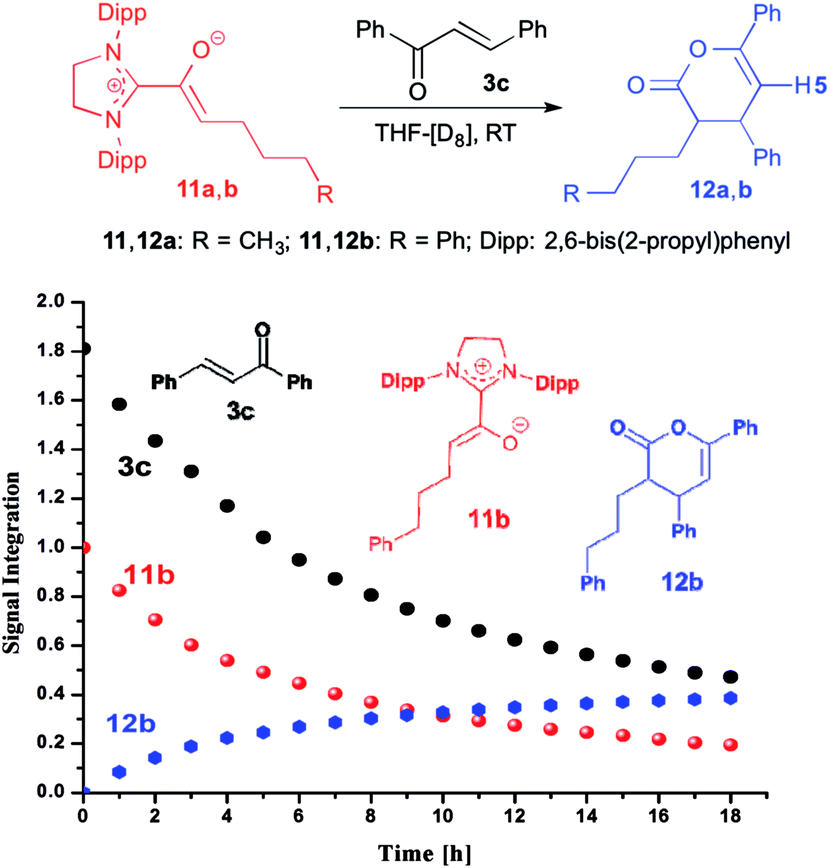
When we added E-chalcone (3c) to the pre-formed azolium enolate 11b, the concentrations of both 11b and 3c decreased simultaneously over time (Fig. 4), along with the appearance of the unsaturated δ-lactone 12b (trans:cis 5.8:1). The latter is characterized by a 1H NMR doublet at δ = 5.97 ppm [3JHH = 4.4 Hz, 1H, H5 (trans)] and a doublet at δ = 6.21 ppm [3JHH = 6.5 Hz, 1H, H5 (cis)] (see ESI† for the NMR identification of the lactone 12b). In the case of the azolium enolate 11a, reaction with E-chalcone (3c) gave the analogous unsaturated δ-lactone 12a (trans:cis 11:1), characterized by a 1H NMR doublet at δ = 5.98 ppm [3JHH = 4.4 Hz, 1H, H5 (trans)] and a doublet at δ = 6.22 ppm [3JHH = 6.6 Hz, 1H, H5 (cis)] (see ESI† for the full NMR identification of 12a).
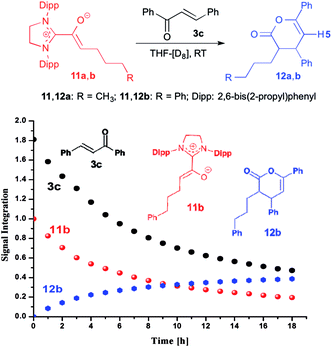 |
| | Fig. 4 Top: formation of the γ,δ-unsaturated lactones 12a,b from the azolium enolates 11a,b and E-chalcone (3c); bottom: time course of the conversion of the azolium enolate 11b to the γ,δ-unsaturated lactone 12b. | |
Conclusion
We have reported (i) the selective generation and characterization of a number of hitherto postulated diamino dienol and azolium enolate reaction intermediates, by interaction of the N-heterocyclic carbene SIPr with various α,β-unsaturated aldehydes. (ii) The homoenolate and enolate equivalents thus prepared were stable enough for NMR-spectroscopic characterization, but still reactive enough for further transformations when exposed to electrophilic reaction partners: exposure of diamino dienols to Michael acceptors gave hitherto postulated addition products stable enough for NMR and even X-ray crystallographic characterization. Heating of the latter completed the reaction cycle, affording trisubstituted cyclopentenes. (iii) In the same manner, the postulated reaction of diamino dienol intermediates with aldehydes to γ-butyrolactones could be verified experimentally. (iv) The tautomerization of primarily formed diamino dienols to azolium enolates, the postulated precursors of γ,δ-unsaturated δ-lactones, was monitored by 1H NMR in two cases. Subsequent exposure of the azolium enolates to E-chalcone as Michael acceptor indeed gave the corresponding γ,δ-unsaturated δ-lactones, thus proving the postulated C–C bond formation at Cβ of the azolium enolate intermediate. We are convinced that the mechanistic information disclosed herein will promote the understanding of other existing NHC-catalyzed transformations, and the design of novel ones.
Acknowledgements
This work was supported by the Fonds der Chemischen Industrie and by BASF SE.
Notes and references
- For selected reviews on NHC-carbene organocatalysis, see:
(a) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696–707 CrossRef CAS PubMed;
(b) A. Grossmann and D. Enders, Angew. Chem., 2012, 124, 320–332 (
Angew. Chem., Int. Ed.
, 2012
, 51
, 314–325
) CrossRef PubMed;
(c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC;
(d)
P.-C. Chiang and J. W. Bode, Science of Synthesis: Asymmetric Organocatalysis, ed. B. List, Thieme, Stuttgart, 2012, vol. 1, pp. 639–672 Search PubMed;
(e) J. Douglas, G. Churchill and A. D. Smith, Synthesis, 2012, 2295–2309 CAS;
(f) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336–5346 RSC.
-
(a) R. Breslow, J. Am. Chem. Soc., 1957, 79, 1762–1763 CrossRef CAS;
(b) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
-
(a) J. Kaeobamrung, M. C. Kozlowski and J. W. Bode, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20661–20665 CrossRef CAS PubMed;
(b) P.-C. Chiang and J. W. Bode, TCI MAIL, 2011, 149, 2–17 Search PubMed.
- Z. Fu, H. Sun, S. Chen, B. Tiwari, G. Li and Y. R. Chi, Chem. Commun., 2013, 49, 261–263 RSC.
-
(a) C. Burstein and F. Glorius, Angew. Chem., 2004, 116, 6331–6334 (
Angew. Chem., Int. Ed.
, 2004
, 43
, 6205–6208
) CrossRef PubMed;
(b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS PubMed;
(c) Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, Nat. Chem., 2013, 5, 835–839 CrossRef CAS PubMed.
-
(a) V. Nair, S. Vellalath, M. Poonoth, R. Mohan and E. Suresh, Org. Lett., 2006, 8, 507–509 CrossRef CAS PubMed;
(b) J. Dugal-Tessier, E. A. O'Bryan, T. B. H. Schroeder, D. T. Cohen and K. A. Scheidt, Angew. Chem., 2012, 124, 5047–5051 (
Angew. Chem., Int. Ed.
, 2012
, 51
, 4963–4967
) CrossRef PubMed.
- Z.-D. Wang, F. Wang, X. Li and J.-P. Cheng, Org. Biomol. Chem., 2013, 11, 5634–5641 CAS.
- J. Kaeobamrung and J. W. Bode, Org. Lett., 2009, 11, 677–680 CrossRef CAS PubMed.
-
(a) M. He and J. W. Bode, Org. Lett., 2005, 7, 3131–3134 CrossRef CAS PubMed;
(b) X. Zhao, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 12466–12469 CrossRef CAS PubMed.
- M. He and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 418–419 CrossRef CAS PubMed.
-
(a) V. Nair, S. Vellalath, M. Poonoth and E. Suresh, J. Am. Chem. Soc., 2006, 128, 8736–8737 CrossRef CAS PubMed;
(b) P.-C. Chiang, J. Kaeobamrung and J. W. Bode, J. Am. Chem. Soc., 2007, 129, 3520–3521 CrossRef CAS PubMed;
(c) B. Cardinal-David, D. E. A. Raup and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 5345–5347 CrossRef CAS PubMed.
-
(a) S. S. Sohn and J. W. Bode, Org. Lett., 2005, 7, 3873–3876 CrossRef CAS PubMed;
(b) A. Chan and K. A. Scheidt, Org. Lett., 2005, 7, 905–908 CrossRef CAS PubMed.
-
(a) B. Maji and H. Mayr, Angew. Chem., 2013, 125, 11370–11374 (
Angew. Chem., Int. Ed.
, 2013
, 52
, 11163–11167
) CrossRef PubMed;
(b) Y.-R. Zhang, L. He, X. Wu, P.-L. Shao and S. Ye, Org. Lett., 2008, 10, 277–280 CrossRef CAS PubMed;
(c) N. Duguet, C. D. Campbell, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2008, 6, 1108–1113 RSC;
(d) L. He, H. Lv, Y.-R. Zhang and S. Ye, J. Org. Chem., 2008, 73, 8101–8103 CrossRef CAS PubMed;
(e) X.-N. Wang, P.-L. Shao, H. Lv and S. Ye, Org. Lett., 2009, 11, 4029–4031 CrossRef CAS PubMed;
(f) C. Concellón, N. Duguet and A. D. Smith, Adv. Synth. Catal., 2009, 351, 3001–3009 CrossRef PubMed;
(g) T.-Y. Jian, P.-L. Shao and S. Ye, Chem. Commun., 2011, 47, 2381–2383 RSC.
-
(a) M. He, G. J. Uc and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 15088–15089 CrossRef CAS PubMed;
(b) M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418–8420 CrossRef CAS PubMed;
(c) Y. Li, X.-Q. Wang, C. Zheng and S.-L. You, Chem. Commun., 2009, 5823–5825 RSC;
(d) V. Nair, R. R. Paul, K. C. S. Lakshmi, R. S. Menon, A. Jose and C. R. Sinu, Tetrahedron Lett., 2011, 52, 5992–5994 CrossRef CAS PubMed;
(e) X. Fang, X. Chen and Y. R. Chi, Org. Lett., 2011, 13, 4708–4711 CrossRef CAS PubMed;
(f) X. Zhao, K. E. Ruhl and T. Rovis, Angew. Chem., 2012, 124, 12496–12499 (
Angew. Chem., Int. Ed.
, 2012
, 51
, 12330–12333
) CrossRef PubMed;
(g) J. Mo, R. Yang, X. Chen, B. Tiwari and Y. R. Chi, Org. Lett., 2013, 15, 50–53 CrossRef CAS PubMed;
(h) S. E. Allen, J. Mahatthananchai, J. W. Bode and M. C. Kozlowski, J. Am. Chem. Soc., 2012, 134, 12098–12103 CrossRef CAS PubMed.
-
(a) L. Hao, C. W. Chuen, R. Ganguly and Y. R. Chi, Synlett, 2013, 24, 1197–1200 CrossRef CAS PubMed;
(b) L. Hao, Y. Du, H. Lv, X. Chen, H. Jiang, Y. Shao and Y. R. Chi, Org. Lett., 2012, 14, 2154–2157 CrossRef CAS PubMed;
(c) S. Chen, L. Hao, Y. Zhang, B. Tiwari and Y. R. Chi, Org. Lett., 2013, 15, 5822–5825 CrossRef CAS PubMed.
- Mayr and Maji studied the O- vs. C-alkylation of azolium enolates, prepared by addition of carbenes to ketenes, see ref. 13a.
-
(a) M. Regitz, J. Hocker and B. Weber, Angew. Chem., 1970, 82, 394–395 (
Angew. Chem., Int. Ed. Engl
, 1970
, 9
, 375–375
) CrossRef PubMed;
(b) L. Weber, U. Lassahn, H.-G. Stammler and B. Neumann, Eur. J. Inorg. Chem., 2005, 4590–4597 CrossRef CAS PubMed;
(c) Y.-G. Lee, J. P. Moerdyk and C. W. Bielawski, J. Phys. Org. Chem., 2012, 25, 1027–1032 CrossRef CAS PubMed;
(d) M. Hans, J. Wouters, A. Demonceau and L. Delaude, Chem. - Eur. J., 2013, 19, 9668–9676 CrossRef CAS PubMed.
-
(a) A. Berkessel, S. Elfert, V. R. Yatham, J.-M. Neudörfl, N. E. Schlörer and J. H. Teles, Angew. Chem., 2012, 124, 12537–12541 (
Angew. Chem., Int. Ed.
, 2012
, 51
, 12370–12374
) CrossRef PubMed;
(b) A. Berkessel, V. R. Yatham, S. Elfert and J.-M. Neudörfl, Angew. Chem., 2013, 125, 11364–11369 (
Angew. Chem., Int. Ed.
, 2013
, 52
, 11158–11162
) CrossRef PubMed.
- Bode et al. have published later that the formation of intermediate III proceeds via cross-benzoin reaction between diamino dienol I and enone, followed by an oxy-Cope rearrangement, see ref. 11b.
- As pointed out by one of the referees, Chi et al. hypothesize in ref. 5c that upon deprotonation, azolium enolates can be transformed to homoenolates. However, for the azolium enolate 11a, exposure to equimolar amounts of DBU did not result in any transformation.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, and X-ray crystallographic data of compounds 4b-Et and 4b-Me. CCDC 1014843 and 1014844. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01027f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a