
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homochiral self-assembly of biocoordination polymers: anion-triggered helicity and absolute configuration inversion†
Nadia
Marino
ab,
Donatella
Armentano
*a,
Emilio
Pardo
*c,
Julia
Vallejo
c,
Francesco
Neve
a,
Leonardo
Di Donna
 a and
Giovanni
De Munno
a
a and
Giovanni
De Munno
a
aDipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, 87036, Arcavacata di Rende, Cosenza, Italy. E-mail: donatella.armentano@unical.it
bDepartment of Chemistry, Syracuse University Syracuse, NY 13244-4100, USA
cDepartament de Química Inorgànica, Instituto de Ciencia Molecular (ICMOL), Universitat de València, 46980 Paterna, València, Spain
First published on 30th April 2015
Abstract
The different natures of the weakly coordinating anions – triflate or perchlorate – in the Cu2+-mediated self-assembly of cytidine monophosphate nucleotide play a fundamental role in the homochiral resolution process, yielding one-dimensional copper(II) coordination polymers of opposite helicity that can be easily inverted, in a reversible way, by changing the nature of the anion as revealed by circular dichroism experiments both in solution and in the solid state.
Introduction
New generations of metal complexes containing ligands from the biological world are attracting continuous interest in the attempt to develop new materials.1–3 The powerful self-assembly features of biomolecules – which may have the ability to bridge metal ions with multiple possible coordination modes2,3 – offer the possibility to obtain both discrete zero-dimensional (0D) metal complexes and also coordination polymers of higher dimensionality (1D–3D) with fascinating architectures.4–6 Among the plethora of interesting properties that a coordination polymer can show, chirality has attracted intense attention from many research groups. In particular, the appearance of homochirality in biological systems, which is likely related to the origin of life,7 is still largely unclear. A chiral coordination complex or polymer can be obtained either in a rational way, by a judicious choice of chiral enantiopure ligands or “chiral auxiliaries” capable of transmitting their “chiral information” to the stereochemistry of the metal atoms,8 or serendipitously, when so-called spontaneous resolution processes9 occur. In this regard, interesting works have been reported recently pointing towards external factors as responsible for these spontaneous resolution processes (e.g. stirring,10a rotational and magnetic forces,10b,cetc.). Nevertheless, despite the light shed by these studies, further work is needed to fully understand this phenomenon, which could also be helpful to understand chemical processes of fundamental biological importance, such as the chirality switching experienced by DNA and proteins upon external stimuli.11 In this perspective, examples of metal complexes whose helicity can be inverted by external stimuli (pH, temperature, guest molecules, etc.) have been reported.12 In particular, some of them show helicity inversion in the presence of achiral anions,13 leading to intriguing potential applications in anion recognition.Nucleotides, the basic constituents of nucleic acids like RNA or DNA, thus emerge as valuable ligands for the construction of a unique class of biocoordination polymers (bioCPs) with tailored architectures and tunable properties.
In the framework of our current research focused on the reactivity of first-row transition metal ions toward ligands from the biological world, such as cytidine nucleoside (H2cyd), we have recently shown that nucleoside-containing 3D metal complexes can be used as building blocks for the rational design of nucleoside-bridged high-nuclearity coordination compounds and high-dimensionality coordination polymers.3 We observed unprecedented metal-nucleoside coordination modes and excellent chiral induction, which accounted for the formation of octanuclear calixarene-like3a as well as dodecanuclear globular-shaped complexes,3b together with the first example of a 3D copper(II)–cytidine coordination polymer.3c
Aiming at further exploring the potential role of this type of ligands as chiral inducers, and inspired to contribute to a better understanding of the driving forces behind supramolecular aggregations as a prerequisite for the design and construction of molecular arrays, we have more recently considered the cytidine 5′-monophosphate (CMP) nucleotide (Scheme 1).14 As a ligand, CMP has received relatively little consideration, always affording structurally characterized transition metal complexes that are either dimeric or, more often, polymeric in nature.15 On the other hand, CMP is offering good prospects as a chiral inducer in supramolecular 1D assemblies.16
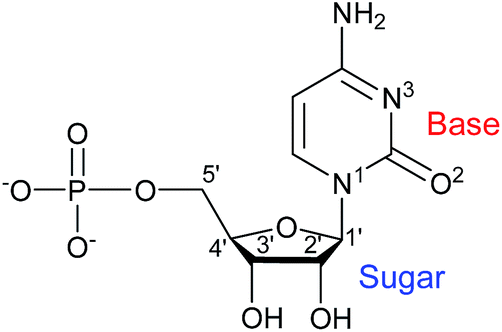 | ||
| Scheme 1 Chemical structure of the ligand CMP with selected atomic numbering. | ||
In this paper, we show a fascinating example of anion-mediated homochiral resolution in polymeric metallo-helices, reporting on two quasi-identical CMP-based homochiral 1D biopolymers of opposite helicity and the respective formulas {[Cu5(bpy)5(OH)(H2O)2(CMP)2(ClO4)](ClO4)4·9H2O}n (1P) and {[Cu15(bpy)15(OH)3(H2O)7(CMP)6(CF3SO3)](CF3SO3)14·15H2O}n (2M),17a which are built through the simultaneous self-assembly of the CMP nucleotide, the 2,2′-bipyridine (bpy) ligand and Cu(X)2·6H2O [where X = ClO4− (1P) or CF3SO3− (2M)] in aqueous solution. Interestingly, 1P and 2M can be rapidly interconverted by exchanging the anion, in a reversible manner (vide infra), with the corresponding inversion of the copper(II) absolute configuration and helicity.
Results and discussion
1P and 2M crystallize in the chiral space groups P212121 and P21 of the orthorhombic and monoclinic systems, respectively, their absolute configuration being reliably assigned. The structure of 1P consists of single-stranded helices containing the repeating unit [Cu5(bpy)5(H2O)2(OH)(CMP)2(ClO4)]4+ (including a single, weakly-coordinating ClO4− ion) (Fig. 1, left and Fig. 2), perchlorate counterions and a large amount of lattice water molecules. On the other hand, fifteen crystallographically independent copper atoms are present in the repeating cationic unit of 2M, which could be alternatively formulated17a as {[Cu5(bpy)5(H2O)2(OH)(CMP)2(CF3SO3)][Cu5(bpy)5(H2O)3(OH)(CMP)2][Cu5(bpy)5(H2O)2(OH)(CMP)2]}14+ (see Fig. 1, right and Fig. 2). The single-stranded helices are arrayed in a right-handed (1P) or left-handed (2M) fashion, with similar helical pitches [16.431 Å (the a axis value) for 1P and 16.538 and 16.785 Å (ca. 1/3 of the b axis value) for 2M].17b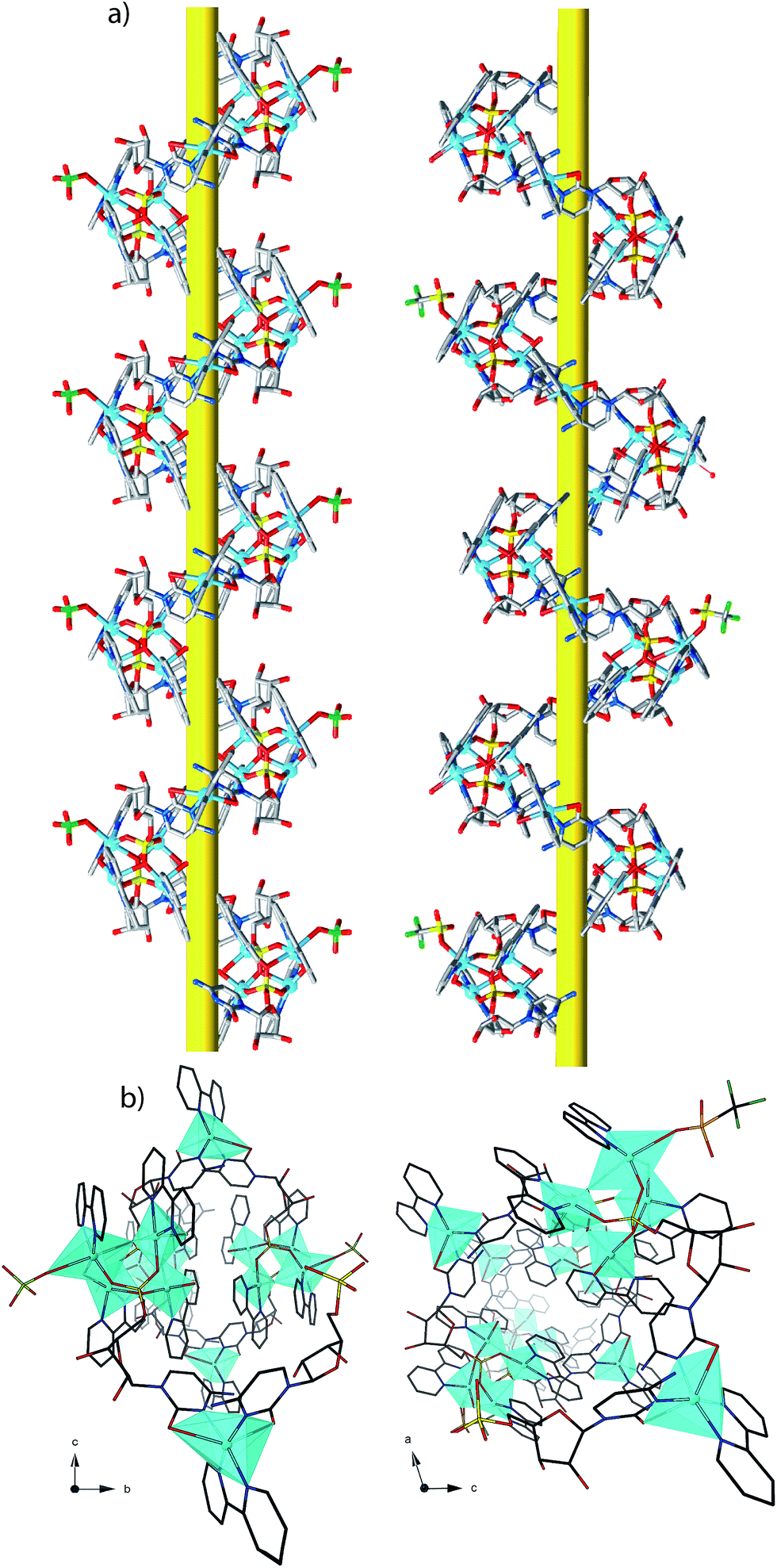 | ||
| Fig. 1 Side view (a) and top view (b) of the cationic copper(II) chains of 1P (left) and 2M (right). | ||
 | ||
| Fig. 2 Schematic view for 1P (a) and 2M (b) with the metal atom numbering scheme [symmetry code (1P): a = x − 1/2, −y + 3/2, −z + 2; b = x + 1/2, −y + 3/2, −z + 2; (2M): a = −x + 1, y − 1/2, −z + 1; b = −x + 1, y + 1/2, −z + 1]. | ||
Each helix of 1P and 2M contains CMP ligands coordinated through the oxygen atoms of the phosphate groups and via N(3) and the exocyclic O(2) of the nucleobase as bridges (Fig. 3). Pairs of μ4-phosphate groups connect four copper(II) ions, giving rise to butterfly-shaped tetranuclear cores of the type [Cu4(μ4-PO4)2(μ-OH)], also supported by a bridging hydroxo group (Fig. 2, 3a and S1†). The remaining copper atoms, chelated by the cytosine base of the nucleotides, (Fig. 2 and 3b), constitute the connectors of the CMP ligands, i.e. the chiral inducers of the overall helical structure. In fact, they exhibit an octahedral geometry in both 1P and 2M but with opposite C(Δ) or A(Λ) propeller chirality.18,19 Since the role of such connectors is pivotal, each helix consists of a single strand of alternating tetranuclear cores and chiral connectors (Fig. 3c), leading to helices with P (1P) or M (2M) chirality.
 | ||
| Fig. 3 Tetranuclear cores (a), Cu(II) connectors (b) and a portion of the single-stranded helix of alternating tetranuclear cores (c) for 1P (left) and 2M (right). C/A are general chiral descriptors while Δ/Λ refer to axial chirality.19 | ||
Interestingly, within the cores only one of the four metal ions is further anion-linked achieving an octahedral geometry. In fact, while in 1P merely the perchlorate anion is weakly coordinated to one copper atom in four, in 2M competition between the water solvent and the triflate ion actually leads to three different tetranuclear cores. Two of them [core I (Fig. 3b, right and S2†) and core III (Fig. S4†)], present an extra coordination site either occupied by a triflate anion or by a water molecule. Finally, in the third one (cluster II in Fig. S3†), neither anions nor solvent molecules are linked to the would-be octahedral copper, further proving the lability of the extra anion/solvent coordination. At this point, one may immediately notice that contrary to the general expectation based on the slightly lower coordinating ability of the perchlorate ion,20 coverage of the bound triflate along the polymeric chain is consistently smaller. Thus, fewer anions are directly linked to the metal centres of the helix in 2M with respect to 1P (Fig. 1 and 2), with both electronic and structural consequences.
In the search for the driving force behind the opposite supramolecular helical chirality in 1P and 2M, we found that the ClO4− anion coordinated in 1P, is hydrogen-bonded to the bridging hydroxo group within the tetranuclear unit (Fig. 3a, left), suggesting that the copper ions showing a C chirality can be stabilized more effectively than the corresponding A form (Fig. 3, right). On the contrary, the corresponding triflate-coordinated copper ion in 2M (cluster I, Fig. 3b and S2†), shows an A configuration.
In general, the coordinated anions in 2M are not at all or scarcely involved (see Fig. S4†) in intramolecular H-bonds with the hydroxo group of the tetranuclear core. In cluster I, for instance, the hydroxo group is engaged with a free CF3SO3−. Thus our hypothesis is that the different nature, size and shape of the CF3SO3− anion represent the major reasons for the A absolute configuration of the anion-coordinated metal centres in 2M. The geometry around the sulphur atom, combined with the potential steric clash of the CF3 group with the closest free ribose moiety of CMP, most likely causes a destabilization of the C form. For that reason the coordinated CF3SO3− is not involved in intramolecular H-bonding, and the hydroxide group of the tetranuclear core is at this time engaged by a free CF3SO3−.
Remarkably, all metal ions within the tetranuclear cores and connectors, exhibit the same chirality as the anion-coordinated metal center (i.e. Cu(I)), confirming thus the transmission of the chiral properties induced by the anions to the coordinated metal center through weak interactions. Each homochiral asymmetric fragment of the helices [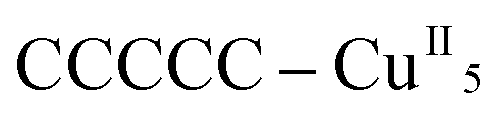 (1P) and
(1P) and 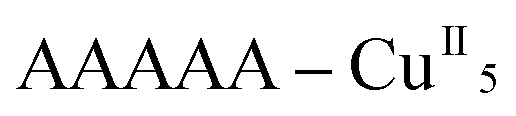 (2M)] then has opposite chirality. These structural features suggest that the anions play a key role in the self-assembly process that leads to the chain formation with opposite handedness.
(2M)] then has opposite chirality. These structural features suggest that the anions play a key role in the self-assembly process that leads to the chain formation with opposite handedness.
The pseudo-cylindrical helices reveal further different structural features depending on the anions, forming a pillar of metal ions with the shortest intrahelical Cucores⋯Cuconnectors separations varying in the range 10.21–12.00 Å in 1P and 8.78–12.53 Å in 2M, with a clear putative role of the different anions in the induction of prominently different arrangements of the copper ions along the helical chains. Notably, in 1P the sugar moieties of the CMP ligands exhibit the C(3′) – exo conformation while in 2M they show the C(2′) – endo conformation. The conformation of C(4′)–C(5′) is gauche–gauche for both compounds. The chiral centers of the CMP ligand C1′, C2′, C3′ and C4′ in the helices have the configurations R, R, S, R.
The packing of 1P and 2M produces small voids inside and outside each chain, (Fig. S5†) where the lattice water molecules and ClO4− (1P) or CF3SO3− (2M) anions reside as blocks of a 3D H-bonded grid. The potential solvent and anion accessible areas around the chains account for 31.8% (1P), and 37.7% (2M) of the unit cell volume.
In order to confirm the enantiopurity of the sample in a given synthesis, and also to ensure the reproducibility of the results, solid circular dichroism (CD) experiments were carried out for crystals of five different syntheses. The solid CD spectra of 1P and 2M in the visible region (Fig. 4a) confirm the absolute configuration of the chiral metal centers. An almost mirror image21 can be observed for 1P and 2M. Thus, 1P exhibits a broad maximum positive Cotton effect at 644 nm whereas 2M exhibits a maximum negative Cotton effect at ca. 690 nm with a similar intensity to that of 1P. Both bands are attributed to d–d transitions as a result of the chirality induced effect on the copper(II) ions. These opposite Cotton effects further confirm that the presence of the different ClO4− or CF3SO3− anions induces opposite chirality on the metal centers of 1P and 2M. Fig. S6† shows identical Cotton effects for both 1P and 2M in the UV region, that is, negative and positive Cotton effects at ca. 220 and 280 nm, respectively. These former bands have already been reported for the CMP ligand,15 and, ultimately, confirm the presence of D-ribose in its enantiopure natural form in both 1P and 2M. H2O/CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solution CD experiments, which are depicted in Fig. S7,† show similar positive and negative Cotton effects, confirming that homochiral species with opposite absolute configurations for the copper(II) ions are also present in solution.
:1 v/v) solution CD experiments, which are depicted in Fig. S7,† show similar positive and negative Cotton effects, confirming that homochiral species with opposite absolute configurations for the copper(II) ions are also present in solution.
 | ||
| Fig. 4 (a) CD spectra of 1P (blue) and 2M (red) in the visible region as KBr pellets (ca. 1 mg of complex/100 mg of KBr). (b) Evolution of the CD spectra of a H2O/CH3CN (1:1 v/v) solution of 1P (blue line) (10−3 M) upon the addition of LiCF3SO3 (0 eq., blue line; 2 eq. green line; 4 eq. orange line; 6 eq. red line) followed by the addition of LiClO4 (2 eq. dashed green line; 4 eq. dashed blue line). | ||
Challenged by these results, and in order to check the reversible nature of this anion-mediated chiral resolution process, we monitored the anion exchange in H2O–CH3CN mixture (1:1 v/v) solutions of 1P and 2M by measuring the CD spectra after controlled additions of LiCF3SO3 and LiClO4 salts, respectively. Fig. 4b shows the evolution of the CD spectra of a 1P solution in H2O/CH3CN (1:1, v/v) with increasing [CF3SO3−]. The intensity of the initial positive Cotton effect decreases with the addition of LiCF3SO3 to become finally negative. Then, the reversible nature of the process was verified when the positive Cotton effect was recovered by adding LiClO4 (dashed lines in Fig. 4b). This reversible and fast (going to completeness in a few minutes) switching in solution, achieved by changing the nature of the anion, is most likely due to the labile coordination of both the ClO4− and CF3SO3− anions to the Cu(II) centre of the cores; thus they are undoubtedly exchanged.
In order to achieve a better understanding of the stable oligomeric species in solution – whose absolute configuration can be dynamically switched by changing the nature of the achiral anion as shown by the CD spectra – ESI-MS and related tandem mass spectroscopy (MS/MS) experiments were carried out for solutions of 1P and 2M. The presence of stable oligomers in solution can be inferred by the electrospray experiments, which show dimeric species for 1P and both trimeric and dimeric ones for 2M (see Schemes S1 and S2 and Fig. S8–S11†).
These results prompted us to speculate that the switching of the absolute configuration of the copper(II) ions of the oligomeric species lies at the origin of the helix inversion in the final solid-state coordination polymers. In order to confirm our hypothesis, we did a final test. Crystals of 1P were dissolved in water/acetonitrile, an excess of LiCF3SO3 salt was added to the solution, and the product was allowed to precipitate. The powder X-ray diffraction pattern of the polycrystalline solid that appeared is shown in Fig. S12† confirming the crystallization of the pure enantiomer 2M. The application of the same procedure to crystals of 2M gave a similar outcome, as reported in the ESI† material.
Conclusions
In summary, we report herein a remarkable example of anion-triggered homochiral induction that yields two copper(II) 1D coordination polymers of opposite chirality depending on the anion used: ClO4− (1P) or CF3SO3− (2M). Interestingly, we have also shown that the absolute configuration at the copper(II) ions and the sense of the helix (or at least a fragment of it), can be dynamically switched – in a similar manner to what happens in biological DNA – by using achiral anions of different natures. The labile anion coordination accounts for the switching of the copper ion configuration and the consequent helicity inversion of the entire chain (or oligomer). In these blocks the orientation of the nucleobase rings is such as to place the carbonyl pointing toward the copper atoms, with its oxygen atom occupying the elongated axial positions, and it is therefore easily flexible and removable on demand.Although a few examples of polynuclear complexes and coordination polymers exhibiting switching of chirality triggered by achiral anions have been reported,12,13 evidence of coordination bio-helix inversion is unprecedented. Achieving control over the structure of a nucleotide-based helix holds great potential for developing stimuli-responsive materials matching the level of sophistication of biological systems, with potential applications in memory devices, biomimetic materials, specific ion sensors and molecular recognition.12 In order to get deeper insight into the rational design of chiral systems, current efforts are devoted to further investigating the putative templating role of these anions in novel examples of chiral coordination polymers with other nucleotide-based ligands. Exploration of the coordination flexibility of the CMP and other nucleotides to yield chiral metal-organic frameworks (MOFs) is a second important goal of our research.
Acknowledgements
This work was supported by MIUR (Italy), the MINECO (Spain) (Projects CTQ2013-46362-P and CTQ2013-44844-P), the Generalitat Valenciana (Spain) (Project PROMETEOII/2014/070). J. V. thanks the MICINN for a contract. Thanks are also extended to the Ramón y Cajal Program (E. P.) and to the European Commission, FSE (Fondo Sociale Europeo) and Calabria Region (N. M.).Notes and references
- (a) H. Yang, K. L. Metera and H. F. Sleiman, Coord. Chem. Rev., 2010, 254, 2403 CrossRef CAS PubMed; (b) A. Singh, M. Tolev, M. Meng, K. Klenin, O. Plietzsch, C. I. Schilling, T. Muller, M. Nieger, S. W. Wenzel and C. Richert, Angew. Chem., Int. Ed., 2011, 50, 3227 CrossRef CAS PubMed; (c) T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. N. Nguyen and E. Stulz, Chem. Soc. Rev., 2011, 40, 138 RSC; (d) J.-L. H. A. Duprey, Y. Takezawa and M. Shionoya, Angew. Chem., Int. Ed., 2013, 52, 1212 CrossRef CAS PubMed; (e) T. Grancha, J. Ferrando-Soria, J. Cano, F. Lloret, M. Julve, G. De Munno, D. Armentano and E. Pardo, Chem. Commun., 2013, 49, 5942 RSC.
- (a) R. A. Smaldone, R. S. Forgan, H. Furukawa, J. J. Gassensmith, A. M. Z. Slawin, O. M. Yaghi and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49(122), 8812 CrossRef PubMed; (b) Y. Liu and Z. Tang, Chem.–Eur. J., 2012, 18, 1030 CrossRef CAS PubMed; (c) A. D'Urso, S. Nardis, G. Pomarico, M. E. Fragalà, R. Paolesse and R. Purrello, J. Am. Chem. Soc., 2013, 135, 8632 CrossRef PubMed.
- (a) D. Armentano, T. F. Mastropietro, M. Julve, R. Rossi, P. Rossi and G. De Munno, J. Am. Chem. Soc., 2007, 129, 2740 CrossRef CAS PubMed; (b) D. Armentano, N. Marino, T. F. Mastropietro, J. Martínez-Lillo, J. Cano, M. Julve, F. Lloret and G. De Munno, Inorg. Chem., 2008, 47, 10229 CrossRef CAS PubMed; (c) N. Marino, D. Armentano, T. F. Mastropietro, M. Julve, F. Lloret and G. De Munno, Cryst. Growth Des., 2010, 10, 1757 CrossRef CAS; (d) N. Marino, D. Armentano, T. F. Mastropietro, M. Julve, G. De Munno and J. Martínez-Lillo, Inorg. Chem., 2013, 52, 11934 CrossRef CAS PubMed.
- I. Imaz, M. Rubio-Martínez, J. An, I. Solé-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287 RSC , and references therein.
- (a) M. J. Rauterrkus and B. Krebs, Angew. Chem., Int. Ed., 2004, 43, 1300 CrossRef PubMed; (b) J. A. R. Navarro, E. Freisinger and B. Lippert, Eur. J. Inorg. Chem., 2000, 147 CrossRef CAS; (c) J. Galy, A. Mosset, I. Grenthe, I. Puigdoménech, B. Sjöberg and F. Hultén, J. Am. Chem. Soc., 1987, 109, 380 CrossRef CAS; (d) K. Yamanari, R. Ito, S. Yamamoto and A. Fuyuhiro, Chem. Commun., 2001, 1414 RSC; (e) E. G. Bardaji, F. Freisinger, B. Costisella, C. A. Schalley, W. Bruning, M. Sabat and B. Lippert, Chem.–Eur. J., 2007, 13, 6019 CrossRef CAS PubMed.
- (a) B. Lippert, Coord. Chem. Rev., 2000, 200–202, 487 CrossRef CAS; (b) C. Price, A. Shipman, N. H. Rees, M. R. J. Elsegood, A. J. Edwards, W. Clegg and A. Houlton, Chem.–Eur. J., 2001, 7, 1194 CrossRef CAS; (c) C. Price, B. R. Horrocks, A. Mayeux, M. R. J. Elsegood, W. Clegg and A. Houlton, Angew. Chem., Int. Ed., 2002, 41, 1047 CrossRef CAS; (d) D. Choquesillo-Lazarte, M. D. P. Brandi-Blanco, I. García-Santos, J. M. Gonzalez-Perez, A. Castiñeiras and J. Niclos-Gutierrez, Coord. Chem. Rev., 2008, 252, 1241 CrossRef CAS PubMed.
- A. Guijarro and M. Yus, The Origin of Chirality in the Molecules of Life, Royal Society of Chemistry, Cambridge, UK, 2008 Search PubMed.
- (a) J. Crassous, Chem. Commun., 2012, 48, 9687 RSC; (b) C. Capici, Y. Cohen, A. D'Urso, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi, R. Purrello, S. Slovak and V. Villari, Angew. Chem., Int. Ed., 2011, 50, 11956 CrossRef CAS PubMed; (c) R. Randazzo, A. Mammana, A. D'Urso, R. Lauceri and R. Purrello, Angew. Chem., Int. Ed., 2008, 47, 9879 CrossRef CAS PubMed.
- (a) H. Amouri and M. Gruselle, in Chirality in Transition Metal Chemistry: Molecules, Supramolecular Assemblies and Materials, ed. H. Amouri and M. Gruselle, John Wiley & Sons, Ltd, Chichester, UK, 2008, ch. 4 Search PubMed; (b) I. Bernal, Inorg. Chim. Acta, 1985, 96, 99 CrossRef CAS; (c) E. Pardo, C. Train, G. Gontard, K. Boubekeur, O. Fabelo, H. Liu, B. Dkhil, F. Lloret, K. Nakagawa, H. Tokoro, S. Ohkoshi and M. Verdaguer, J. Am. Chem. Soc., 2011, 133, 15328 CrossRef CAS PubMed.
- (a) J. M. Ribo, J. Crusats, F. Sagues, J. Claret and R. Rubires, Science, 2001, 292, 2063 CrossRef CAS PubMed; (b) L. D. Barron, Nat. Chem., 2012, 4, 150 CrossRef CAS PubMed; (c) N. Micali, H. Engelkamp, P. G. Van Rhee, P. C. M. Christianen, L. Monsù Scolaro and J. C. Maan, Nat. Chem., 2012, 4, 201 CrossRef CAS PubMed.
- M. A. Fuertes, V. Cepeda, C. Alonso and J. M. Pérez, Chem. Rev., 2006, 106, 2045 CrossRef CAS PubMed.
- H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977 RSC.
- (a) H. Miyake, K. Yoshida, H. Sugimoto and H. Tsukube, J. Am. Chem. Soc., 2004, 126, 6524 CrossRef CAS PubMed; (b) A. Gerus, K. Ślepokura and J. Lisowski, Inorg. Chem., 2013, 52, 12450 CrossRef CAS PubMed; (c) N. Ousaka, Y. Takeyama and E. Yashima, Chem.–Eur. J., 2013, 19, 4680 CrossRef CAS PubMed; (d) J. Suk, V. R. Naidu, X. Liu, M. S. Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938 CrossRef CAS PubMed; (e) H. Miyake, M. Hikita, M. Itazaki, H. Nakazawa, H. Sugimoto and H. Tsukube, Chem.–Eur. J., 2008, 14, 5393 CrossRef CAS PubMed; (f) H. Miyake, H. Kamon, I. Miyahara, H. Sugimoto and H. Tsukube, J. Am. Chem. Soc., 2008, 130, 792 CrossRef CAS PubMed.
- N. Marino, D. Armentano, C. Zanchini and G. De Munno, CrystEngComm, 2014, 16, 8286 RSC.
- (a) K. Aoki, J. Chem. Soc., Chem. Commun., 1979, 589 RSC; (b) G. R. Clark and J. D. Orbell, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1815 CrossRef; (c) J. K. Shiba and R. Bau, Inorg. Chem., 1978, 17, 3484 CrossRef CAS; (d) S. Louie and R. Bau, J. Am. Chem. Soc., 1977, 99, 3874 CrossRef CAS; (e) K. Aoki, J. Chem. Soc., Chem. Commun., 1976, 748 RSC; (f) K. Aoki, Biochim. Biophys. Acta, 1976, 447, 379 CrossRef CAS.
- P. Zhou, J. Yao, C. Sheng and H. Li, CrystEngComm, 2013, 15, 8430 RSC.
- (a) In fact, in our best model for 2M a water molecule competes against a triflate anion for the coordination to the Cu(11) atom. This model showed the atom Cu(11) in cluster III being coordinated either to a water molecule or to a triflate ion with a probability of about 70% vs. 30%, respectively (see ESI†), giving {[Cu15(bpy)15(OH)3(H2O)6.7 (CMP)6(CF3SO3)1.3](CF3SO3)13.7·15H2O}n as the formula for 2M; (b) The two slightly different values of helical pitch in 2M are due to the asymmetry of the three tetranuclear cores in the asymmetric unit that is comprised of two helical turns.
- (a) A. Von Zelewsky, Stereochemistry of Coordination Compounds, Wiley, Chichester, 1996 Search PubMed; (b) E. C. Constable, Chem. Soc. Rev., 2013, 42, 1637 RSC.
- According to IUPAC rules, the Δ/Λ nomenclature for metal complexes exhibiting axial chirality should be used only for octahedral species. Given the combined presence of five-coordinate and six-coordinate metal centres in our complexes, and in order to avoid two different conventions for the stereogenic metal centres, we adopted the more general C/A chiral descriptors. See Nomenclature of Inorganic Chemistry. IUPAC Recommendations 2005 at http://old.iupac.org/publications/books/rbook/Red_Book_2005.pdf.
- Both the triflate and perchlorate ions are considered not very weak among the weakly coordinating anions, with negative coordinating ability indices of −0.4 for CF3SO3− and −0.6 for ClO4− (as defined according to R. Diaz-Torres and S. Alavarez, Dalton Trans., 2011, 40, 10742). The coordinating ability index of ClO4− drops to −1.1 (comparable to that of BF4−) when the analysis is restricted to compounds synthesized in aqueous solution, likely due to unfavorable competition with water.
- Strictly speaking, the Cotton effects of 1P and 2M in the visible region are not exactly mirror images. This can be easily understood taking into account that, although the copper(II) ions of 1P and 2M show opposite chirality, their coordination surroundings are not exactly the same as they include different coordinated anions (ClO4− and CF3SO3− respectively).
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and physical characterization data of 1P and 2M, additional structural description, UV-Vis and CD spectra (Fig. S1–S7), crystallographic refinement details for 1P and 2M (Table S1), selected bond distances and angles for 1P and 2M (Tables S2–S5), ESI(+)-MS and ESI(+)-MSMS spectra (Fig. S8–S11 and Schemes S1 and S2) and PXRD (Fig. S12). CCDC 1046609 and 1046610. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01089f |
| This journal is © The Royal Society of Chemistry 2015 |