
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed diamination of unactivated alkenes with hydroxylamines†
Kun
Shen
and
Qiu
Wang
*
Department of Chemistry, Duke University, Durham, NC 27708–0346, US. E-mail: qiu.wang@duke.edu
First published on 18th May 2015
Abstract
A copper-catalyzed regio- and stereoselective diamination of unactivated alkenes has been developed with O-acylhydroxylamines as electrophilic nitrogen sources and oxidants. This method provides the first example of metal-catalyzed alkene diamination for directly installing an electron-rich amino group and extends the diamination scope for the synthesis of diverse 1,2-diamines. It offers a rapid and efficient approach to construct a wide range of 1,2-diamines that are an important structural motif in organic synthesis, medicines, catalysts and ligands.
Introduction
The 1,2-diamine moiety is widely represented in bioactive compounds, synthetic building blocks, catalysts, ligands, and medicines.1 Alkene diamination reactions that can directly add two amino groups across a double bond provide a straightforward route to the synthesis of 1,2-diamines and are therefore of great interest.2 A number of elegant transition-metal-catalyzed intra- and intermolecular diamination reactions have been reported (Scheme 1).3–5 Oxidative diaminations of alkenes have been developed with Pd, Ni, Au or Cu as a catalyst using N,N′-disubstituted ureas as nitrogen sources (Scheme 1, A).6 The Shi group employed diaziridines as both nitrogen sources and oxidants and established the Cu- and Pd-catalyzed intermolecular alkene diamination reactions (Scheme 1, B).7 Recently, Chemler reported a copper-catalyzed diamination with free sulfonamides or anilines in the second intermolecular C–N bond-forming step in the presence of an oxidant (Scheme 1, C),8 and the Michael group reported a Pd(II)-catalyzed alkene diamination using electrophilic N-fluorobenzenesulfonimide in the intermolecular amination step as its nitrogen source and oxidant (Scheme 1, D).9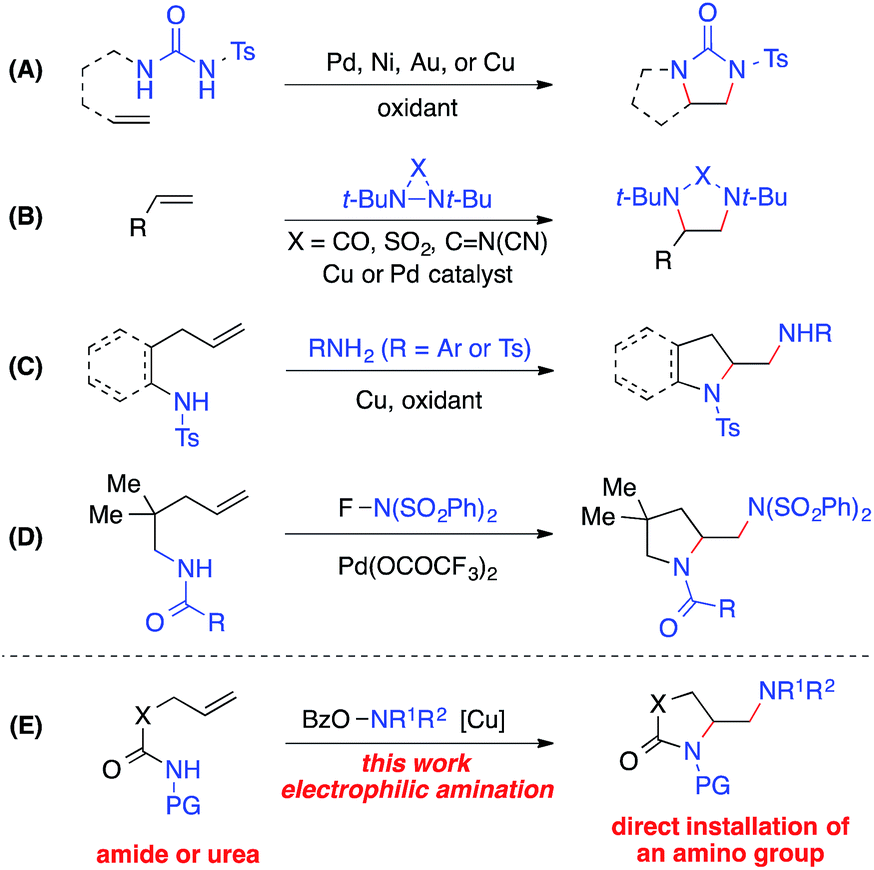 | ||
| Scheme 1 Metal-catalyzed alkene diaminations. | ||
Despite these important advances in alkene diamination, the direct installation of an amino group in metal-catalyzed diamination has not been reported,5 likely because electron-rich free amines would lead to strong coordination to the metal and subsequent catalyst poisoning.2,3 So far, the introduction of an amino group has to be derived from nitrogen sources that are compatible with metal-catalyzed diaminations, such as ureas, sulfonamides, and anilines. Thus, the resulting amino group is restricted to primary and secondary amines, in many cases, an NH2 group.2,3 Furthermore, most intramolecular diamination reactions provided pyrrolidine-containing diamines. Few examples starting from unsaturated amide precursors were achieved for the synthesis of lactam-containing diamination products.8b Therefore, it is of great value to develop new alkene diaminations that can directly incorporate an amino group for the synthesis of more diverse 1,2-diamine skeletons.
Herein we report a copper-catalyzed alkene diamination that achieves, for the first time, the direct installation of an amino group via an intermolecular electrophilic amination step. Recently, our group reported the direct amination of sp2 and sp3 C–H bonds10via copper-catalyzed electrophilic amination using hydroxylamine as an amine source.11,12 We postulated that such an intermolecular electrophilic amination, in conjunction with copper-catalyzed aminocyclization, would offer an attractive alkene diamination strategy to achieve the direct addition of an amino group (Scheme 1, E). Particularly advantageous is the use of O-acylhydroxylamines, readily available from diverse amino precursors, with the dual role of an amine source and oxidant in the proposed diamination reaction. It bypasses the poisoning interference of a free amine group with the catalyst, offers a source for diverse amino groups, and eliminates the need of excess external oxidants required in an oxidative diamination reaction. Thus, the development of this new method will provide a rapid and efficient approach to 1,2-diamino skeletons, especially those containing cyclic and acyclic tertiary amines or lactam-based 1,2-diamines that would be inaccessible by other metal-catalyzed diaminations. Furthermore, the wide applicability of this strategy renders it highly valuable for the synthesis of important 1,2-diamines in catalysis, biological studies, and medicinal chemistry.
Results and discussion
Our studies began with the diamination of unsaturated amide 1a with O-benzoyl hydroxylmorpholine 2a (Table 1). Preliminary studies found that the desired diamination product was observed in 68% yield in the presence of Cu(OAc)2 catalyst in toluene at 80 °C (Table 1, entry 1). Among different solvents, MTBE was best for the formation of the desired product 3aa (entries 2–5). When different copper catalysts were examined, Cu(OAc)2 remained most effective, providing 3aa in 80% yield (entries 5–10). The control experiment proved that no product was formed in the absence of a copper catalyst (entry 11). During the studies, we recognized that the protecting group (PG) played an important role in this reaction. Similar to N-methoxy amide 1a, N-benzyloxy amide 1b smoothly reacted with hydroxylamine 2a, giving diamination product 3ba in 74% yield. However, similar substrates bearing other N-protecting groups such as the tosyl, benzyl, or the free amide did not afford the desired products (entries 13–15). These results suggested the critical role of alkoxyl group13 on the nitrogen, which might coordinate and stabilize the alkyl-copper intermediate resulting from the alkene aminocupration step.|
|
||||
|---|---|---|---|---|
| Entry | 1 | Catalyst | Solvent | 3, yieldb [%] |
| a Reaction conditions: 1a (0.20 mmol, 1.0 equiv.), 2a (1.2 equiv.), catalyst (10 mol%), K2CO3 (2.0 equiv.), solvent (1 mL), 2 h. b Yields were determined by 1H NMR with CH2Br2 as an internal standard. c The isolation yield was indicated in the parenthesis. d Not detected by GC/MS or 1H NMR. PG = protecting group. DCE = 1,2-dichloroethane. MTBE = methyl tert-butyl ether. | ||||
| 1 | 1a, PG = OMe | Cu(OAc)2 | Toluene | 3aa, 68 |
| 2 | 1a | Cu(OAc)2 | DCE | 3aa, 74 |
| 3 | 1a | Cu(OAc)2 | THF | 3aa, 48 |
| 4 | 1a | Cu(OAc)2 | CH3CN | 3aa, 70 |
| 5 | 1a | Cu(OAc) 2 | MTBE | 3aa, 84 (80) |
| 6 | 1a | Cu(CF3COO)2 | MTBE | 3aa, 60 |
| 7 | 1a | Cu(OTf)2 | MTBE | 3aa, 46 |
| 8 | 1a | Cu(acac)2 | MTBE | 3aa, 24 |
| 9 | 1a | CuCl2 | MTBE | 3aa, 39 |
| 10 | 1a | CuOAc | MTBE | 3aa, 60 |
| 11 | 1a | — | MTBE | 3aa, 0d |
| 12 | 1b, PG = OBn | Cu(OAc)2 | MTBE | 3ba, 81 (74)c |
| 13 | 1c, PG = Ts | Cu(OAc)2 | MTBE | 3ca, 0d |
| 14 | 1d, PG = Bn | Cu(OAc)2 | MTBE | 3da, 0d |
| 15 | 1e, PG = H | Cu(OAc)2 | MTBE | 3ea, 0d |
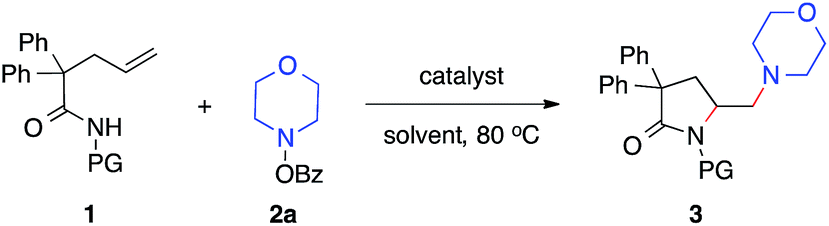
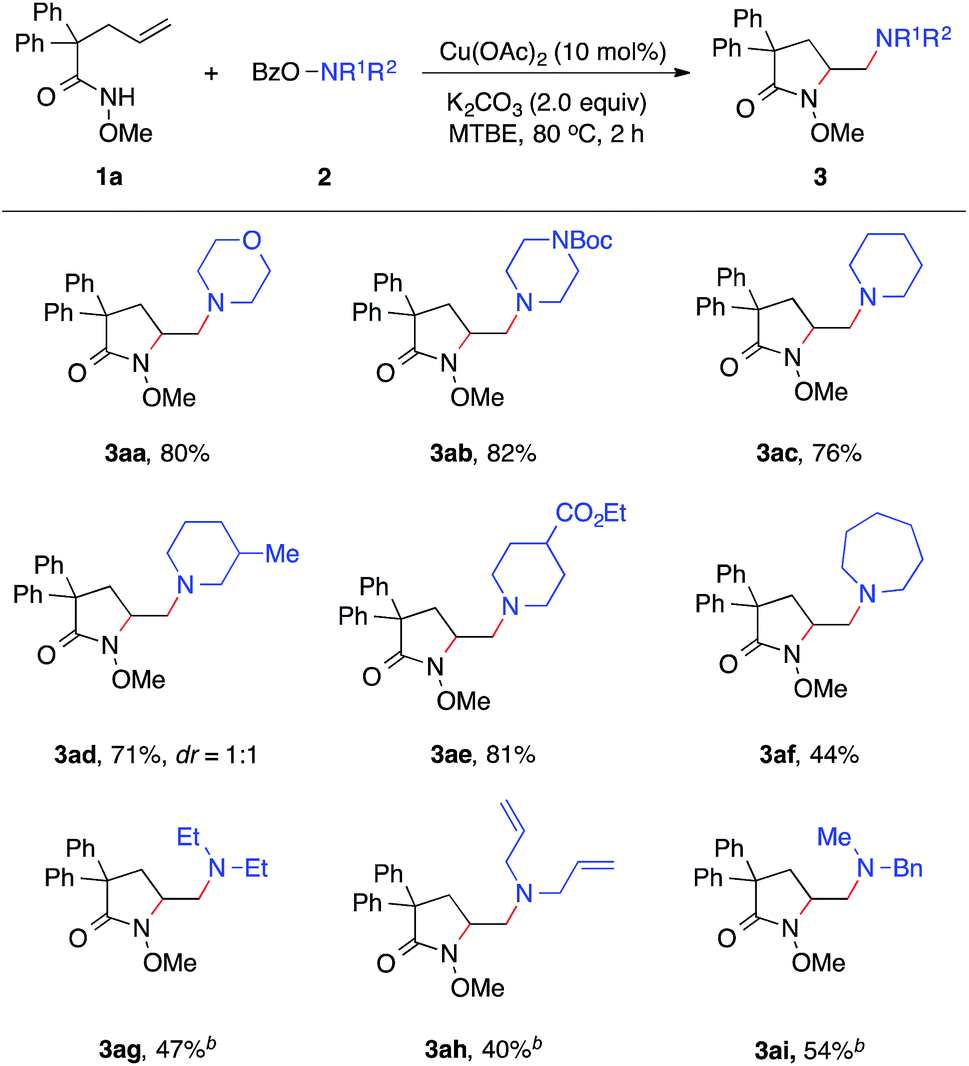
With established diamination conditions, we examined the scope of this alkene diamination using different hydroxylamines (Table 2). O-Benzoylhydroxylamines 2a–2e, derived from 6-membered cyclic amines such as morpholine, N-Boc piperazine and piperidines, all readily participated in the diamination reaction and gave corresponding 1,2-diamines 3aa–3ae in high yields. Azepane-derived 7-membered O-benzoylhydroxylamine 2f was also a viable substrate and afforded 3af in 44% yield. The reactions with O-benzoylhydroxylamines derived from acyclic amines, such as N,N-diethylamine, N,N-diallylamine and N-methyl-N-benzylamine, were found to be more effective under the conditions with a higher catalyst loading and elevated temperature.14 Nonetheless, all underwent the diamination reaction and successfully formed the corresponding 1,2-diamine products 3ag–3ai. Notably, the compatibility with benzyl and allyl group in these reactions offered opportunities for further transformations. For example, the cleavage of the allyl group or benzyl group can afford either a primary amine or a secondary amine.15
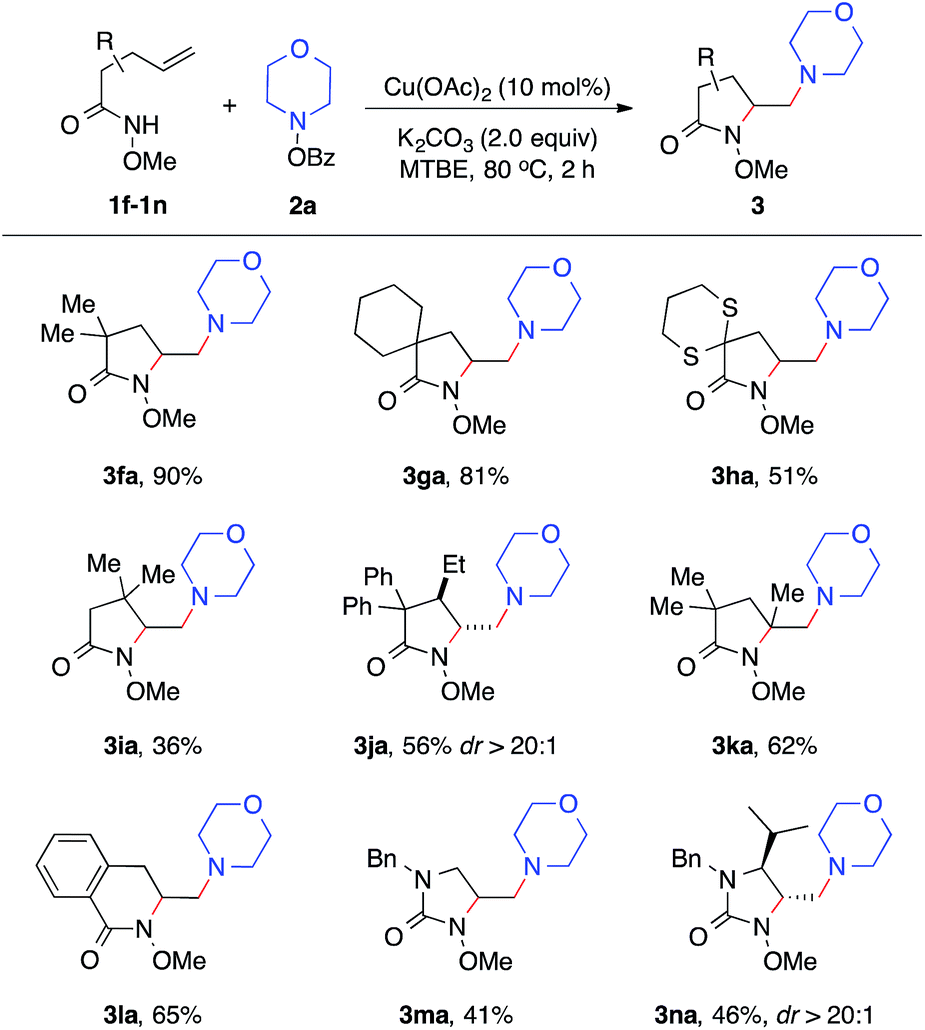
We next examined the alkene scope of the diamination reaction using hydroxylamine 2a (Table 3). Similar to 1a, monosubstituted alkenes 1f–1j bearing different substituents on the alkenyl chain all underwent smooth 5-exo cyclization and afforded the γ-substituted amino lactam products 3fa–3ja in modest to high yields. In addition, disubstituted alkene 3k also readily provided 3ka in 62% yield. The formation of 6-membered lactam 3la was also effective. Urea-based alkenes 1m–1n proved to be viable substrates for the diamination reaction and successfully provided desired amino-imidazolidinones 3ma–3na. It is important to note that this diamination reaction occurs with high diastereoselectivity, as observed in the formation of major products 3ja and 3na.
| a Reaction conditions: 1a (0.30 mmol, 1.0 equiv.), 2 (1.2 equiv.), Cu(OAc)2 (10 mol%), K2CO3 (2.0 equiv.), MTBE (1.5 mL), 80 °C, 2 h. |
|---|
|
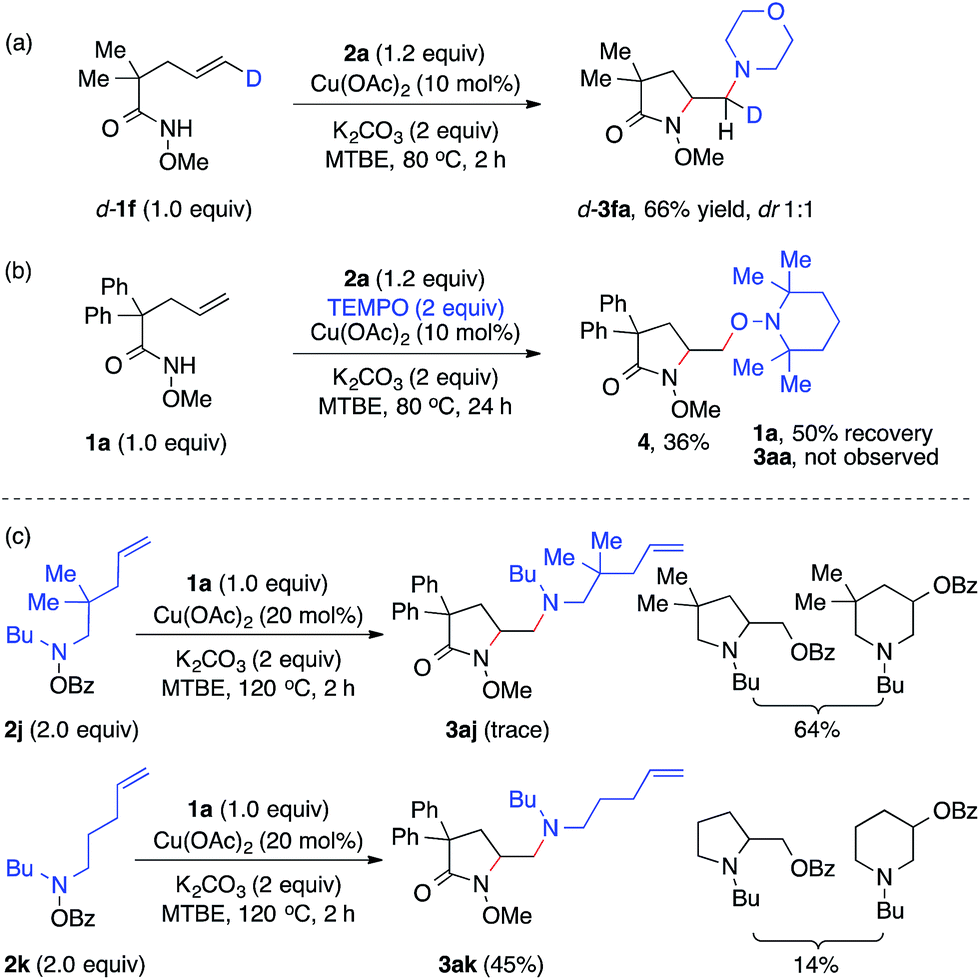
To study the nature of the intermolecular C–N bond formation step with hydroxylamines, the trans-d-substituted alkenyl amide d-1f was subjected to the standard diamination reaction, giving a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of d-substituted diaminated diastereomers d-3fa in 66% yield (Scheme 2, a). The loss of stereochemistry implies the radical nature of the alkyl-Cu complex that results from the intramolecular aminocupration step.16 Next when the reaction of model substrates 1a and 2a was performed in the presence of a radical scavenger TEMPO (Scheme 2, b), aminooxygenation product 4 was isolated in 36% yield and no diamination product 3aa was observed, indicating the radical intermediate was trapped by TEMPO.
:1 mixture of d-substituted diaminated diastereomers d-3fa in 66% yield (Scheme 2, a). The loss of stereochemistry implies the radical nature of the alkyl-Cu complex that results from the intramolecular aminocupration step.16 Next when the reaction of model substrates 1a and 2a was performed in the presence of a radical scavenger TEMPO (Scheme 2, b), aminooxygenation product 4 was isolated in 36% yield and no diamination product 3aa was observed, indicating the radical intermediate was trapped by TEMPO.
 | ||
| Scheme 2 Mechanism investigations. | ||
To investigate the involvement of the hydroxylamine-derived amino radical in the diamination reaction, we examined the reactions of alkene 1a using hydroxylamines 2j and 2k, both of which contain a tethered olefin for possible amino-radical-initiated cyclization (Scheme 2, c). The reaction with 2j only provided a trace amount of desired diamination product 3aj with a significant amount of aminooxygenation products.11b However, the reaction with 2k was much more effective and offered diamination product 3ak in 45% yield. These results suggested that amino radicals might be generated under the standard diamination conditions and the diamination reaction would be less competitive for those hydroxylamines that might undergo side reactions, such as facile aminocyclization (2j).
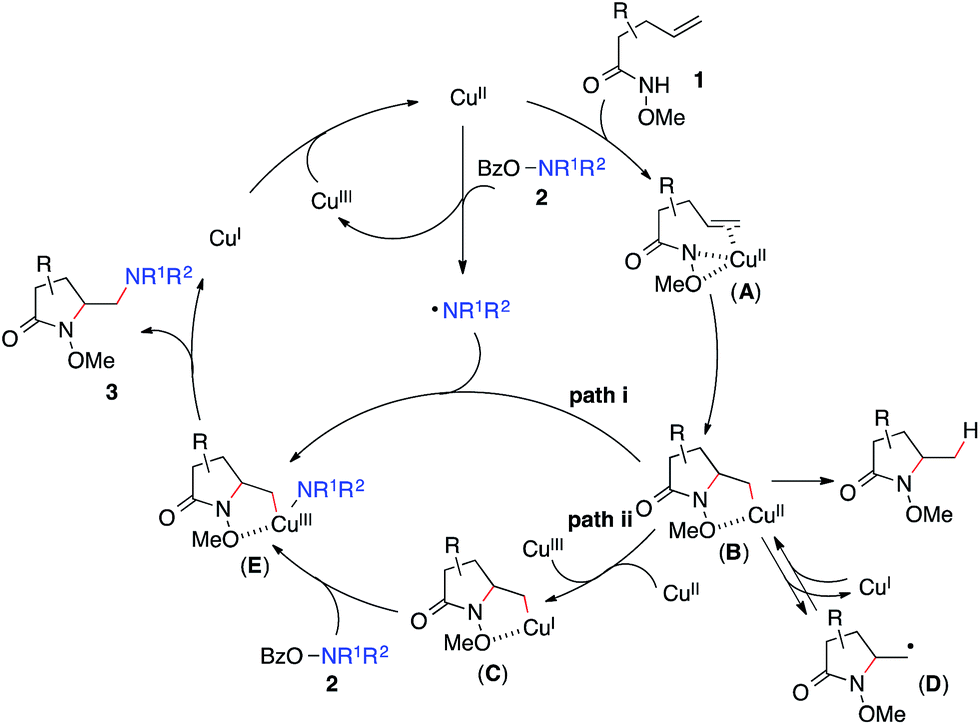
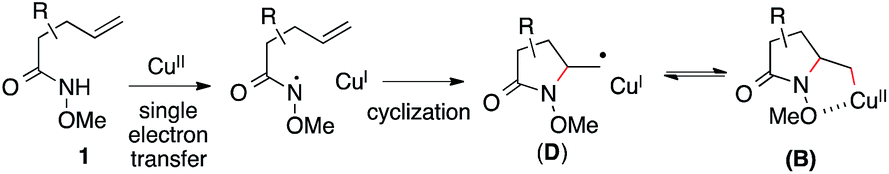
Based on these results, Scheme 3 outlines the possible pathways involved in this copper-catalyzed diamination reaction. First, the intramolecular aminocupration of alkene 1 will occur upon activation by a copper catalyst to form alkyl-Cu(II) complex B.17 We propose that the intermediate B could associate with the amino radical, which is generated from O-benzoylhydroxylamine 2 in the presence of a copper catalyst, to form Cu(III)–complex E (path i). Alternatively, the intermediate B may be reduced to Cu(I)–complex C, which can undergo a direct electrophilic amination with hydroxylamine 2 to form the same intermediate E (path ii). In either path, E would readily undergo reductive elimination, providing the diaminated product 3 and regenerating the Cu(II)-catalyst.18 The alkoxyl group of the amide, which was found to be critical for the diamination reaction (Table 1, entries 5 and 12–15), would contribute to stabilizing the alkyl-copper intermediates B and C, therefore facilitating the intermolecular amination. Furthermore, radical intermediate D may form from the intermediate B upon a reversible C–Cu(II) homolysis, thus abolishing the stereochemical control in the intermolecular amination step (Scheme 2, a).17 Besides the desired intermolecular amination, direct H-abstraction of B may occur to produce the hydroamination byproduct.
 | ||
| Scheme 3 Proposed mechanism. | ||
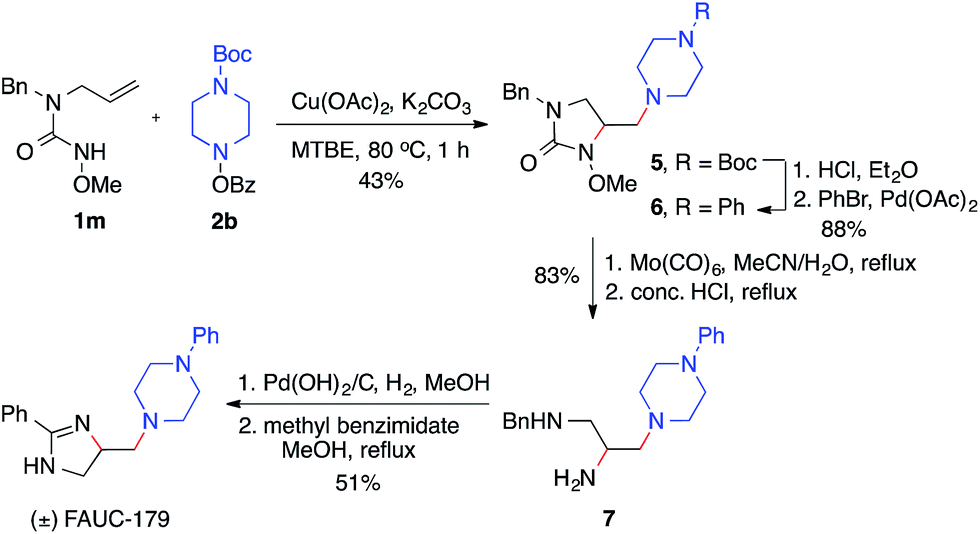
The utility of this diamination reaction for the synthesis of valuable diamine-containing agents was demonstrated by the rapid synthesis of FAUC-179, a selective dopamine D4 receptor partial agonist.19 As shown in Scheme 4, the preparation of FAUC-179 was readily achieved by using the diamination reaction of the simple alkene 1m, followed by removal of protecting groups and subsequent functionalizations. It is noteworthy that the methoxy protecting group on the nitrogen necessary for the diamination reaction can be easily cleaved by Mo(CO)6. This example also highlights the applicability of this diamination method in the synthesis of 1,2,3-triamines with distinct substitutions (e.g.7), which would be challenging for other diamination methods.
 | ||
| Scheme 4 Synthesis of FAUC-179. | ||
Conclusions
In summary, a copper-catalyzed intramolecular diamination reaction of unactivated alkenes with O-benzoylhydroxylamines has been developed. It is the first metal-catalyzed alkene diamination that enables the direct incorporation of an electron-rich amino group. The method offers a rapid and efficient approach to construct diverse 1,2-diamine skeletons, including biologically and medicinally important γ-lactams and imidazolidinones. Further studies of the reaction mechanism and the development of an enantioselective diamination procedure are currently underway in our laboratory.Acknowledgements
We acknowledge financial support of Duke University to this work.Notes and references
- (a) D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2581–2627 CrossRef; (b) S. R. S. S. Kotti, C. Timmons and G. G. Li, Chem. Biol. Drug Des., 2006, 67, 101–114 CrossRef CAS PubMed.
- Recent reviews on olefin diaminations, see: (a) V. G. Aranda, J. Barluenga and F. Aznar, Synthesis, 1974, 7, 504–505 CrossRef; (b) J. E. Backvall, Tetrahedron Lett., 1978, 19, 163–166 CrossRef; (c) S. De Jong, D. G. Nosal and D. J. Wardrop, Tetrahedron, 2012, 68, 4067–4105 CrossRef CAS PubMed; (d) K. Muñiz and C. Martiínez, J. Org. Chem., 2013, 78, 2168–2174 CrossRef PubMed.
- Recent reviews for metal-catalyzed olefin diaminations, see: (a) F. Cardona and A. Goti, Nat. Chem., 2009, 1, 269–275 CrossRef CAS PubMed; (b) R. M. de Figueiredo, Angew. Chem., Int. Ed., 2009, 48, 1190–1193 CrossRef CAS PubMed.
- Recent examples of metal-free diaminations: (a) C. Roben, J. A. Souto, Y. Gonzalez, A. Lishchynskyi and K. Muñiz, Angew. Chem., Int. Ed., 2011, 50, 2729–2731 CrossRef PubMed; (b) P. Chávez, J. Kirsch, C. H. Hövelmann, J. Streuff, M. Martínez-Belmonte, E. C. Escudero-Adán, E. Martin and K. Muñiz, Chem. Sci., 2012, 3, 2375–2382 RSC; (c) J. A. Souto, Y. González, A. Iglesias, D. Zian, A. Lishchynskyi and K. Muñiz, Chem. –Asian J., 2012, 7, 1103–1111 CrossRef CAS PubMed; (d) K. Muñiz, Pure Appl. Chem., 2013, 85, 755–761 CrossRef; (e) C. Röben, J. A. Souto, E. C. Escudero-Adán and K. Muñiz, Org. Lett., 2013, 15, 1008–1011 CrossRef PubMed; (f) R. M. Romero, T. H. Wöste and K. Muñiz, Chem. –Asian J., 2014, 9, 972–983 CrossRef CAS PubMed; (g) P. Mizar, A. Laverny, M. El-Sherbini, U. Farid, M. Brown, F. Malmedy and T. Wirth, Chem. –Eur. J., 2014, 20, 9910–9913 CrossRef CAS PubMed; (h) J. Zhang, W. Wu, X. Zhang, G. Zhang, S. Xu and M. Shi, Chem. –Asian J., 2015, 10, 544–547 CrossRef CAS PubMed; (i) J. Zhang, X. Zhang, W. Wu, G. Zhang, S. Xu and M. Shi, Tetrahedron Lett., 2015, 56, 1505–1509 CrossRef CAS PubMed; (j) H. Chen, A. Kaga and S. Chiba, Org. Lett., 2014, 6136–6139 CrossRef CAS PubMed.
- Examples for transition-metal-free alkene diaminations involving electron-rich amine groups, see: (a) C. H. Muller, R. Frohlich, C. G. Daniliuc and U. Hennecke, Org. Lett., 2012, 14, 5944–5947 CrossRef PubMed; (b) K. B. Hong and J. N. Johnston, Org. Lett., 2014, 16, 3804–3807 CrossRef CAS PubMed; (c) G. X. Ortiz Jr., B. Kang and Q. Wang, J. Org. Chem., 2014, 79, 571–581 CrossRef PubMed. Other related syntheses of 1,2-diamines incorporating aliphatic amine groups: (d) A. R. Ickes, S. C. Ensign, A. K. Gupta and K. L. Hull, J. Am. Chem. Soc., 2014, 136, 11256–11259 CrossRef CAS PubMed; (e) M. J. MacDonald, D. J. Schipper, P. J. Ng, J. Moran and A. M. Beauchemin, J. Am. Chem. Soc., 2011, 133, 20100–20103 CrossRef CAS PubMed.
- (a) J. Streuff, C. H. Hövelmann, M. Nieger and K. Muñiz, J. Am. Chem. Soc., 2005, 127, 14586–14587 CrossRef CAS PubMed; (b) K. Muñiz, J. Am. Chem. Soc., 2007, 129, 14542–14543 CrossRef PubMed; (c) K. Muñiz, J. Streuff, P. Chavez and C. H. Hovelmann, Chem. –Asian J., 2008, 3, 1248–1255 CrossRef PubMed; (d) K. Muñiz, C. Hövelmann, J. Streuff and E. Campos-Gomez, Pure Appl. Chem., 2008, 80, 1089–1096 CrossRef; (e) K. Muñiz, C. H. Hovelmann and J. Streuff, J. Am. Chem. Soc., 2008, 130, 763–773 CrossRef PubMed; (f) A. Iglesias, E. G. Perez and K. Muñiz, Angew. Chem., Int. Ed., 2010, 49, 8109–8111 CrossRef CAS PubMed; (g) C. Martínez, L. Martínez, J. Kirsch, E. C. Escudero-Adàn, E. Martin and K. Muñiz, Eur. J. Org. Chem., 2014, 2014, 2017–2021 CrossRef PubMed; (h) H. Li and R. A. Widenhoefer, Org. Lett., 2009, 11, 2671–2674 CrossRef CAS PubMed; (i) G. L. J. Bar, G. C. Lloyd-Jones and K. I. Booker-Milburn, J. Am. Chem. Soc., 2005, 127, 7308–7309 CrossRef CAS PubMed; (j) S. M. Fu, H. H. Yang, G. Q. Li, Y. F. Deng, H. F. Jiang and W. Zeng, Org. Lett., 2015, 17, 1018–1021 CrossRef CAS PubMed.
- (a) B. G. Zhao, W. C. Yuan, H. F. Du and Y. A. Shi, Org. Lett., 2007, 9, 4943–4945 CrossRef CAS PubMed; (b) B. G. Zhao, H. F. Du and Y. Shi, J. Am. Chem. Soc., 2008, 130, 7220–7221 CrossRef CAS PubMed; (c) B. G. Zhao, X. G. Peng, S. L. Cui and Y. A. Shi, J. Am. Chem. Soc., 2010, 132, 11009–11011 CrossRef CAS PubMed; (d) B. G. Zhao, X. G. Peng, Y. G. Zhu, T. A. Ramirez, R. G. Cornwall and Y. Shi, J. Am. Chem. Soc., 2011, 133, 20890–20900 CrossRef CAS PubMed; (e) R. G. Cornwall, B. G. Zhao and Y. A. Shi, Org. Lett., 2013, 15, 796–799 CrossRef CAS PubMed.
- (a) M. T. Bovino and S. R. Chemler, Angew. Chem., Int. Ed., 2012, 51, 3923–3927 CrossRef CAS PubMed; (b) F. C. Sequeira, B. W. Turnpenny and S. R. Chemler, Angew. Chem., Int. Ed., 2010, 49, 6365–6368 CrossRef CAS PubMed; (c) B. W. Turnpenny and S. R. Chemler, Chem. Sci., 2014, 5, 1786–1793 RSC.
- (a) P. A. Sibbald and F. E. Michael, Org. Lett., 2009, 11, 1147–1149 CrossRef CAS PubMed; (b) P. A. Sibbald, C. F. Rosewall, R. D. Swartz and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 15945–15951 CrossRef CAS PubMed; (c) E. L. Ingalls, P. A. Sibbald, W. Kaminsky and F. E. Michael, J. Am. Chem. Soc., 2013, 135, 8854–8856 CrossRef CAS PubMed.
- (a) S. L. McDonald and Q. Wang, Angew. Chem., Int. Ed., 2014, 53, 1867–1871 CrossRef CAS PubMed; (b) S. L. McDonald, C. E. Hendrick and Q. Wang, Angew. Chem., Int. Ed., 2014, 53, 4667–4670 CrossRef CAS PubMed; (c) S. L. McDonald and Q. Wang, Chem. Commun., 2014, 50, 2535–2538 RSC; (d) S. L. McDonald and Q. Wang, Synlett, 2014, 25, 2233–2238 CrossRef CAS PubMed.
- (a) S. Seko and N. Kawamura, J. Org. Chem., 1996, 61, 442–443 CrossRef CAS; (b) M. Noack and R. Göttlich, Chem. Commun., 2002, 536–537 RSC; (c) A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680–5681 CrossRef CAS PubMed; (d) A. M. Berman and J. S. Johnson, J. Org. Chem., 2005, 70, 364–366 CrossRef CAS PubMed; (e) A. M. Berman and J. S. Johnson, J. Org. Chem., 2006, 71, 219–224 CrossRef CAS PubMed; (f) K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 2395–2397 CrossRef CAS PubMed; (g) N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 2860–2863 CrossRef CAS PubMed; (h) N. Matsuda, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 11827–11831 CrossRef CAS PubMed; (i) S. L. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746–15749 CrossRef CAS PubMed; (j) Y. Miki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1498–1501 CrossRef CAS PubMed; (k) R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, J. Am. Chem. Soc., 2012, 134, 6571–6574 CrossRef CAS PubMed; (l) R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, Angew. Chem., Int. Ed., 2012, 51, 3953–3956 CrossRef CAS PubMed; (m) M. H. Nguyen and A. B. Smith, Org. Lett., 2013, 15, 4872–4875 CrossRef CAS PubMed.
- (a) E. J. Yoo, S. Ma, T.-S. Mei, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7652–7655 CrossRef CAS PubMed; (b) Z. Dong and G. B. Dong, J. Am. Chem. Soc., 2013, 135, 18350–18353 CrossRef CAS PubMed; (c) C. Grohmann, H. Wang and F. Glorius, Org. Lett., 2013, 15, 3014–3017 CrossRef CAS PubMed; (d) A. M. Berman and J. S. Johnson, Synthesis, 2005, 11, 1799–1801 Search PubMed; (e) T. J. Barker and E. R. Jarvo, Angew. Chem., Int. Ed., 2011, 50, 8325–8328 CrossRef CAS PubMed; (f) T. J. Barker and E. R. Jarvo, Synthesis, 2011, 3954–3964 CAS; (g) C. Grohmann, H. G. Wang and F. Glorius, Org. Lett., 2012, 14, 656–659 CrossRef CAS PubMed; (h) K.-H. Ng, Z. Zhou and W.-Y. Yu, Org. Lett., 2012, 14, 272–275 CrossRef CAS PubMed; (i) M. Shang, S. H. Zeng, S. Z. Sun, H. X. Dai and J. Q. Yu, Org. Lett., 2013, 15, 5286–5289 CrossRef CAS PubMed; (j) C. B. Huehls, A. J. Lin and J. Yang, Org. Lett., 2014, 16, 3620–3623 CrossRef CAS PubMed.
- The O-alkyl protecting group of amides is readily cleaved. See the ESI† for deprotection conditions.
- See the ESI† for more details.
- When O-benzoyl-N-n-butylhydroxylamine was used in this reaction, hydroamination product was isolated in 61% yield, suggesting that in this case the H-abstraction occurred faster than the desired intermolecular amination and this hydroxylamine might become the source for the H-abstracton.
- M. C. Paderes, L. Belding, B. Fanovic, T. Dudding, J. B. Keister and S. R. Chemler, Chem. –Eur. J., 2012, 18, 1711–1726 CrossRef CAS PubMed.
- The amidyl radical cyclization catalyzed by copper is another possible pathway for the formation of the intermediate B
. - The direct trapping of radical D can't be excluded for the intermolecular C–N bond forming step.
- J. Einsiedel, H. Hubner and P. Gmeiner, Bioorg. Med. Chem. Lett., 2001, 11, 2533–2536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and experimental procedures. See DOI: 10.1039/c5sc00897b |
| This journal is © The Royal Society of Chemistry 2015 |