
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple electrochemical sensing of attomolar proteins using fabricated complexes with enhanced surface binding avidity†
Chao
Li
a,
Xiaoxi
Li
a,
Luming
Wei
a,
Muyun
Liu
a,
Yangyang
Chen
b and
Genxi
Li
*ab
aState Key Laboratory of Pharmaceutical Biotechnology, Department of Biochemistry, Nanjing University, 210093, China. E-mail: genxili@nju.edu.cn
bLaboratory of Biosensing Technology, School of Life Sciences, Shanghai University, 200444, China
First published on 5th May 2015
Abstract
Various strategies have been proposed for the detection of disease protein biomarkers; however, most methods are too expensive, cumbersome or limited in sensitivity for clinical use. Here, we report that a fabricated complex can be used as a powerful tool to detect trace proteins in complex samples. In this strategy, a DNA–protein complex that comprises of one target molecule and two or more deoxyribozyme-containing probes can exhibit autonomous cleavage behavior on the surface of the substrate DNA modified electrode. In the meantime, the complex can remove the cleaved DNA fragment from the electrode surface by taking advantage of the proximity effect. The proposed approach allows one-step and highly sensitive detection of a variety of targets based on the changes of the direct electrochemical readout. Moreover, this method may also have considerable advantages over the commonly reported DNA amplification-assisted immunoassays, particularly in terms of assay simplicity and cost, which may hold great potential for application in resource-constrained regions.
Introduction
Simple and cost-effective methods for detecting protein biomarkers at ultralow concentrations are of great importance for early disease diagnosis. However, since there is no polymerase chain reaction (PCR)-like technique to amplify protein molecules as there is for nucleic acids, the analyzing of proteins of low abundance still faces great challenges. To circumvent this problem, the coupling of specific ligands (antibody, aptamer, etc.) with DNA amplification-assisted strategies such as immune-PCR,1,2 rolling circle amplification (RCA),3,4 liposome-PCR,5 and nanoparticle-based barcode6,7 has been developed and recognized as a powerful tool to improve the sensitivity for protein detection. Nevertheless, these methods still require the help of expensive reagents and instruments, complex experimental procedures, and skilled labour, which may limit their applications in resource-constrained countries. Therefore, it is urgent to develop simple and cost-effective approaches for ultrasensitive protein detection.Here, we demonstrate for the first time that a DNA enzyme (DNAzyme)-based mechanical device, a fabricated complex which autonomously cleaves the substrate modified electrode surface, can be used to detect disease biomarkers with high sensitivity and selectivity in complex real samples. DNA walkers, a class of nucleic acid nanomachines that move along a linear track or a two-dimensional surface, have received considerable attention.8–11 Among them, one kind of DNA walker that makes use of deoxyribozyme to cleave the substrate strands is particularly attractive due to its advantage of running autonomously.12–14 This kind of DNA walker has also been explored to perform some machine-like tasks in cargo transport and biosynthesis.15,16 However, how to render these nanomachines more specific to the task still remains elusive. With a different perspective, we envision that this fabricated complex may be utilized as a signal translator and allow high sensitivity, specificity and efficiency toward protein detection with a very low cost due to (1) the advantages of DNAzyme itself such as excellent catalytic activity, inherent signal amplification achieved by multiple enzymatic turnovers, better thermal stability and economics than their counterpart protein enzymes, (2) autonomous cleavage of the complex, which makes it possible to realize one-step and washing-free detection and minimize the operational difficulty, and (3) easy integration with the kind of detecting platform aroused from the reliable working ability for both homogenous solution and heterogeneous interface.
Results and discussion
The principle of the fabricated complex-based electrochemical assay (FCEA) is shown in Scheme 1. The complex comprises of one target molecule as an inert body and at least two proximity probes as functional elements, which contain a specific affinity ligand for binding to the target protein and an 8–17 DNAyzme tail sequence. The metal ion-dependent DNAzyme used here can bind and cleave oligonucleotide substrates into two shorter products that have lower affinities for the DNAzyme and, finally, result in release of the products.17,18 Electrochemistry is adopted as the signal readout owing to the fact that it is a simple, portable, and economical platform.19–21 Moreover, the tuneable and convenient surface modification makes it easy to obtain a reliable working platform. As shown in Scheme 1, a short oligonucleotide, as the substrate of the DNAzyme, is first immobilized onto a gold electrode through an alkanethiol linker at one end of the strand, while the other end is labelled with a redox probe (methylene blue, MB), which can provide a better shelf life and performance in serum compared to other electrochemical species (e.g., ferrocene) according to a previous report.22 The DNAzyme is designed to be complementary to the surface-tethered DNA strands with a predesigned low melting temperature (Tm < 25 °C). In the absence of the target protein, the proximity probes cannot associate with the surface-tethered strands because the complementary fragment is too short to promote effective annealing. However, in the presence of the target protein, the binding of the target molecule to the proximity probes simultaneously brings the tail sequences into close proximity, with their local concentration increased dramatically, to allow the DNAzyme sequences to hybridize with the surface-tethered substrates.23–25 Then, the DNAzyme effectively cleaves the surface-tethered substrate DNA into two short pieces, resulting in removal of the redox label from the electrode surface, thus reducing electron transfer efficiency. After cleavage, the single dissociated probe will quickly explore neighbouring sites until it finds another intact substrate to reattach to and cleave. This ensures that the fabricated complex continuously cleaves the substrate DNA on the electrode surface and eliminates many redox labels, thus leading to considerable signal amplification eventually.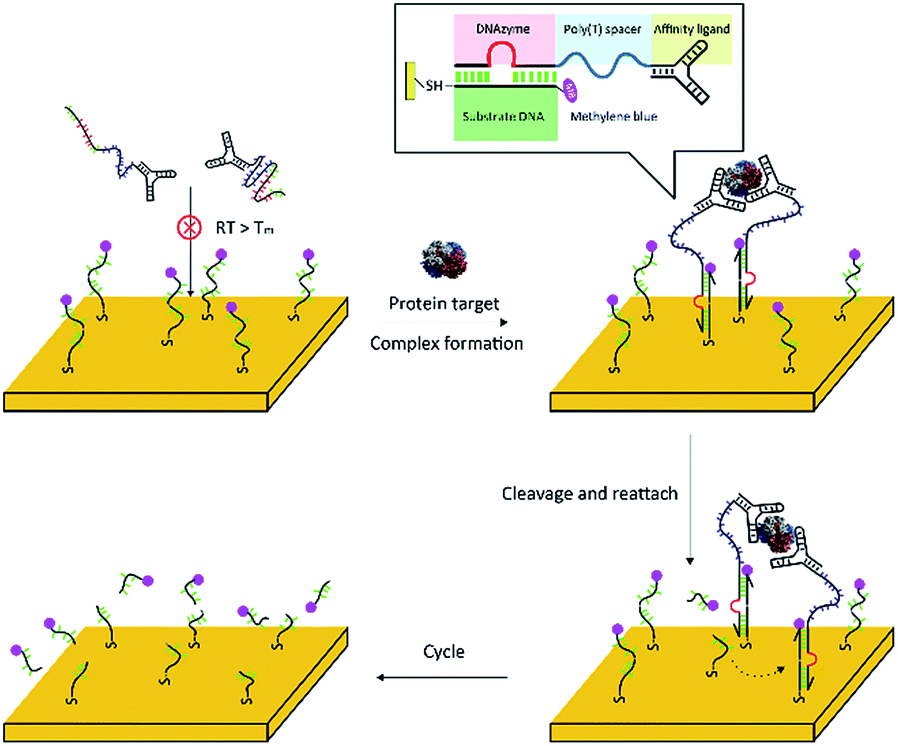 | ||
| Scheme 1 Schematic illustration of the principle of the fabricated complex-based electrochemical assay (FCEA), where here a two-probe complex is taken as an example. | ||
To test the feasibility of our strategy, we have first designed a four-proximity probe complex that consists of one streptavidin molecule and four biotin labelled proximity legs. We believe that this complex may be easily formed and have extremely high work efficiency owing to the robust interaction of the streptavidin–biotin system (Kd = 10−15 M) and its polycatalytic behavior. Fig. 1A depicts the cyclic voltammetry (CV) voltammogram of a gold electrode modified with the substrate oligonucleotides. In the absence of streptavidin, a typical redox peak of MB is observed in the CV curve, evidencing that the cleavage events resulting from the free proximity probes will be performed with extremely low efficiency. After incubation of the electrode with 1 pg mL−1 streptavidin and 1 nM proximity probe for 1 h, the MB peak pair disappears, thereby demonstrating the occurrence of cleavage events aroused from the fabricated complexes. In addition to CV, we have also used electrochemical impedance spectroscopy (EIS) to characterize the surface, since EIS is highly sensitive to the changes occurring at the electrode surface.26,27 As shown in Fig. 1B, the EIS of the bare electrode exhibits an almost straight line. After conjugation of the substrate oligonucleotides, electron transfer resistance (Ret) increases significantly because of the formation of a negatively charged DNA layer on the electrode surface. Furthermore, streptavidin results in further increase in Ret owing to association of the complex onto the electrode surface. Nevertheless, after incubation of the electrode with Pb2+-containing buffer, the electrochemical impedance dramatically decreases, indicating that the target-induced complex can cleave surface-tethered DNA and the DNA fragments are dissociated from the electrode surface eventually. To directly confirm that the cleavage is indeed streptavidin induced, we have also modified a tetramethylrhodamine (TAMRA) dye labelled DNA onto glass slides and conducted fluorescence studies. Similar to the results of the electrochemistry studies, the fluorescence at the glass surface only changes when the slide is incubated with streptavidin and proximity probes simultaneously (Fig. 1C). In addition, to better understand the kinetic process of this sensor, the progression of a typical experiment using surface plasmon resonance (SPR) is shown in Fig. 1D. Taken together, all of the measurements strongly support that a small number of fabricated complexes can soundly work on the solid surface.
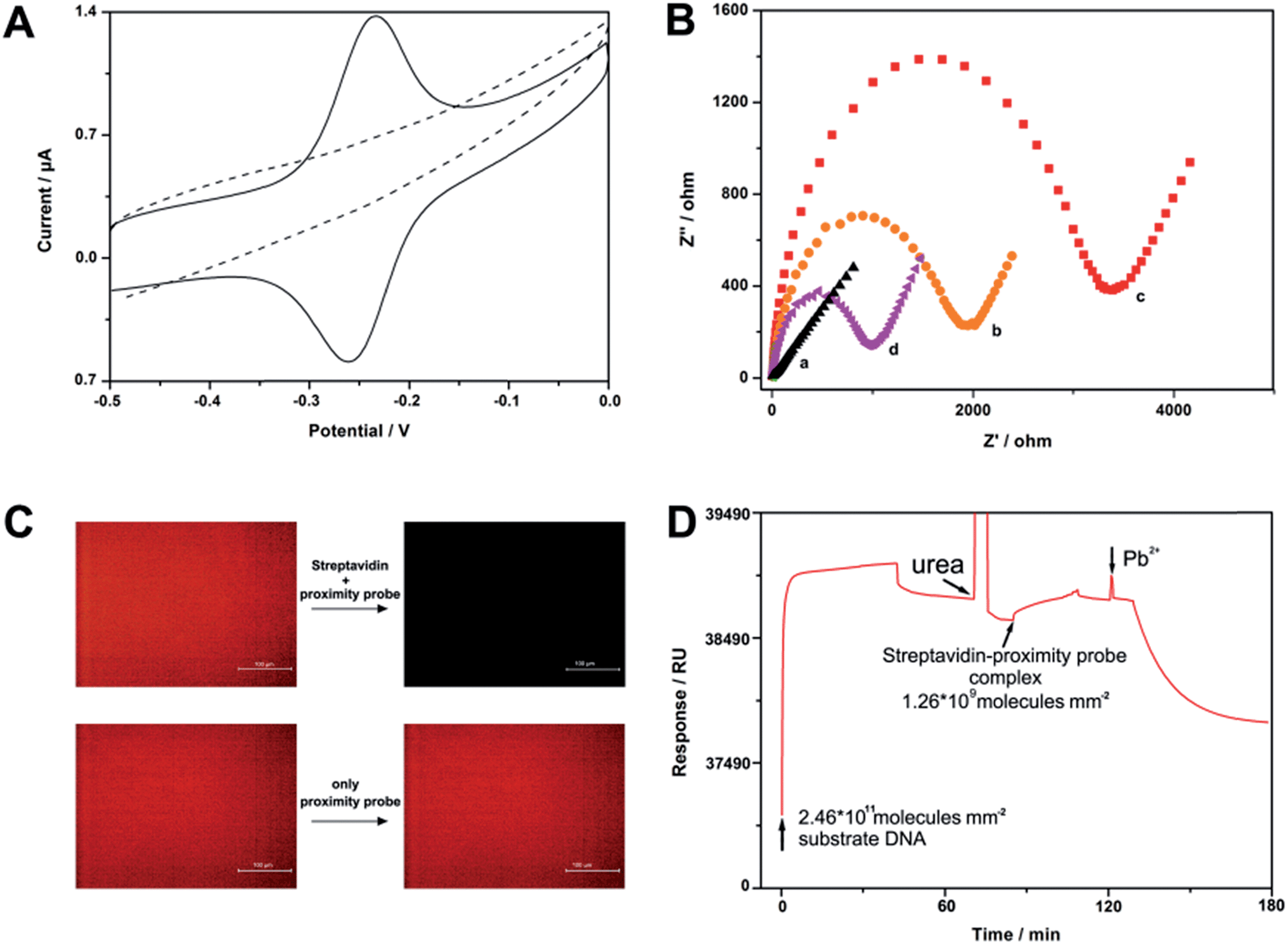 | ||
| Fig. 1 Proof of the feasibility of the FCEA. (A) CV curves of the sensor in 10 mM Tris–HCl before (solid line) and after (dashed line) reaction with 1 pg mL−1 of streptavidin. Potential scanning rate is 100 mV s−1. (B) EIS of (a) the bare electrode, (b) the electrode immobilized with substrate DNA, and the modified electrode after (c) incubation with 1 pg mL−1 streptavidin and 1 nM proximity probe for 30 min and (d) further incubation with Pb2+ containing buffer. Of note, no MB modified substrate DNA is used here. (C) Fluorescence study of the sensor with or without incubation with streptavidin. (D) Time course of substrate DNA modification, fabricated complex deposition and product formation and release. First, a substrate is attached, resulting in an increase in mass. Next, a nonspecifically bound substrate is desorbed by urea (leaving the substrate at Δ = 2311.1 RU, or 2.46 × 1011 molecules per mm2); then fabricated complexes are attached (Δ = 229.8 or 1.26 × 109 molecules per mm2). Finally, cleavage is initiated by the addition of a Pb2+-containing buffer and is visualized by a release of the product and a decrease in mass on the gold chip. Response units (RU) can be converted to mass using the standard formula 1000 RU = 1 ng mm−2. All experiments were performed using a “5 + 6” substrate DNA modified electrode and the corresponding proximity probe (1 nM). | ||
A key concept to our success in this study is the binding-induced cleavage event that is accelerated upon target binding and minimized in the absence of target binding. For this purpose, the following factors may play major roles in controlling the rate of the whole reaction and can potentially influence the final performance of the sensor: the length of the substrate DNA/DNAzyme duplex, the concentration and length of the proximity probes, the reaction time, the temperature and the salt concentration. As shown in Fig. 2A, we have first optimized the design of the proximity oligonucleotide that consists of a recognition ligand, spacer and DNAzyme tail. In this study, the 3′ side of the DNAzyme is fixed to be 5 nucleotides (nt) long, and the 5′ side ranges from 5 nt to 10 nt (more details see Table S1†). In principle, a shorter DNAzyme sequence can significantly reduce nonspecific cleavage events, but a too short sequence may fail to obtain specific signal intensities, while an excess length of DNA duplex will yield a strong false electrochemical signal, resulting from bypassing the effect of the target molecules. Therefore, we have chosen a tail sequence that has 11 nt complementary to substrate DNA.
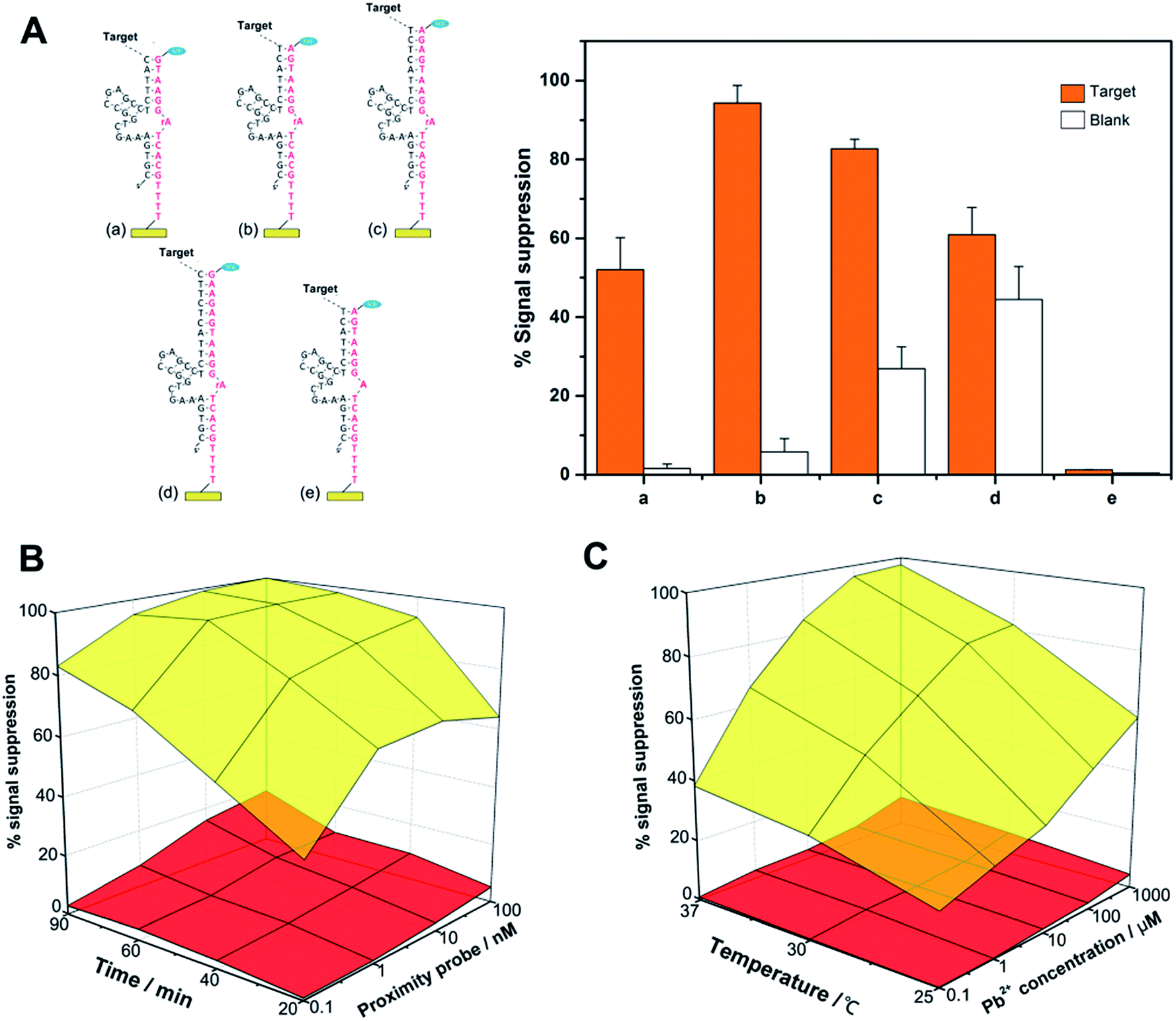 | ||
| Fig. 2 Optimization of reaction conditions for the FCEA. (A) Four designs of proximity probes and the corresponding substrate DNA, showing 5 + (a) 5, (b) 6, (c) 8 and (d) 10 base pairing sequences. Meanwhile, all-deoxyribose substrate strand (e) fails to respond. The blank groups are used as negative controls where no protein is included to determine background noise. (B) Effect of proximity probe concentration and time of reaction on the signal and background. (C) Influence of Pb2+ concentration and temperature. The upper, yellow layers connect the signals recorded in reactions with 1 pg mL−1 streptavidin, while the lower, red layers represent the target-independent background. Relative sensor response (%) is shown because this is more reproducible electrode-to-electrode than the absolute current change. | ||
Next, we employed different concentrations of proximity probe (0.1, 1, 10, and 100 nM) to incubate with a fixed amount of streptavidin or with no streptavidin for 20, 40, 60, or 90 min. As shown in Fig. 2B, increased detection sensitivity can be obtained if streptavidin-independent cleavage events are limited by using a minimal amount of probe, and an extended incubation time hardly caused an increase for both signal and background. Therefore, a proximity probe concentration of 1 nM was selected for the subsequent experiments, and the whole reaction can be completed in 60 min.
We have also examined the effects of incubation temperature and the concentration of Pb2+, which may directly influence the kinetics of strand binding and the cleavage of DNAzyme. From Fig. 2C, it is known that these two factors can act as a ‘lubricant’ for the fabricated complex, whose performance is significantly enhanced by increasing the incubation temperature (e.g., 37 °C) and the Pb2+ concentration (e.g., 100 μM).
After optimization of the assay parameters, we have thus made use of square-wave voltammetry (SWV) to quantitatively determine different concentrations of streptavidin. Fig. 3A shows typical SWV curves with a sharp reduction peak appearing at −0.27 V, and the peak current gradually decreases along with the increase of streptavidin concentration. Fig. 3B (red plot) represents the relative signal response of current intensity as a function of streptavidin concentration, which clearly shows that the current intensity increases when the concentration of streptavidin decreases in the range between 1.5 aM and 15 fM. Conversely, control experiments performed with bovine serum albumin (BSA) have a negligible effect on the current intensity (green plot). Since the sample volume for the current assay is 100 μL, the minimum amount of target protein needed for detection is 1.5 amol (or 93 molecules). This excellent sensitivity may be derived from two aspects. First, the enzymatic multiple turnover of DNAzyme is the fundamental guarantee of sensitivity. Second, there is no need to cleave all of the substrates to obtain substantial signal response, since a detectable current signal cannot be obtained as long as the surface-tethered electrochemical species reduce to a certain extent. Streptavidin has also been analyzed in the presence of human serum (HS), fetal calf serum (FCS), and saliva. These experimental results can demonstrate the applicability of the assay to a complex biological fluid (Fig. 3C). Furthermore, we have also found that the salt concentration (e.g., [NaCl]) can be adjusted to achieve an optimal dynamic range for detection (Fig. 3D), which is probably due to the shielding effect on the negative charge of the DNA that makes the fabricated complexes more easy to diffuse and cleave on the electrode surface.
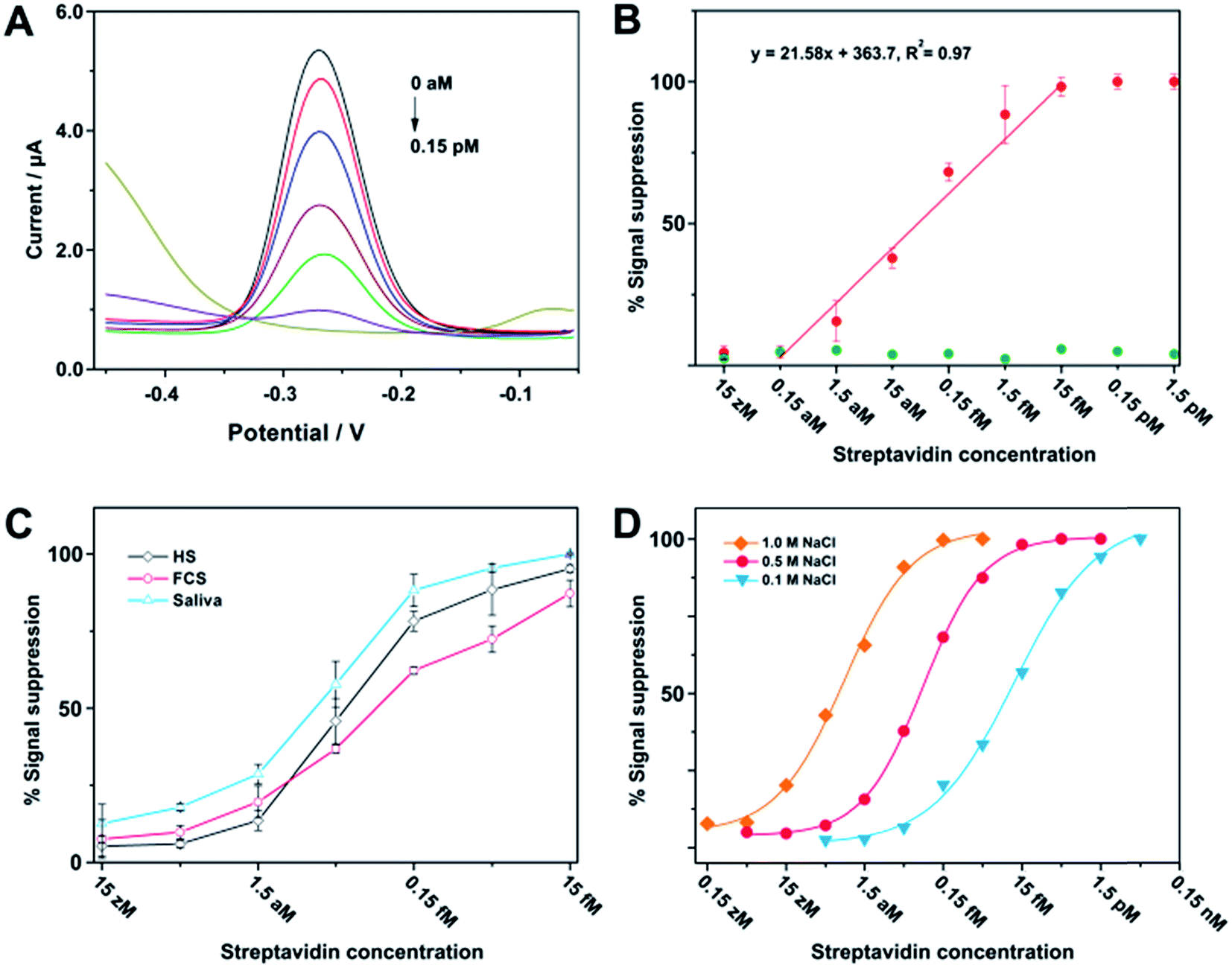 | ||
Fig. 3 Sensor performances for streptavidin detection. (A) Voltammograms obtained with SWV showed that the decrease in faradaic current is proportional to serial dilution of streptavidin (from top to bottom: 0, 1.5, 15, 150, 1500, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 150000 aM). (B) A dose–response curve for the detection of streptavidin (red) and BSA (green) in PBS. (C) Detection of streptavidin in the presence of complex biological fluids containing HS, FCS, and human saliva. (D) Streptavidin detection in PBS buffer with different salt concentrations. 000, 150000 aM). (B) A dose–response curve for the detection of streptavidin (red) and BSA (green) in PBS. (C) Detection of streptavidin in the presence of complex biological fluids containing HS, FCS, and human saliva. (D) Streptavidin detection in PBS buffer with different salt concentrations. | ||
Encouraged by the outstanding sensitivity of our FCEA, we then tested the general applicability of our strategy to the analysis of several clinically relevant proteins: platelet derived growth factor BB (PDGF-BB), vascular endothelial growth factor (VEGF), prostate-specific antigen (PSA), carcino embryonic antigen (CEA), α-fetoprotein (AFP), and troponin I. It is known that VEGF, PSA, CEA and AFP are biomarkers correlating to the presence of a cancerous tumor in the body,26 and troponin I is presently the gold-standard molecular marker for the diagnosis of acute myocardial infarction (AMI).27 In the meanwhile, we have also made use of the emerging and traditional ligands (aptamer vs. antibody) to demonstrate the versatility of our method, since PDGF-BB has a pair of specific aptamers, while the other proteins employ their corresponding antibodies for recognition. As shown in Fig. 4, all the assays perform equally well no matter whether DNA aptamer or antibody is used as the specific ligand, and the LOD for all the six analytes in the 100 μL samples is in the attomolar sensitivity range, for which the blank signals are all restricted to under 25% (see Table S2†). In contrast, there is no obvious signal response when only one aptamer or antibody-containing proximity probe is used, emphasizing the role of proximity-dependent hybridization (see Fig. S1†). Considering the most detected protein molecules only employ a pair of ligands, we have fabricated the complex with two catalytic units instead of using a streptavidin-based complex for the detection of the six disease-related protein molecules, so they have a lower work efficiency than those assembled by streptavidin, leading to a comprised sensitivity to some extent. Nevertheless, compared with commercial enzyme-linked immunosorbent assay (ELISA) kits, the fabricated complex-based assays still exhibit 1 × 103 to 1 × 104 times higher sensitivities and a wider dynamic range. Meanwhile, their sensitivities can also be compared favorably with other reported sensors based on DNA-assisted amplification strategies.1–4
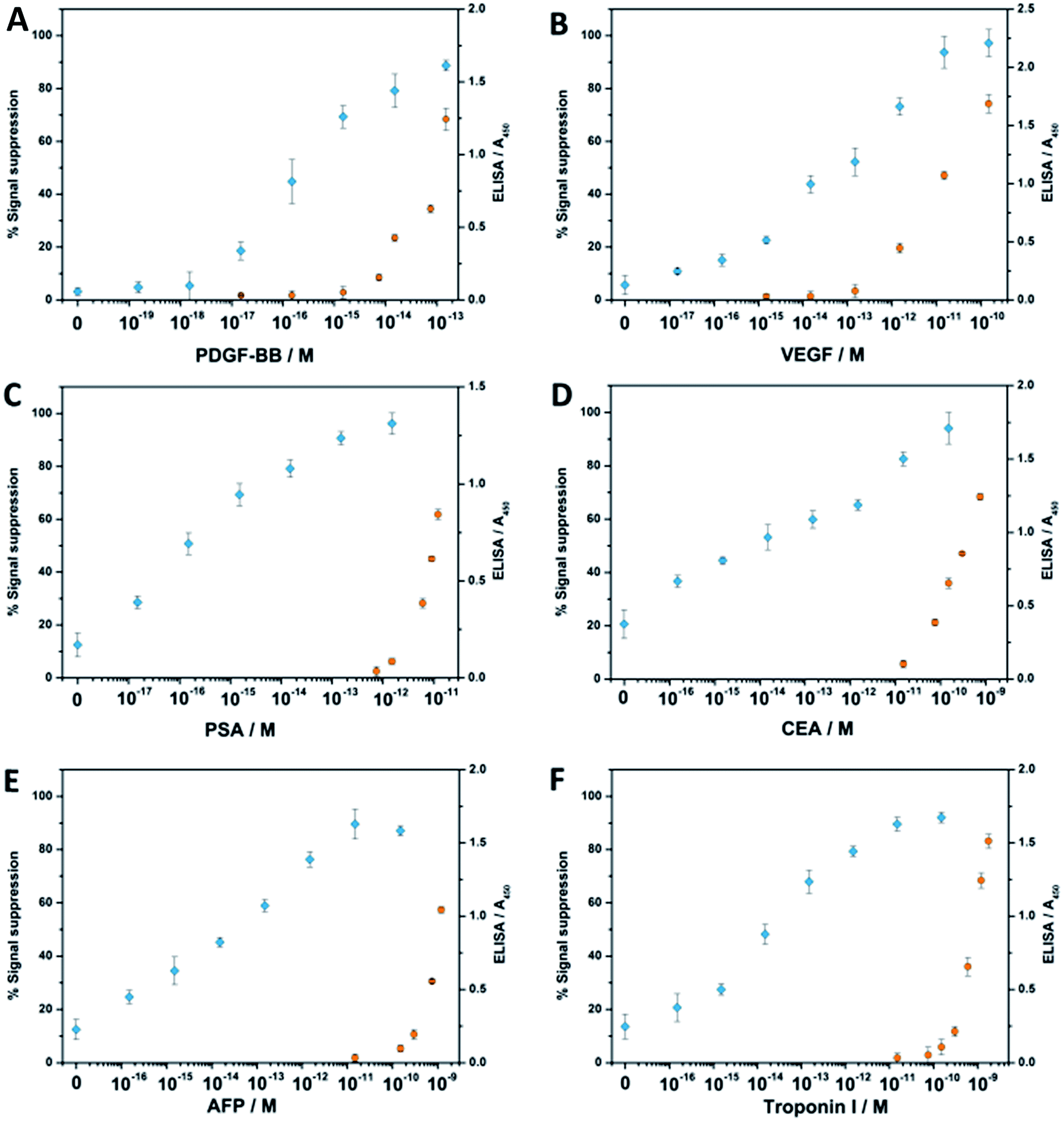 | ||
| Fig. 4 Comparison of the FCEA (blue) with sandwich ELISA (orange) for the detection of PDGF-BB, VEGF, PSA, CEA, AFP, and troponin I. All analyses were performed in the presence of 50% blood serum. The y axes display the relative signal response for the FCEA or absorbance at 450 nm for the ELISA. All measurements were performed at least in triplicate. Error bars indicate SD. | ||
We then investigated the assay performance for clinical diagnoses of 15 clinically positive AMI patients (see Table S3†). In the fabricated complex-based assay (Fig. 5A), only one AMI patient serum (no. 2) was not positively detected, thereby indicating 93.3% sensitivity. However, the commercial ELISA kits detect true positive signals for only 10 of the AMI patients (Fig. 5B). Moreover, nearly half of the signals (no. 1, 4, 8, and 13) are very close to the clinical cutoff value (indicated by the horizontal dotted line). The poor results obtained by ELISA are not surprising, because troponin I itself is easily subject to degradation by proteases and the clinical specificity of the ELISA kit is reported by the supplier to be 87.5%. Meanwhile, the ELISA needs more time and only has moderate detection sensitivity, making it not suitable for early detection of AMI.
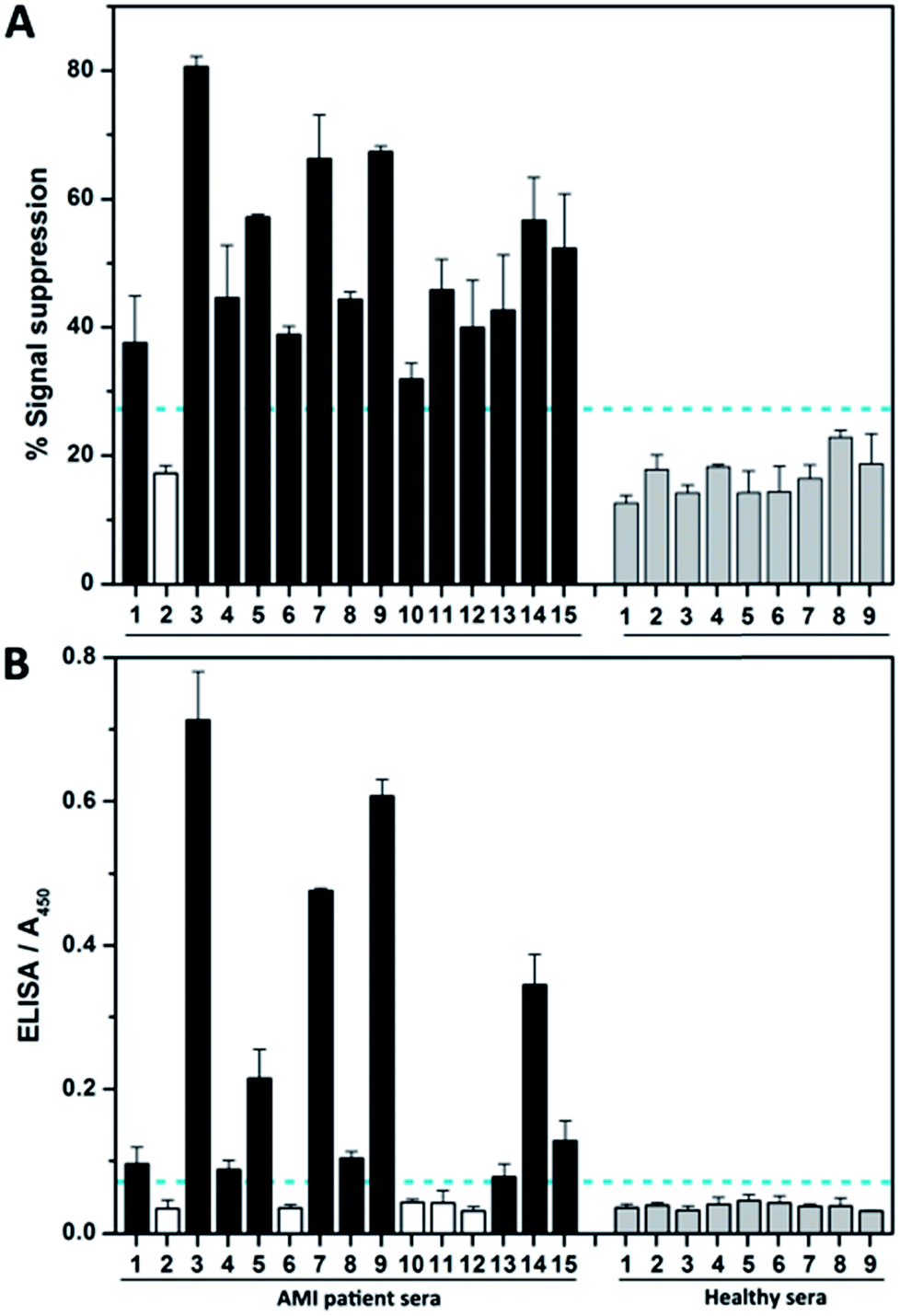 | ||
| Fig. 5 Clinical detection of troponin I. Clinical samples collected from 15 AMI patients and 9 healthy individuals (control) were used to evaluate the detection performance of (A) the FCEA proposed in this work and (B) the conventional HRP-based ELISA. The FCEA only fails to detect one patient, but the ELISA-based diagnosis fails to detect five patients (no. 2, 6, 10, 11, 12) and reveals four ambiguous signals (no. 1, 4, 8, 13) close to the clinical cutoff value (horizontal dotted line). The cutoff value of each assay was determined according to the IUPAC definition: average of the detection signals from healthy sera (negative control samples) + 3 × standard deviation of negative control signals from healthy sera. | ||
Conclusions
In summary, we have proposed a simple and highly sensitive method for protein assay, named as FCEA, based on the fabrication of a DNA–protein complex and its cleavage on the surface of an electrode immobilized with the substrate DNA. In this method, the fabricated complex, by taking advantage of the principle of proximity-dependent DNA hybridization, can remove the electrochemical signal molecules from the electrode surface. The experimental results presented here have also validated the detection capabilities of the proposed method and open new avenues for developing DNA fabrication-based detection strategies. Compared with other DNA amplification-based immunoassays, our method has two significant advantages: (1) it abandons the expensive and cumbersome thermal cycling protocol, making the proposed method cost-efficient and easy for use in clinical applications; (2) it fully makes use of DNA-based nanotechnology and interfacial sensors to develop a novel powerful tool to detect a variety of protein targets, increasing the assay sensitivity and clinical specificity as compared to the conventional ELISA assay.Acknowledgements
This work is supported by the National Natural Science Foundation of China (grant nos 21235003, J1103512), and the National Science Fund for Distinguished Young Scholars (grant no. 20925520).Notes and references
- C. M. Niemeyer, M. Adler and R. Wacker, Trends Biotechnol., 2005, 23, 208–216 CrossRef CAS PubMed.
- T. Sano, C. L. Smith and C. R. Cantor, Science, 1992, 258, 120–122 CAS.
- B. Schweitzer, S. Roberts, B. Grimwade, W. P. Shao, M. J. Wang, Q. Fu, Q. P. Shu, I. Laroche, Z. M. Zhou, V. T. Tchernev, J. Christiansen, M. Velleca and S. F. Kingsmore, Nat. Biotechnol., 2002, 20, 359–365 CrossRef CAS PubMed.
- L. Zhou, L.-J. Ou, X. Chu, G.-L. Shen and R.-Q. Yu, Anal. Chem., 2007, 79, 7492–7500 CrossRef CAS PubMed.
- J. T. Mason, L. X. Xu, Z. M. Sheng and T. J. O'Leary, Nat. Biotechnol., 2006, 24, 555–557 CrossRef CAS PubMed.
- J. M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS PubMed.
- S. I. Stoeva, J. S. Lee, J. E. Smith, S. T. Rosen and C. A. Mirkin, J. Am. Chem. Soc., 2006, 128, 8378–8379 CrossRef CAS PubMed.
- J. Bath and A. J. Turberfield, Nat. Nanotechnol., 2007, 2, 275–284 CrossRef CAS PubMed.
- M. K. Beissenhirtz and I. Willner, Org. Biomol. Chem., 2006, 4, 3392–3401 CAS.
- J. S. Shin and N. A. Pierce, J. Am. Chem. Soc., 2004, 126, 10834–10835 CrossRef CAS PubMed.
- W. B. Sherman and N. C. Seeman, Nano Lett., 2004, 4, 1203–1207 CrossRef CAS.
- Y. Tian, Y. He, Y. Chen, P. Yin and C. D. Mao, Angew. Chem., Int. Ed., 2005, 44, 4355–4358 CrossRef CAS PubMed.
- R. Pei, S. K. Taylor, D. Stefanovic, S. Rudchenko, T. E. Mitchell and M. N. Stojanovic, J. Am. Chem. Soc., 2006, 128, 12693–12699 CrossRef CAS PubMed.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206–210 CrossRef CAS PubMed.
- T.-G. Cha, J. Pan, H. Chen, J. Salgado, X. Li, C. Mao and J. H. Choi, Nat. Nanotechnol., 2014, 9, 39–43 CrossRef CAS PubMed.
- Y. He and D. R. Liu, Nat. Nanotechnol., 2010, 5, 778–782 CrossRef CAS PubMed.
- Y. Lu and J. Liu, Curr. Opin. Biotechnol., 2006, 17, 580–588 CrossRef CAS PubMed.
- C. Li, L. Wei, X. Liu, L. Lei and G. Li, Anal. Chim. Acta, 2014, 831, 60–64 CrossRef CAS PubMed.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS PubMed.
- C. Li, Z. Wang, T. Gao, A. Duan and G. Li, Chem. Commun., 2013, 49, 3760–3762 RSC.
- H. Li, Y. Huang, B. Zhang, D. Yang, X. Zhu and G. Li, Theranostics, 2014, 4, 701–707 CrossRef CAS PubMed.
- D. Kang, X. Zuo, R. Yang, F. Xia, K. W. Plaxco and R. J. White, Anal. Chem., 2009, 81, 9109–9113 CrossRef CAS PubMed.
- S. Fredriksson, M. Gullberg, J. Jarvius, C. Olsson, K. Pietras, S. M. Gustafsdottir, A. Ostman and U. Landegren, Nat. Biotechnol., 2002, 20, 473–477 CrossRef CAS PubMed.
- H. Zhang, F. Li, B. Dever, C. Wang, X.-F. Li and X. C. Le, Angew. Chem., Int. Ed., 2013, 52, 10698–10705 CrossRef CAS PubMed.
- Y.-L. Zhang, Y. Huang, J.-H. Jiang, G.-L. Shen and R.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 15448–15449 CrossRef CAS PubMed.
- J. A. Ludwig and J. N. Weinstein, Nat. Rev. Cancer, 2005, 5, 845–856 CrossRef CAS PubMed.
- J. E. Adams, G. S. Bodor, V. G. Davilaroman, J. A. Delmez, F. S. Apple, J. H. Ladenson and A. S. Jaffe, Circulation, 1993, 88, 101–106 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc00891c |
| This journal is © The Royal Society of Chemistry 2015 |