
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining triazole ligation and enzymatic glycosylation on solid phase simplifies the synthesis of very long glycoprotein analogues†
Mathieu
Galibert
,
Véronique
Piller
,
Friedrich
Piller
,
Vincent
Aucagne
* and
Agnès F.
Delmas
Centre de Biophysique Moléculaire, CNRS UPR 4301, Rue Charles Sadron, 45071 Orléans Cedex 2, France. E-mail: aucagne@cnrs-orleans.fr
First published on 14th April 2015
Abstract
The solid-phase chemical assembly of a protein through iterative chemoselective ligation of unprotected peptide segments can be followed with chemical and/or enzymatic transformations of the resulting immobilized protein, the latter steps thus benefitting from the advantages provided by the solid support. We demonstrate here the usefulness of this strategy for the chemo-enzymatic synthesis of glycoprotein analogues. A linker was specifically designed for application to the synthesis of O-glycoproteins: this new linker is readily cleaved under mild aqueous conditions compatible with very sensitive glycosidic bonds, but is remarkably stable under a wide range of chemical and biochemical conditions. It was utilized for solid-supported N-to-C peptidomimetic triazole ligation followed by enzymatic glycosylation, ultimately leading to a very large MUC1-derived glycoprotein containing 160 amino acid residues, 24 α-GalNAc moieties linked to Ser and Thr, and 3 triazoles as peptide bond mimetics.
Introduction
Solid-supported chemoselective ligations of unprotected biomolecule segments1,2 have opened a route towards high-yielding and rapid synthesis of proteins and protein analogues,1 peptide–PNA conjugates,2a and more recently very long oligonucleotides.2b The main benefit of such an iterative strategy relies on the use of simple draining processes as alternatives for the laborious intermediate chromatographic purification and lyophilization steps. In addition, post-ligation synthetic transformations of the resulting immobilized biomacromolecule can also benefit from the advantages provided by the solid support. This has been demonstrated for disulfide formation1d or cysteine desulfurization,1i,3 but the extension to other reactions like enzyme-mediated transformations or chemical conjugation with probes remains to be explored.The chemo-enzymatic synthesis of glycoproteins is a fast growing area,4 and we sought to combine solid phase ligations of peptide fragments with enzymatic glycosylation5,6 to provide a simplified access to complex glycoproteins. We herein focus on O-glycoproteins, identified as particularly demanding targets considering the narrow chemical compatibilities of O-glycans, which are sensitive to base-mediated β-elimination but also to acid hydrolysis, particularly the biologically-relevant sialylated compounds.
Besides protein elongation from the C-terminus to the N-terminus (C-to-N),1a–d,g–ii.e. in the same direction as solid-phase peptide synthesis (SPPS), “reversed” N-to-C ligation methodologies1a,e,f benefit from a self-purification effect (Fig. 1):7 provided that SPPS elongation includes an acetylation step before the removal of each Fmoc group, the truncated N-acetylated contaminants of the peptide segments can be eliminated by a simple draining after each ligation step. Paramount to such solid-phase ligation strategies are linkers to tether the starting unprotected (glyco)peptide segment to a water-compatible solid support (ligation resin, Fig. 1). For N-to-C protein assembly, a hetero-bifunctional N-terminal linker is introduced during SPPS as the Nα-protection group of the last amino acid. After cleavage from the SPPS resin and concomitant removal of the side-chain protective groups, the linker is used for chemoselective immobilization of the starting unprotected peptide segment on the ligation resin. After the ligation-based assembly, the linker must allow for a traceless and quantitative release of the protein, while being stable to a wide range of conditions. We recently introduced an azide-functionalized N3-Esoc1e,f linker (1, Fig. 2) that conveniently enables immobilization of the starting peptide segment by either CuI-catalyzed azide/alkyne cycloaddition1e (CuAAC) or strain-promoted azide/alkyne cycloaddition1f (SPAAC), forming a bio- and chemically-stable triazole linkage. However, the base treatment necessary for its cleavage is not compatible with O-glycopeptides (ESI, Fig. S31†). Other currently available linkers also call for harsh release procedures, either under basic,1a,e,f or acidic1b,g–i conditions. New linkers cleavable under milder conditions and thus compatible with a wider range of post-translational or other modifications are still seriously needed to further push back the limits of (glyco)protein chemical synthesis.
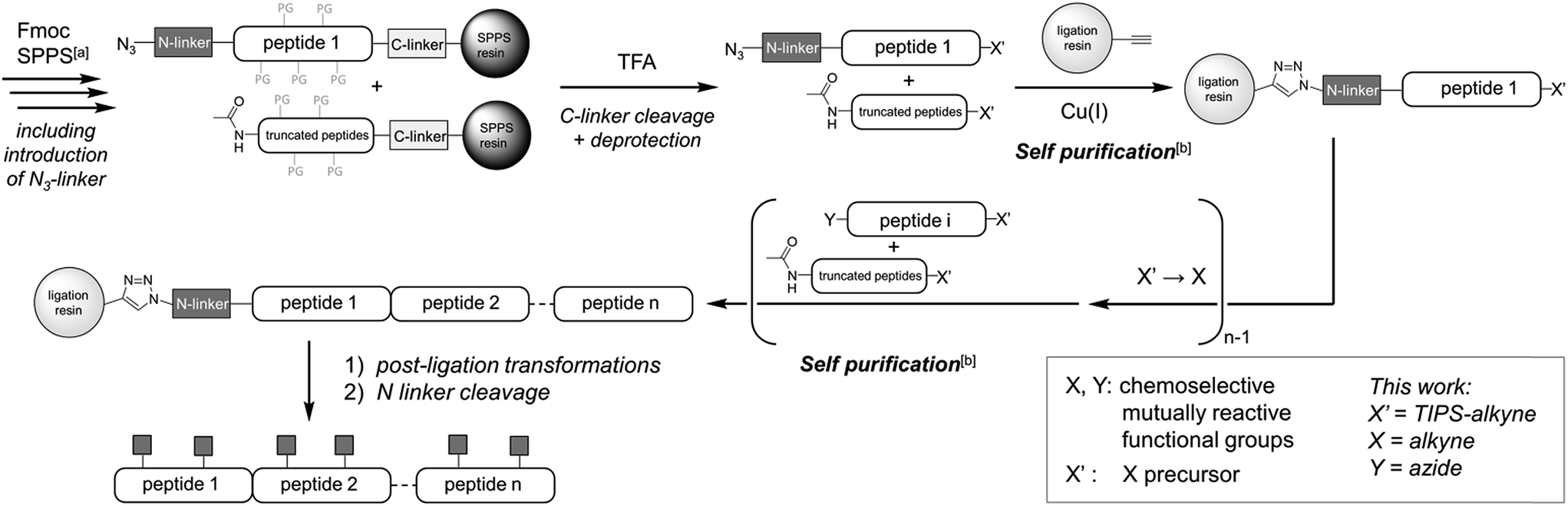 | ||
| Fig. 1 Overall description of the strategy, highlighting the self-purification nature1e of the solid-supported N-to-C assembly. a The SPPS process includes an acetylation-mediated capping step after each amino acid coupling step. b Subsequent click-chemistry can discriminate the target peptide having an azido group at the N-terminus from the undesired truncated acetylated byproducts. | ||
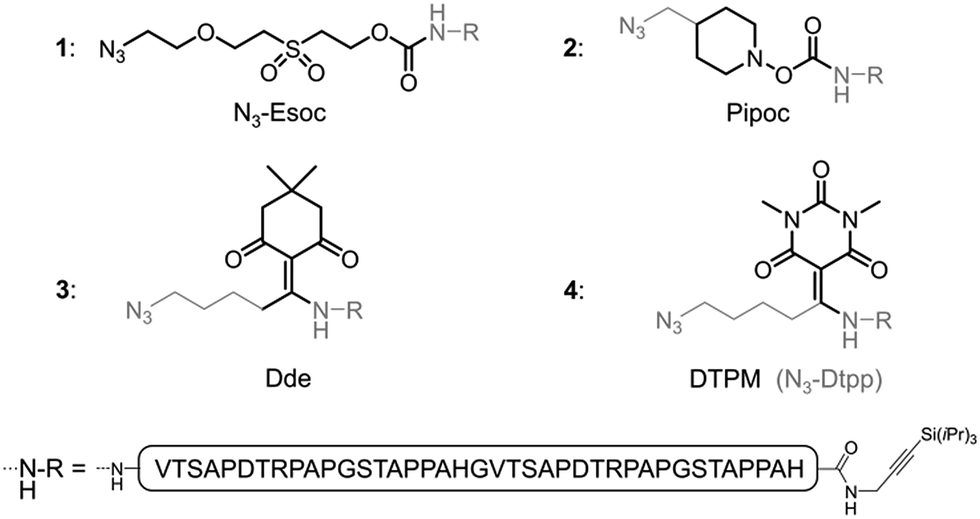 | ||
| Fig. 2 Linker-containing peptides evaluated in this work. | ||
We report here on the development of a promising N-terminal linker cleavable under mild neutral conditions and compatible with both NCL and peptidomimetic triazole ligation (PTL) as well as a wide range of chemical and biochemical treatments. Its usefulness is illustrated here by the synthesis of high molecular weight (up to ∼20 kDa)8 triazole-containing glycoprotein analogues, using a unprecedented combination of successive solid phase ligations and enzymatic glycosylations, ending with the release of glycoprotein analogues. Only one final HPLC purification step leads to well defined compounds mainly composed of single glycoforms.
Results and discussion
We chose as a model the human mucin MUC1, which has been widely used as a benchmark O-glycoprotein for chemical ligation-based methodological developments.1e,h,9 The extracellular domain of MUC1 is mainly constituted of identical tandem repeats of 20 amino acids (aa), VT2S3APDT7 RPAPGS13T14APPAHG, with 5 potential Ser/Thr O-glycosylation sites. This glycoprotein, and its glycopeptide derivatives, represents a major target for the development of anti-tumour vaccines,10 as it is overexpressed in most epithelial cancers, decorated with short tumour-specific O-glycans (α-GalNAc, β-Gal-1,3-α-GalNAc, or sialyl α-GalNAc).Optimization of an N-terminal linker adapted to the synthesis of O-glycopeptides and O-glycoproteins
Inspired by existing amine protecting groups, we selected three scaffolds (Fig. 2) that could be compatible with O-glycoproteins: Pipoc,11 cleavable under mild reducing conditions, and enamine-based Dde12 and DTPM,13 cleavable upon treatment with nitrogen nucleophiles. We prepared azide-functionalized derivatives suitable for immobilization on a solid support through CuAAC or SPAAC. The resulting hetero-bifunctional linkers were installed during SPPS at the N-terminus of a protected MUC1 double tandem repeat sequence. Peptides were also equipped at their C-termini with a silyl-protected alkyne14 for further solid phase triazole ligations.The stabilities of the new azido-linkers (peptides 2–4) were systematically screened in solution under a representative set of conditions (ESI, Table S1†), and compared to N3-Esoc (peptide 1). Unexpectedly, the Pipoc-based linker was not stable in the TFA cocktail used to cleave and deprotect peptide 2 after SPPS, making it unfit for our purpose. Surprisingly, we also observed slow cleavage of the Dde-derived linker (peptide 3) under mildly acidic and even neutral aqueous conditions.15 In contrast, the DTPM-based linker (peptide 4) was perfectly stable under acidic conditions as well as to a wide range of chemical treatments, including particularly harsh sodium methoxide-based deacetylation of chemically-introduced glycans, NCL and PTL conditions, and TBAF- or Ag-mediated alkyne desilylation. We thus concentrated our efforts on this promising 1-azido-5-[1,3-dimethyl-2,4,6(1H,3H,5H)-trioxopyrimidine-5-ylidene]pentyl linker, hereafter referred as N3-Dtpp.
Though perfectly stable towards nucleophiles such as thiolates (NCL conditions and cysteine), a large excess of sodium methoxide or primary amines (Tris buffer), Dtpp was quantitatively cleaved by 1 M aqueous hydrazine (pH 9.5). However, along with linker cleavage we observed the concomitant formation of two by-products showing an increase in mass of +2 Da and +4 Da. We suspected a diimide-based reduction of the silylalkyne into the corresponding Z-alkene and alkane,16 which was confirmed using a simple model alkyne (see ESI p S37†). Optimized, milder cleavage conditions using a 1 M aqueous hydroxylamine solution containing 100 mM sodium ascorbate,17 either buffered at neutral pH or without buffer (pH 8.5), cleanly cleaved the linker within a few hours.
Application to the synthesis of O-glycopeptides through solid-phase enzymatic glycosylation
With a reliable linker cleavage procedure established, we wanted to test N3-Dtpp with a more challenging example, such as glycopeptide 5 bearing a silylalkyne at its C-terminus and two acetate-protected Thr(Ac3-α-D-GalNAc) groups chemically introduced as building blocks during SPPS (Fig. 3). 5 was treated with excess sodium methoxide in methanol for 1 h to effect the chemoselective deacetylation of the glycan moieties, cleanly leading to glycopeptide 6 without affecting the Dtpp. Crude 6 was subsequently treated with TBAF to remove the C-terminal tri-isopropylsilyl (TIPS) alkyne protecting group, without showing any trace of sugar release by β-elimination or linker cleavage. Finally, N3-Dtpp was easily cleaved by aqueous hydroxylamine. No trace of degradation of the resulting unprotected glycopeptide 8 was observed, even under prolonged cleavage conditions.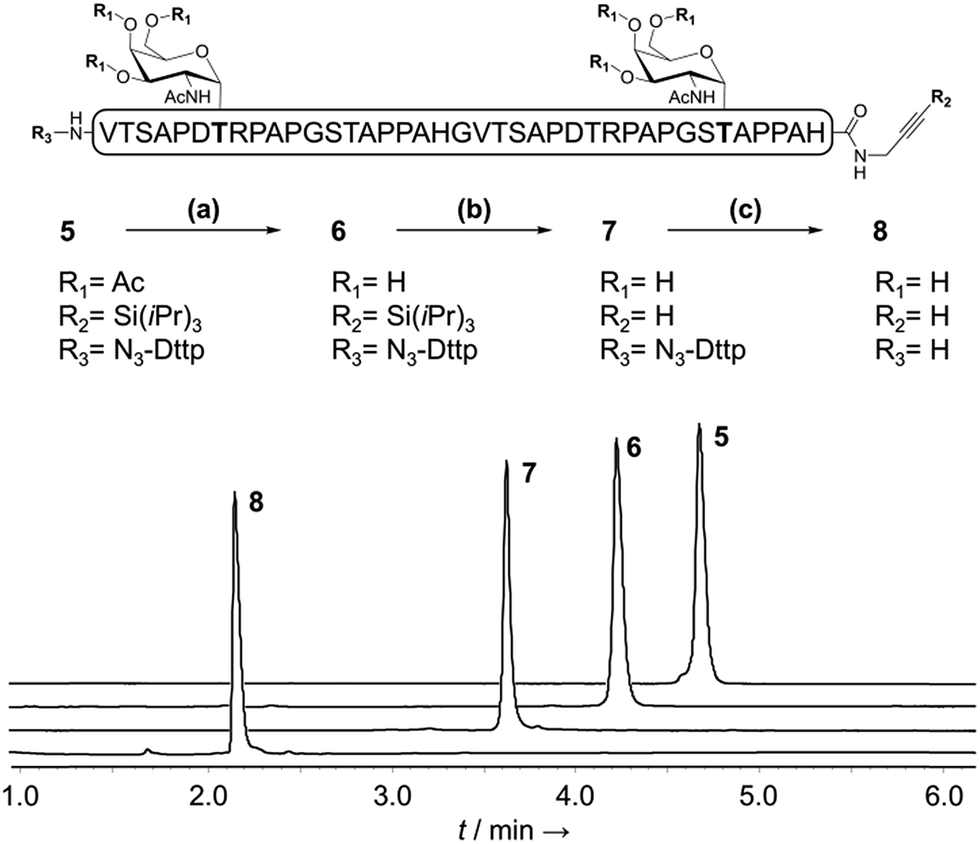 | ||
| Fig. 3 Sequential chemoselective deprotections of purified glycopeptide 5 to give 8. (a) MeONa/MeOH (b) TBAF/DMF (c) aqueous NH2OH. The RP-HPLC traces (λ = 214 nm) correspond to the successive crude deprotection mixtures. | ||
Having demonstrated the chemical robustness of the Dtpp linker we next applied it to the challenge5 of solid-supported enzymatic glycosylation by the recombinant polypeptide α-N-acetyl-galactosaminyl transferase 1 (GalNAc-T1). This widely-used enzyme catalyzes the specific transfer of α-N-acetyl-D-galactosamine (α-GalNAc) from UDP-GalNAc to the hydroxyl groups on the side chains of Ser or Thr. To ensure a complete accessibility to enzymes with a relatively high molecular weight such as GalNAc-T1 (>70 kDa, ∼3 nm diameter), commercially available controlled pore glass (CPG) beads with pore sizes around 100 nm were used as the solid support.18 Crude unglycosylated peptide 4 (∼70% purity) was efficiently immobilized onto alkyne-functionalized CPG 9a through CuAAC then desialylated with TBAF to give supported peptide 10a (Fig. 4). An aliquot of 10a was subsequently treated with hydroxylamine to release peptide 11a into solution. Gratifyingly, 11a was obtained in a much greater purity than substrate 4 due to the self-purification nature of the capture-and-release process, which eliminates truncated acetylated SPPS co-products (22 different truncated peptide impurities identified in crude 4).1e
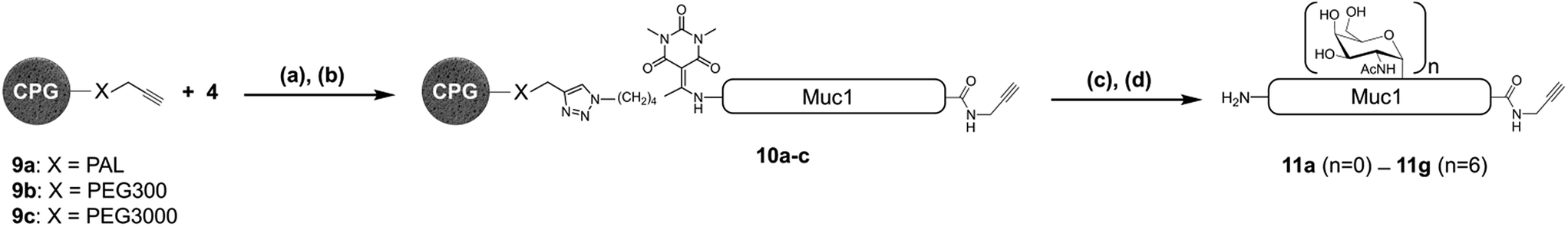 | ||
| Fig. 4 Immobilization and enzymatic glycosylation of a 39 aa solid supported MUC1 peptide. (a) CuBr·Me2S, THPTA, HEPES (pH 7.5)/NMP; (b) TBAF, DMF; (c) UDP-GalNAc, GalNAc-T1; (d) 1 M aqueous NH2OH. Muc1 refers to the same 39 aa sequence as in Fig. 1 and 2. PAL: 5-[3,5-dimethoxy-4-(aminomethyl)phenoxy]pentanoic acid. | ||
In this test, an additional acid-labile PAL linker was inserted between the CPG beads and the alkyne moiety. This double-linker strategy19 was employed to check for any peptide still present on the beads after cleavage of Dtpp. Subsequent TFA treatment did not release any additional peptide, confirming the high efficiency of the hydroxylamine-mediated Dtpp cleavage.20 As a consequence, we did not introduce a PAL linker and used only Dttp cleavage for quality control or final release in all further experiments.21
Although our first assay gave encouraging results, we nevertheless found enzymatic glycosylation of supported peptide 10a to be much less efficient than a solution phase control experiment. However, satisfactory results were obtained by introducing a 3000 Da PEG spacer between the peptide and the solid support22 (10c): after peptide release, LC-MS analysis showed a mixture mainly composed of pentaglycosylated (11f) and hexaglycosylated (11g) peptides, with a product distribution comparable to that obtained in solution (ESI, Tables S6–S8†).23 Only very low amounts (<2%) of compounds containing more than six α-GalNAc could be detected in the mixture. The major product, HPLC-purified hexaglycosylated 11g, was shown to be a single glycoform, its homogeneity being assessed by top-down analysis using electron-transfer dissociation ESI-MS/MS.24,25 The glycosylated amino acid residues were unequivocally mapped to Thr2, Ser13 and Thr14 in the first tandem repeat, and to their counterparts in the second, Thr22, Ser33 and Thr34. The four remaining possible O-glycosylation sites, Ser3, Thr7, Ser23 and Thr27, were clearly shown to be unmodified. These results are in perfect accordance with the sequence specificity of GalNAc-T1: this enzyme is known to be unable to glycosylate these Ser and Thr sites within MUC1 tandem repeats.26 However, the traces of heptaglycosylated products we observed probably correspond to glycosylation, with a very slow kinetic rate, at either one of these four positions. Importantly, this also demonstrates that the GalNAc-T1 tolerates the presence of the Dtpp linker at the N-terminus in close proximity to Thr2.
Considering that the glycosylation of Thr7 and Thr27 could be important for an immune response,10 chemically-synthesized glycopeptide 6 containing two GalNAc units at these positions was immobilized through CuAAC on the optimized support 9c, then desialylated and subjected to enzymatic glycosylation with GalNAc-T1. About eighty percent of the peptide was converted to the octaglycosylated peptide (S21′, ESI Fig. S54†)27: the chemical pre-introduction of GalNAc units significantly increased the enzymatic glycosylation rate. This interesting result may be rationalized by the interaction of the lectin domain of the enzyme28 with the glycopeptide and/or by a conformational change of the peptide induced by the introduction of the two GalNAc residues.29 HPLC purification furnished pure 18 in a 48% yield.
We then considered the possibility of extending the glycans towards more complex O-glycans using appropriate glycosyltransferases. We chose to introduce sialic acid (NeuAc) moieties, as a relevant example. They are found in both O- and N-glycoproteins. Sialylated antigens are of great interest and promise in the cancer vaccine field and are particularly difficult to prepare by purely chemical means.30 In addition, sialyl glycosides are unstable under aqueous acidic conditions. The introduction of sialic acid groups was performed by incubation of supported peptide 15 with the sialyltransferase ST6GalNAc I and CMP-NeuAc under unoptimized conditions. The linker cleavage with hydroxylamine was perfectly compatible with the sialyl moieties. HPLC purification provided di-sialylated glycopeptide 16 in a 38% yield (Fig. 5A). These results highlight the excellent flexibility and robustness of our strategy, which allows both enzymatic and chemical glycosylations to be readily combined.
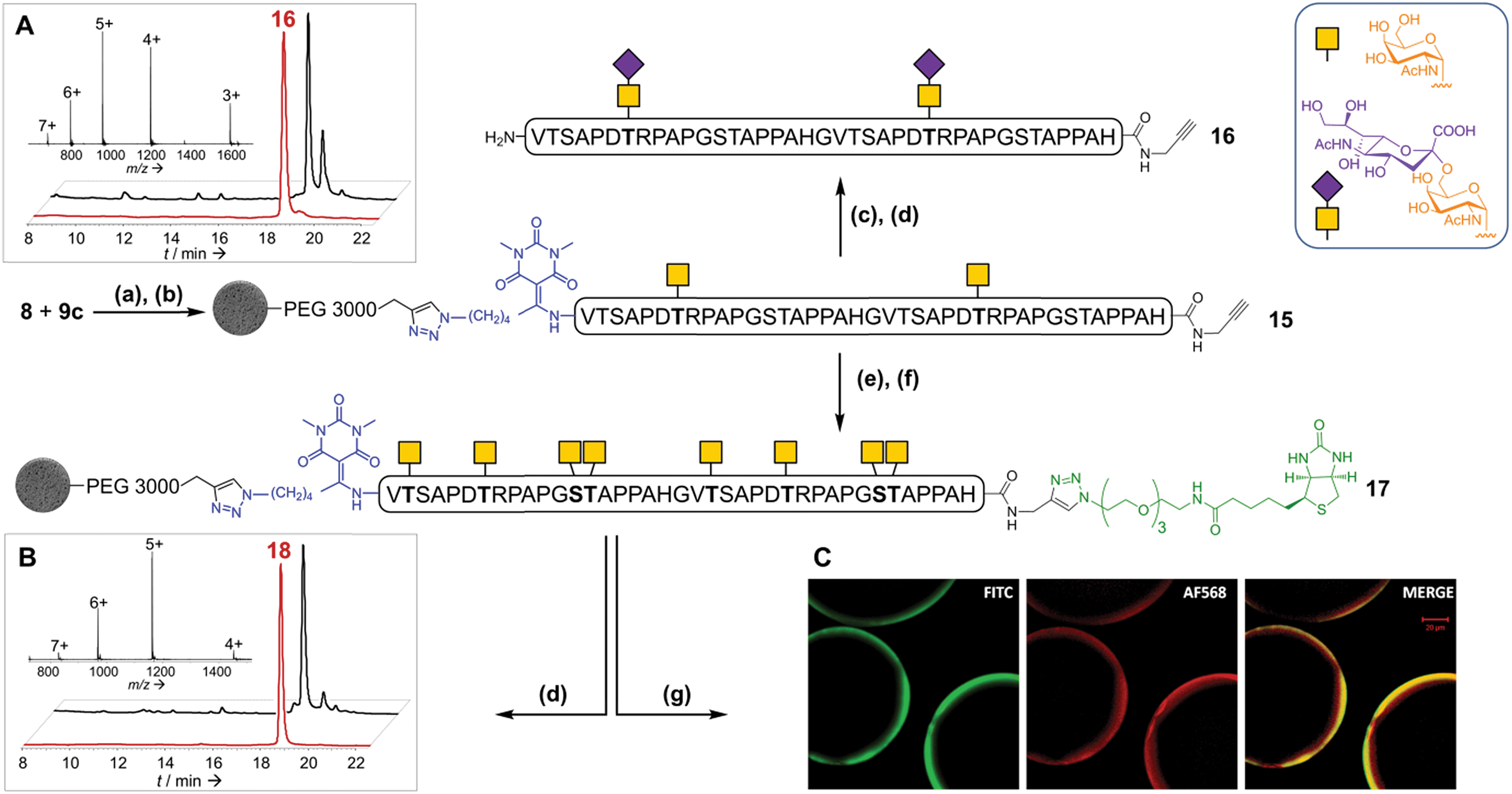 | ||
| Fig. 5 Solid phase glycosylation and conjugation to a biotin probe of peptide 15. (a) CuBr·Me2S, THPTA, HEPES (pH 7.5)/NMP; (b) TBAF, DMF; (c) CMP-NeuAc, ST6GalNAc I; (d) 1 M NH2OH; (e) UDP-GalNAc, GalNAc-T1; (f) N3-PEG3-biotin, CuBr·Me2S, THPTA, HEPES (pH 7.5)/NMP; (g) FITC-conjugated Vicia villosa lectin, Alexa-fluor 568-labelled streptavidin. (A): RP-HPLC profile (λ = 214 nm) of crude (black) and purified (red) glycopeptide 16. Inset: ESI-HRMS spectrum of purified 16. (B): RP-HPLC profile (λ = 214 nm) of crude (black) and purified (red) glycopeptide 18. Inset: ESI-HRMS spectrum of purified 18. (C): Confocal fluorescence microscopy analysis of dually-labelled bead-supported biotinyl-glycopeptide 17. | ||
Application to the combination of solid phase glycosylation and chemoselective ligations
To further illustrate the versatility of our linker, we exploited the C-terminal alkyne for additional chemoselective ligation reactions. Firstly, solid phase CuAAC-based conjugation was used to label the synthetic glycopeptide with a biotin probe, giving compound 18 (Fig. 5B) in extraordinarily high purity considering its chemical complexity (45% isolated yield). We also demonstrated full compatibility of the solid support with imaging techniques typically used for living cells, using a variety of fluorescent antibodies, streptavidin and lectins (ESI Table S8† and Fig. 5C). The latter result suggests that solid-supported glycopeptide mixtures could be directly screened against biologically-relevant targets while still immobilized.Finally, we applied our linker to the synthesis of very long polypeptides through iterative solid-supported triazole ligations. Starting from immobilized peptide 10c, three successive ligation/desilylation cycles, using crude peptide 19 (∼70% purity) featuring both an N-terminal azide group and a C-terminal silylalkyne, gave a very clean 160 residue supported compound 20, as evidenced by LC-MS analysis of the crude released peptide 21 (Fig. 6A). This shows the efficiency of the N-to-C solid-phase chemical ligation strategy. It also illustrates the self-purification effect.1e
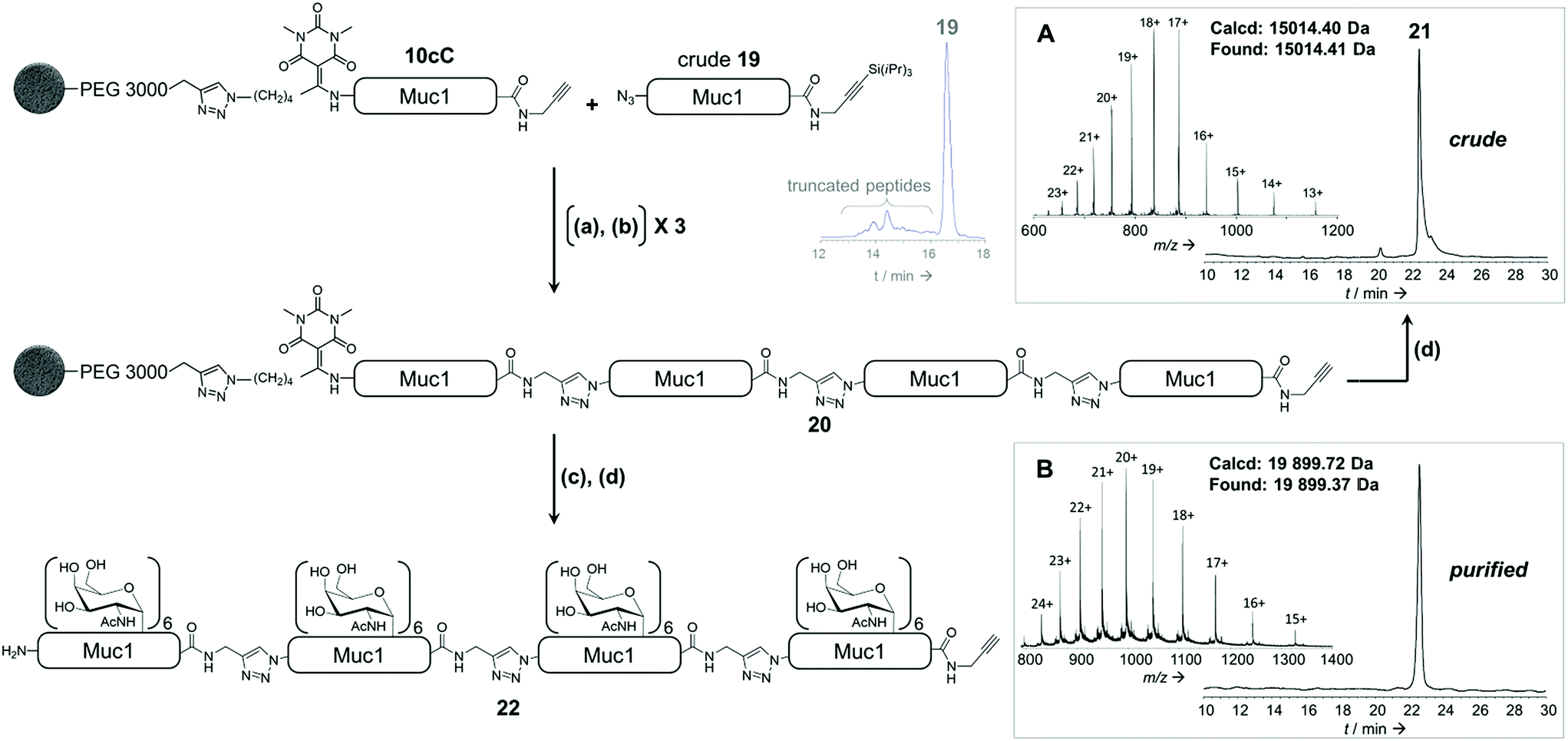 | ||
| Fig. 6 Combination of solid phase peptidomimetic triazole ligation and enzymatic glycosylation. (a) azidopeptide 19, CuBr·Me2S, THPTA, HEPES (pH 7.5)/NMP; (b) TBAF, phenol, DMF; (c) UDP-GalNAc, GalNAc-T1; (d) 1 M NH2OH. (A): RP-HPLC profile (λ = 214 nm) and ESI-HRMS spectrum of crude unglycosylated 160 residue peptide 21. (B): RP-HPLC profile (λ = 214 nm) and ESI-HRMS spectrum of HPLC-purified glycopeptide 22. | ||
Subsequent solid-supported enzymatic glycosylation of 20 with GalNAc-T1, under the conditions optimized for 40-mer 10c, successfully produced the corresponding triazolo-glycoproteins as a mixture of glycoforms, the major ones containing 22, 23 and 24 GalNAc moieties.
Most importantly, the presence of triazoles as amide surrogates did not alter in any way the glycosylation efficiency, as we also rigorously demonstrated with a model triazolopeptide (ESI p S101†). As expected, only low amounts (∼4%) of compounds containing more than 24 GalNAc moieties could be detected, supporting that the triazole-containing glycoprotein analogue 22 bearing 24 saccharide moieties is mainly composed of a single glycoform, glycosylated on the same three sites per tandem repeat that GalNAc-T1 can glycosylate. HPLC purification furnished triazolo-glycoprotein 22 (Fig. 6B) in an overall yield of 6%, taking into account the ligation-mediated assembly, enzymatic glycosylation and purification. Similar results were obtained with 80- and 120-mer compounds containing 12 and 18 α-GalNAc-Ser/Thr moieties and purified in 15% and 10% yields, respectively (ESI, p S115–S119†).
Conclusions
We have designed a new solid phase synthetic strategy, based on multiple successive solid-supported ligations followed by enzymatic glycosylation, applicable to the synthesis of a wide range of glycopeptides and glycoproteins, and potentially to proteins with other post-translational modifications. This strategy is based on a heterobifunctional N-terminal azido linker, N3-Dtpp. Resistant to a wide set of chemical and biochemical transformations, this linker is cleavable under particularly mild neutral aqueous conditions. Its broad applicability was exemplified through the solid phase synthesis of complex mucin-type O-glycopeptides by combining enzymatic and chemical glycosylations, bioconjugation, and also direct fluorescence biochemical screening of the supported peptides with lectins and antibodies, which is promising for further biological applications. We also demonstrated that the linker is compatible with chemical ligation techniques such as peptidomimetic triazole ligations, to enable further solid phase elongation and the synthesis of longer and more complex glycoproteins. The latter point was demonstrated with the synthesis of a very large homogeneous 160 residue triazole-containing glycoprotein analogue through three successive ligations followed by enzymatic glycosylation, thus avoiding multiple solution-phase intermediate purification steps. Application of the linker to other ligation techniques such as NCL, to produce native glycoproteins not incorporating triazoles as backbone modifications, is currently underway and will be reported in due course.Acknowledgements
We thank Dr Guillaume Gabant, Dr Cyril Colas and the CBM and FR2708 federation mass spectrometry platforms for MS and LC-MS analyses, and Dr Ibai Valverde for the synthesis of the Fmoc-Thr(α-Ac3-GalNAc)-OH building block. La Ligue contre le cancer is greatly acknowledged for a postdoctoral fellowship to MG and financial support to VA (local committees 35, 45, 49 and 85).Notes and references
- (a) L. E. Canne, P. Botti, R. J. Simon, Y. Chen, E. A. Dennis and S. B. H. Kent, J. Am. Chem. Soc., 1999, 121, 8720–8727 CrossRef CAS; (b) A. Brik, E. Keinan and P. E. Dawson, J. Org. Chem., 2000, 65, 3829–3835 CrossRef CAS PubMed; (c) G. J. Cotton and T. W. Muir, Chem. Biol., 2000, 7, 253–261 CrossRef CAS PubMed; (d) E. C. B. Johnson, T. Durek and S. B. H. Kent, Angew. Chem., Int. Ed., 2006, 45, 3283–3287 CrossRef CAS PubMed; (e) V. Aucagne, I. E. Valverde, P. Marceau, M. Galibert, N. Dendane and A. F. Delmas, Angew. Chem., Int. Ed., 2012, 51, 11320–11324 CrossRef CAS PubMed; (f) L. Raibaut, H. Adihou, R. Desmet, A. F. Delmas, V. Aucagne and O. Melnyk, Chem. Sci., 2013, 4, 3839–3844 RSC; (g) T. Wang and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11708–11713 CrossRef CAS PubMed; (h) I. E. Decostaire, D. Lelièvre, V. Aucagne and A. F. Delmas, Org. Biomol. Chem., 2014, 12, 5536–5543 RSC; (i) M. Jbara, M. Seenaiah and A. Brik, Chem. Commun., 2014, 50, 12534–12537 RSC.
- (a) M. C. de Koning, D. V. Filippov, G. A. van der Marel, J. H. van Boom and M. Overhand, Eur. J. Org. Chem., 2004, 4, 850–857 CrossRef; (b) J. Qiu, A. H. El-Sagheer and T. Brown, Chem. Commun., 2013, 49, 6959–6961 RSC.
- For a recent example of solid-supported desulfurization following solution phase NCL, see: O. Reimann, C. Smet-Nocca and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2015, 53, 306–310 CrossRef PubMed.
- For recent reviews on glycoprotein synthesis, including chemo-enzymatic approaches, see: (a) L.-X. Wang and M. N. Amin, Chem. Biol., 2014, 21, 51–66 CrossRef CAS PubMed; (b) R. Okamoto, M. Izumi and Y. Kajihara, Curr. Opin. Chem. Biol., 2014, 92–99 CrossRef CAS PubMed; (c) C. Unverzagt and Y. Kajihara, Chem. Soc. Rev., 2013, 42, 4408–4420 RSC.
- Previous reports of enzymatic glycosylation of solid-supported peptides were limited to very short sequences. For pioneering work, see: (a) M. Schuster, P. Wang, J. C. Paulson and C.-H. Wong, J. Am. Chem. Soc., 1994, 116, 1135–1136 CrossRef CAS; (b) O. Seitz and C.-H. Wong, J. Am. Chem. Soc., 1997, 119, 8766–8776 CrossRef CAS and more recently; (c) S. K. Kracun, E. Cló, H. Clausen, S. B. Levery, K. J. Jensen and O. Blixt, J. Proteome Res., 2010, 9, 6705–6714 CrossRef CAS PubMed.
- A recent paper described a soluble polymer as an alternative support for enzymatic glycosylation, followed by enzyme-based release and then further elongation of the glycopeptide through solution phase NCL: C. Bello, K. Farbiarz, J. F. Möller, C. F. W. Becker and T. Schwientek, Chem. Sci., 2014, 5, 1634–1641 RSC.
- For pioneering work on capture and release purification, not including further ligations, see: S. Funakoshi, H. Fukuda and N. Fujii, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6981–6985 CrossRef CAS and ref. 1e for a comprehensive overview.
- For a report of the synthesis of a very long O-glycoprotein by purely chemical means, through multiple repetitive segment condensation in solution, see: H. Hojo, Y. Matsumoto, Y. Nakahara, E. Ito, Y. Suzuki, M. Suzuki, A. Suzuki and Y. Nakahara, J. Am. Chem. Soc., 2005, 127, 13720–13725 CrossRef CAS PubMed.
- (a) G.-A. Cremer, N. Bureaud, D. Lelièvre, V. Piller, F. Piller and A. Delmas, Chem.–Eur. J., 2004, 10, 6353–6360 CrossRef CAS PubMed; (b) G.-A. Cremer, N. Bureaud, V. Piller, H. Kunz, F. Piller and A. F. Delmas, ChemMedChem, 2006, 1, 965–968 CrossRef CAS PubMed; (c) R. J. Payne, S. Ficht, S. Tang, A. Brik, Y. Y. Yang, D. A. Case and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 13527–13536 CrossRef CAS PubMed; (d) R. J. Payne, S. Ficht, W. A. Greenberg and C.-H. Wong, Angew. Chem., Int. Ed., 2008, 47, 4411–4415 CrossRef CAS PubMed; (e) R. Okamoto, S. Souma and Y. Kajihara, J. Org. Chem., 2009, 74, 2494–2501 CrossRef CAS PubMed; (f) B. L. Wilkinson, C. K. Chun and R. J. Payne, Biopolymers, 2011, 96, 137–146 CrossRef CAS PubMed; (g) P. Karmakar, R. S. Talan and S. J. Sucheck, Org. Lett., 2011, 13, 5298–5301 CrossRef CAS PubMed; (h) L. R. Malins, K. M. Cergol and R. J. Payne, ChemBioChem, 2013, 14, 559–563 CrossRef CAS PubMed; (i) D. Al Sheikha, B. L. Wilkinson, G. Santhakumar, M. Thaysen-Andersen and R. J. Payne, Org. Biomol. Chem., 2013, 11, 6090–6096 RSC; (j) C. Xu, H. Y. Lam, Y. Zhang and X. Li, Chem. Commun., 2013, 49, 6200–6202 RSC.
- N. Gaidzik, U. Westerlind and H. Kunz, Chem. Soc. Rev., 2013, 42, 4421–4442 RSC.
- D. Stevenson and G. T. Young, J. Chem. Soc. C, 1969, 2389–2398 RSC.
- (a) B. W. Bycroft, W. C. Chan, S. R. Chhabra and N. D. Hone, J. Chem. Soc., Chem. Commun., 1993, 778–779 RSC; (b) J. J. Díaz-Mochón, L. Bialy and M. Bradley, Org. Lett., 2004, 6, 1127–1129 CrossRef PubMed.
- (a) I. Toth, G. Dekany and B. Kellam, PCT Int. Appl. WO 9915510 A1, 1999; (b) G. Dekany, L. Bornaghi, J. Papageorgioua and S. Taylor, Tetrahedron Lett., 2001, 42, 3129–3132 CrossRef CAS; (c) E. T. da Silva and E. L. Lima, Tetrahedron Lett., 2003, 44, 3621–3624 CrossRef CAS.
- (a) V. Aucagne and D. A. Leigh, Org. Lett., 2006, 8, 4505–4507 CrossRef CAS PubMed; (b) I. E. Valverde, A. F. Delmas and V. Aucagne, Tetrahedron, 2009, 65, 7597–7602 CrossRef CAS; (c) I. E. Valverde, F. Lecaille, G. Lalmanach, V. Aucagne and A. F. Delmas, Angew. Chem., Int. Ed., 2012, 51, 718–722 CrossRef CAS PubMed.
- We have also tested the aqueous stability of a model peptide containing an N-terminal Dde group, and observed a similar slow hydrolysis (ESI p S32†). Contrastingly, a Dde group located on the side chain amine of a lysine derivative (Fmoc-Lys(Dde)-OH) was completely stable under the same conditions.
- B. Rohwedder, Y. Mutti, P. Dumy and M. Mutter, Tetrahedron Lett., 1998, 39, 1175–1178 CrossRef CAS.
- Note that contrary to TIPS-protected alkynes, terminal alkynes were much less sensitive to reduction during Dtpp cleavage and did not require the addition of ascorbate (see ESI data for details†).
- J. Kress, R. Zanaletti, A. Amour, M. Ladlow, J. G. Frey and M. Bradley, Chem.–Eur. J., 2002, 16, 3769–3772 CrossRef.
- S. Mezzato, M. Schaffrath and C. Unverzagt, Angew. Chem., Int. Ed., 2005, 44, 1650–1654 CrossRef CAS PubMed.
- The yield over three steps, CuAAC, desilylation and linker cleavage, was >90%, as evaluated by weight from a HPLC-purified sample of 4, to give 11a. See ESI p S59.†.
- Extended cleavage treatments and solid support washes after Dttp cleavage never showed any trace of further released peptides. We then considered it to be a nearly-quantitative reaction.
- (a) J. Deere, R. F. De Oliveira, B. Tomaszewski, S. Millar, A. Lalaouni, L. F. Solares, S. L. Flitsch and P. J. Halling, Langmuir, 2008, 24, 11762–11769 CrossRef CAS PubMed; (b) S. J. Brooks, L. Coulombel, D. Ahuja, D. S. Clark and J. S. Dordick, Adv. Synth. Catal., 2008, 350, 1517–1525 CrossRef CAS PubMed.
- The enzymatic kinetics were slower for the solid support than in solution, but led to a very similar final product distribution.
- M. Thaysen-Andersen, B. L. Wilkinson, R. J. Payne and N. H. Packer, Electrophoresis, 2011, 32, 3536–3545 CrossRef CAS PubMed.
- Note that contrary to hexaglycosylated 11g, the HPLC peak corresponding to pentaglycosylated 11f is not expected to correspond to a single glycoform, and has not been analyzed by ETD ESI-MS/MS. The same remark remains true for compounds 11b–e, containing one to four α-GalNAc moieties heterogeneously distributed on the six possible O-glycosylation sites for the α-GalNAc transferase that we used.
- (a) V. Bourgeaux, M. Cadène, F. Piller and V. Piller, ChemBioChem, 2007, 8, 37–40 CrossRef CAS PubMed; (b) Y. Kong, H. J. Joshi, K. T. Schjoldager, T. D. Madsen, T. A. Gerken, M. B. Vester-Christensen, H. H. Wandall, E. P. Bennett, S. B. Levery, S. Y. Vakhrushev and H. Clausen, Glycobiology, 2015, 25, 55–65 CrossRef PubMed.
- Only small traces of glycopeptides containing more than 8 GalNAc moieties could be observed by LC-MS analysis of the mixture, supporting that the enzyme does glycosylate the same six Thr and Ser sites as in peptide 11g. In consequence, we did not perform ETD ESI-MS/MS analysis on octaglycosylated 18.
- T. A. Gerken, L. Revoredo, J. J. C. Thome, L. A. Tabak, M. B. Vester-Christensen, H. Clausen, G. K. Gahlay, D. L. Jarvis, R. W. Johnson, H. A. Moniz and K. Moremen, J. Biol. Chem., 2013, 288, 19900–19914 CrossRef CAS PubMed.
- J. Schuman, A. P. Campbell, R. R. Koganty and B. M. Longenecker, J. Pept. Res., 2003, 61, 91–108 CrossRef CAS PubMed.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, characterization and optimization. See DOI: 10.1039/c5sc00773a |
| This journal is © The Royal Society of Chemistry 2015 |