
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of bicyclo[3.n.1]alkanes by chiral phosphoric acid-catalyzed desymmetrizing Michael cyclizations†
Alan R.
Burns‡
ab,
Amaël G. E.
Madec‡
b,
Darryl W.
Low
a,
Iain D.
Roy
a and
Hon Wai
Lam
*ab
aEaStCHEM, School of Chemistry, University of Edinburgh, Joseph Black Building, The King's Buildings, David Brewster Road, Edinburgh, EH9 3FJ, UK.
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: hon.lam@nottingham.ac.uk
First published on 30th April 2015
Abstract
2,2-Disubstituted cyclic 1,3-diketones containing a tethered electron-deficient alkene undergo chiral phosphoric acid-catalyzed desymmetrizing Michael cyclizations to give bridged bicyclic products in high enantioselectivities. Both bicyclo[3.2.1]octanes and bicyclo[3.3.1]nonanes are accessible using this methodology.
Bicyclo[3.n.1]alkanes appear in numerous biologically active natural products (selected examples are shown in Fig. 1) and have widespread applications in organic synthesis.1,2 Although many creative approaches for the synthesis of these structures have been devised,1,2 the preparation of enantiomerically enriched chiral bicyclo[3.n.1]alkanes by asymmetric catalysis currently represents only a small fraction of these methods.3,4 In view of the present level of development, increasing the number of available catalytic enantioselective reactions to access these compounds is an important area of research. Herein, we report a new approach for the enantioselective synthesis of bicyclo[3.2.1]octanes and bicyclo[3.3.1]nonanes by chiral phosphoric acid-catalyzed Michael cyclizations of 1,3-diones onto tethered electron-deficient alkenes. These reactions give products containing three new stereogenic centers, including an all-carbon quaternary center, resulting from the formal enantioselective, desymmetrizing enolization of 2,2-disubstituted cyclic 1,3-diketones. In addition, these reactions further demonstrate the ability of chiral phosphoric acids to promote transformations of unactivated ketones by enolization, which has so far been relatively underexplored.5
 | (1) |
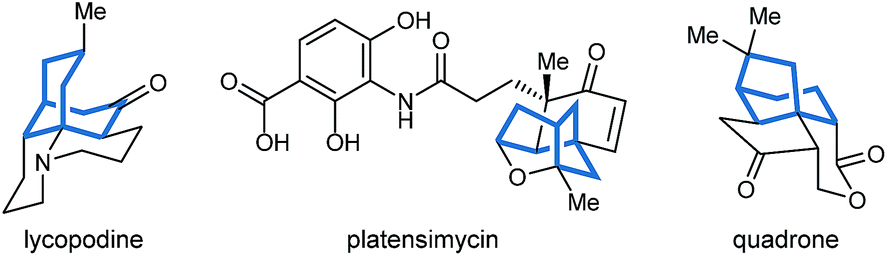 | ||
| Fig. 1 Natural products containing bicyclo[3.n.1]alkanes. | ||
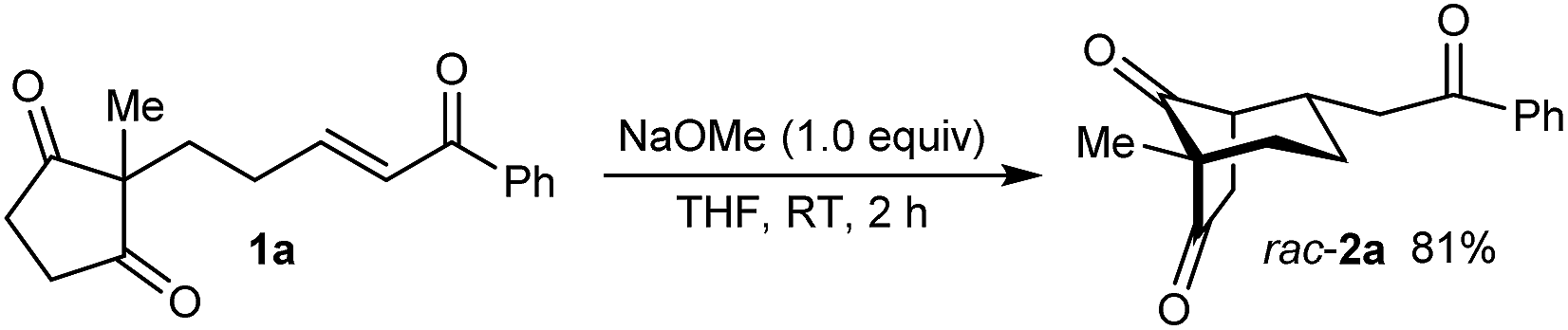
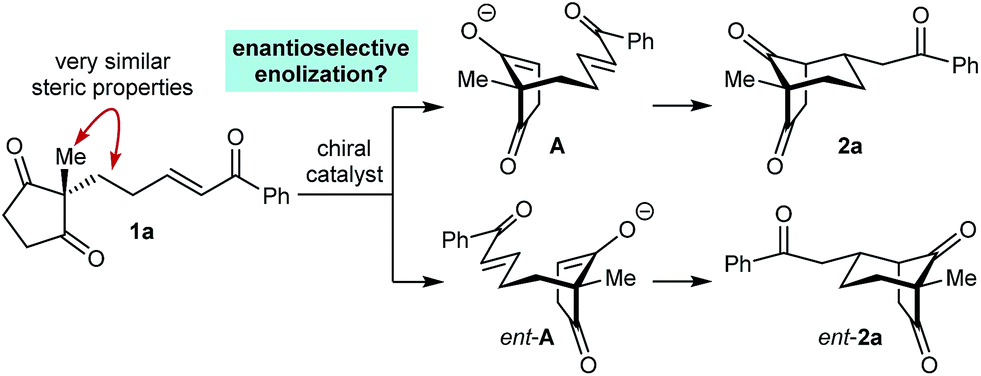
As part of our interest in the catalytic enantioselective desymmetrization of 2,2-disubstituted cyclic 1,3-diketones,6 we observed that enone dione 1a can undergo an intramolecular Michael addition to form the chiral bicyclo[3.2.1]octane rac-2a under basic conditions.7 For example, treatment of 1a with NaOMe in THF at room temperature gave rac-2a in 81% yield (eqn (1)). Following this result, the development of a catalytic enantioselective variant captured our interest. However, a challenging feature of this reaction that differentiates it from the significant majority of catalytic enantioselective Michael reactions described previously8 is that stereogenicity is first generated in the enolization step, rather than the carbon–carbon bond-forming step (Scheme 1).9 Therefore, any chiral catalyst employed has to, at first glance, facilitate the enantioselective, desymmetrizing enolization10 of a 2,2-disubstituted cyclic 1,3-diketone. Due to the two substituents at the prochiral 2-position (methyl versus primary alkyl) possessing very similar steric properties, this task appeared to be far from trivial.
 | ||
| Scheme 1 Asymmetric induction in the formation of 2a. | ||
We hypothesized that a solution to this challenge could be obtained under conditions where enolization of 1a is reversible, potentially enabling rapid interconversion of the enolates A and ent-A. Under such Curtin–Hammett conditions,11 carbon–carbon bond formation could then be the enantiodetermining step. Furthermore, it appeared likely that chiral catalysts capable of binding to both the enolate oxygen atom and the enone carbonyl group would increase the energy difference between diastereomeric transition states, thus maximizing the prospects of achieving high enantioselectivities.
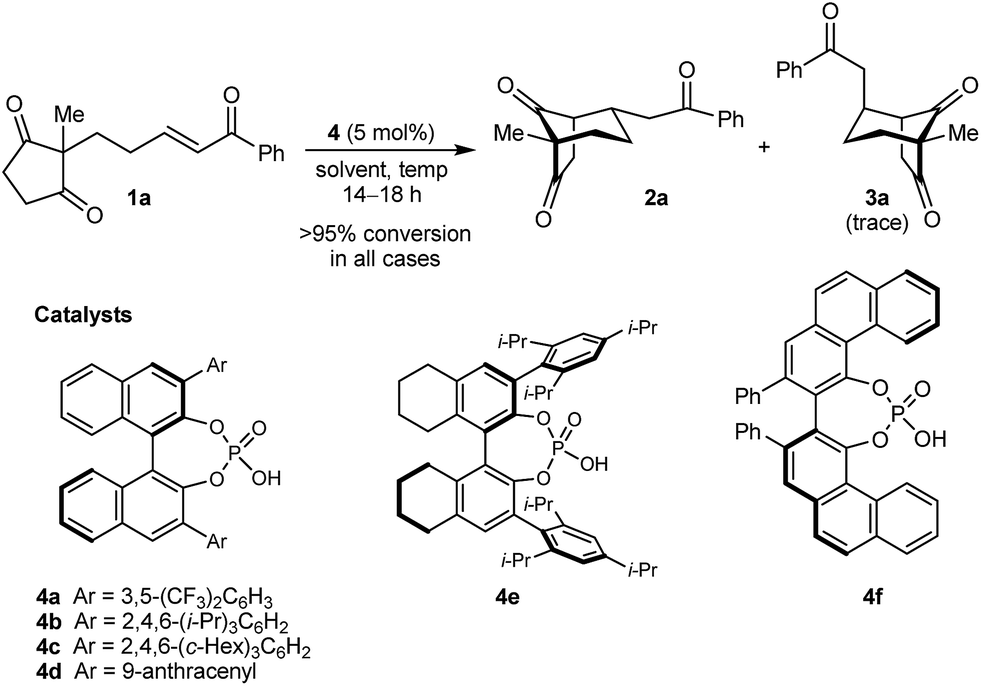
In the last ten years, chiral Brønsted acids, such as phosphoric acids, have emerged as extremely versatile catalysts for a diverse range of transformations.12–14 In many cases, both the Lewis acidity of the hydroxyl group and the Lewis basicity of the phosphoryl group of the catalyst play key roles in the simultaneous activation of electrophile–nucleophile pairs.13 We were therefore hopeful that chiral phosphoric acids would be suitable bifunctional catalysts for the enantioselective cyclization of 1a, and this indeed turned out to be the case (Table 1).15 For example, heating 1a in the presence of various BINOL-derived phosphoric acids 4a–4d (5 mol%) in toluene at 80 °C for 14–18 h led to complete consumption of 1a to form bicyclo[3.2.1]octane 2a as the major product (Table 1, entries 1–4).16 Small traces of a diastereomeric product 3a,16 in which the phenyl ketone-containing substituent occupies an axial position, were detected by TLC analysis, but the exact diastereomeric ratios could not be determined by 1H NMR analysis due to overlapping signals. Furthermore, promising enantioselectivities were obtained with phosphoric acids 4b–4d containing sterically more hindered aryl groups at the 3,3′-positions (entries 2–4). Catalysts 4b and 4c, possessing 2,4,6-trisubstituted aryl groups, gave the best results (entries 2 and 3). Switching to the H8-BINOL scaffold in catalyst 4e was detrimental (entry 5, compare with entry 2), while the vaulted biaryl-derived phosphoric acid 4f gave a low enantioselectivity (entry 6). Due to its commercial availability and relative ease of synthesis compared with 4c, phosphoric acid 4b (TRIP) was selected for further investigations. Changing the solvent to cyclohexane17 and lowering the reaction temperature to 50 °C gave marginally superior results (entries 7 and 8).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | eeb of 2a (%) |
| a Reactions were conducted using 0.05 mmol of 1a. Complete consumption of 1a was observed in all cases by 1H NMR analysis. b Determined by HPLC on a chiral stationary phase. | ||||
| 1 | 4a | Toluene | 80 | 28 |
| 2 | 4b | Toluene | 80 | 89 |
| 3 | 4c | Toluene | 80 | 90 |
| 4 | 4d | Toluene | 80 | 76 |
| 5 | 4e | Toluene | 80 | 74 |
| 6 | 4f | Toluene | 80 | 17 |
| 7 | 4b | Cyclohexane | 80 | 90 |
| 8 | 4b | Cyclohexane | 50 | 91 |
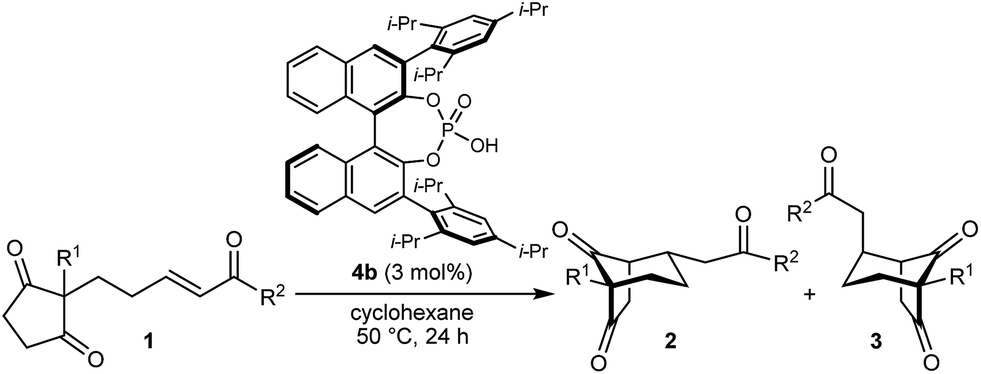
With an effective catalyst and solvent identified, the scope of the reaction with respect to the preparation of bicyclo[3.2.1] octanes was investigated (Table 2). The catalyst loading of 4b could be decreased from 5 mol% to 3 mol% without detriment, and the reactions were complete after 24 h. Under these conditions, a range of enone diones 1a–1k underwent Michael cyclizations to give products 2a–2k in generally high yields and with good to high enantioselectivities (86–95% ee).16 The process is compatible with electron-donating (entries 2 and 3) or electron-withdrawing aryl groups (4 and 5) on the enone carbonyl, as well as 2-naphthyl (entries 6 and 7) or tert-butyl groups (entry 8). Additional Lewis basic heteroatoms in heteroarene substituents such as 2-pyridyl, 2-furyl, or 2-thienyl groups were also tolerated (entries 9–11). Finally, replacement of the methyl group at the 2-position of the cyclic 1,3-diketone with an ethyl group did not affect the efficiency of the reaction (entry 12). In some cases, the reactions were highly diastereoselective, and only one product was detected (entries 2, 3, 6–8, 11, and 12). Although small but appreciable quantities of the minor diastereomeric products 3 were also formed in other cases, these were readily separated from the major isomers, the yields of which remained high (entries 1, 4, 5, 9, and 10). The enantiomeric excesses of the minor product were comparable to those of the major product in some cases (entries 1, 4, and 10), but were lower for substrates containing a 3-chlorophenyl or 2-pyridyl group attached to the enone carbonyl (entries 5 and 9). The process is also amenable to being conducted on a gram-scale. For example, the cyclization of 1f (1.00 g, 3.12 mmol) using 1.6 mol% of phosphoric acid 4b gave 2f as the only observable diastereomer in 84% yield and 90% ee after 90 h (entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Major product 2 | Minor product 3 |
a The reactions were performed with 1a–k (0.20 mmol) in cyclohexane (2 mL). Yields are of pure isolated single diastereomers. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase.
b The minor product was not detected.
c Conducted using 1f (1.00 g, 3.12 mmol) and 1.6 mol% of 4b in cyclohexane/toluene (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 50 °C for 90 h.
d The reaction was conducted in toluene. :1) at 50 °C for 90 h.
d The reaction was conducted in toluene. |
||||
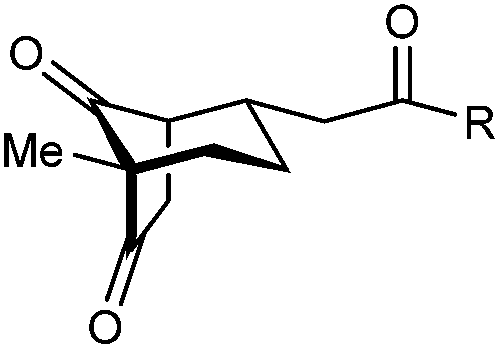
|
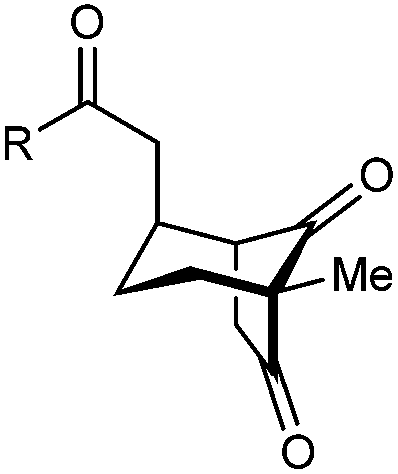
|
|||
| 1 | Me | Ph | 2a 93%, 91% ee | 3a 7%, 87% ee |
| 2 | 4-MeC6H4 | 2b 91%, 92% ee | 3b —b | |
| 3 | 4-MeOC6H4 | 2c 92%, 91% ee | 3c —b | |
| 4 | 4-ClC6H4 | 2d 80%, 94% ee | 3d 11%, 85% ee | |
| 5 | 3-ClC6H4 | 2e 79%, 86% ee | 3e 13%, 66% ee | |
| 6 | 2-Naphthyl | 2f 97%, 91% ee | 3f —b | |
| 7c | 2-Naphthyl | 2f 84%, 90% ee | 3f —b | |
| 8 | t-Bu | 2g 96%, 95% ee | 3g —b | |
| 9d | 2-Pyridyl | 2h 76%, 87% ee | 3h 20%, 29% ee | |
| 10 | 2-Furyl | 2i 80%, 88% ee | 3i 17%, 90% ee | |
| 11 | 2-Thienyl | 2j 97%, 92% ee | 3j —b | |
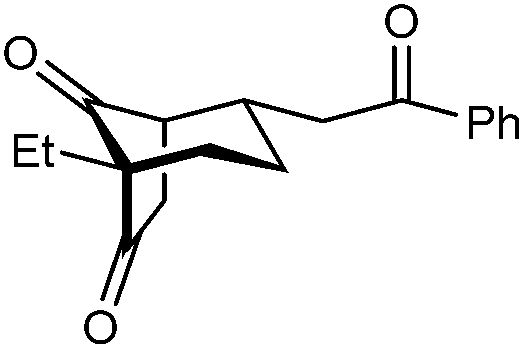
|
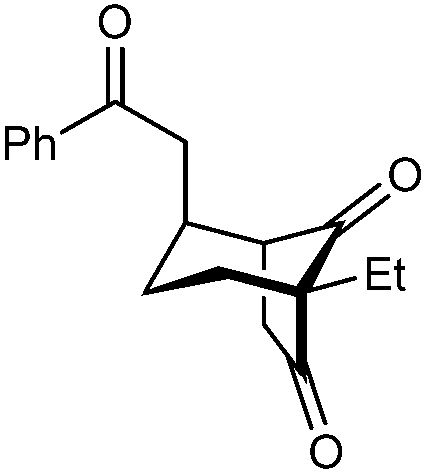
|
|||
| 12 | Et | Ph | 2k 95%, 93% ee | 3k —b |
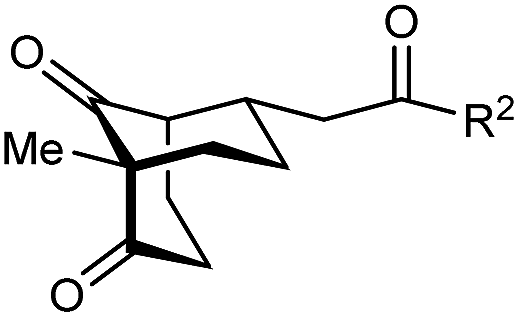
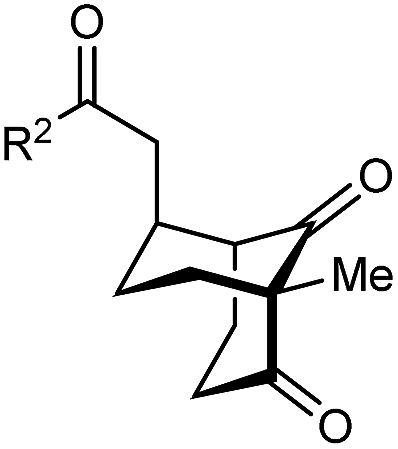
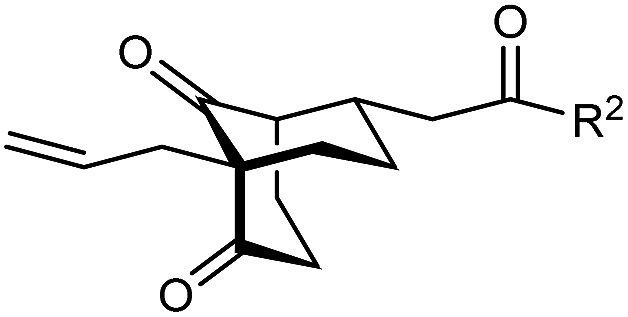
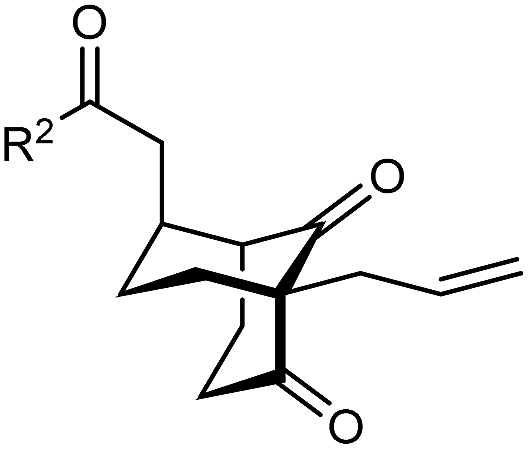
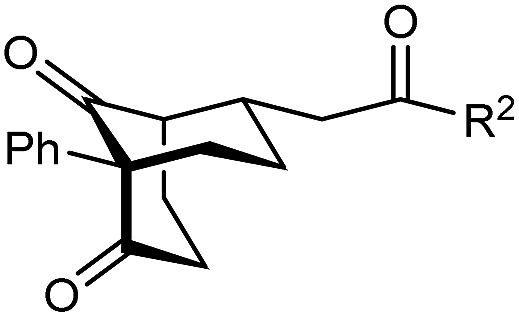
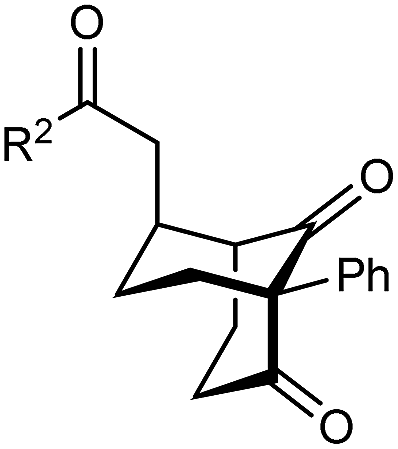
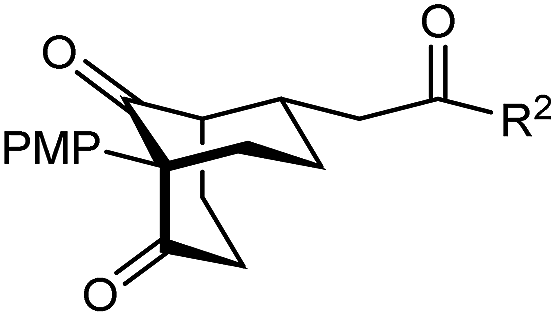
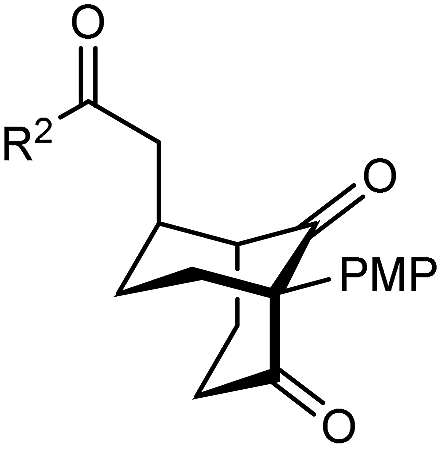
The synthesis of bicyclo[3.3.1]nonanes 6 with good enantioselectivities is also possible using this methodology (Table 3). As with the corresponding bicyclo[3.2.1]octanes, many of these reactions also resulted in diastereomeric products (Table 3, entries 1, 4–9, and 11–13). In general, the enantiomeric excess of the major products were slightly lower compared with those of the bicyclo[3.2.1] octanes 2 (see Table 2), though interestingly, the minor diastereomers 7 were usually formed in higher enantioselectivities. The process remained broadly tolerant of different (hetero)arenes at the enone carbonyl group, including ortho-substituted phenyl groups (entries 8 and 9). A 4-nitrophenyl ketone led to a more modest enantioselectivity for the major product (entry 6). The reaction was also compatible with an alkyl substituent at the enone carbonyl group that possesses enolizable protons; the cyclization of 5l gave products 6l and 7l in 59% combined yield (entry 12). Again, variation of the substituent at the 2-position of the 1,3-diketone was possible, with allyl (entries 13 and 14), phenyl (entries 15 and 16), and para-methoxyphenyl groups (entries 17 and 18) providing good results. In particular, aryl substituents at the 2-position had a beneficial effect on the enantioselectivity compared with the corresponding methyl-substituted analogues. For example, 6o and 6q were obtained in significantly higher enantioselectivity (94% ee, entries 15 and 17) compared with 6a (82% ee, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Major product 6 | Minor product 7 |
| a The reactions were performed with 5a–5r (0.20 mmol) in cyclohexane (2 mL). Yields are of pure isolated single diastereomers. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. b The minor product was not detected. c The reaction was conducted in toluene. d The reaction was conducted in cyclohexane (4 mL). PMP = para-methoxyphenyl. | ||||
|
|
|||
| 1 | Me | Ph | 6a 77%, 82% ee | 7a 14%, 94% ee |
| 2 | 4-MeC6H4 | 6b 95%, 86% ee | 7b —b | |
| 3 | 4-MeOC6H4 | 6c 94%, 87% ee | 7c —b | |
| 4 | 4-FC6H4 | 6d 82%, 86% ee | 7d 13%, 86% ee | |
| 5 | 4-ClC6H4 | 6e 73%, 87% ee | 7e 19%, 96% ee | |
| 6c | 4-NO2C6H4 | 6f 85%, 72% ee | 7f 15%, 88% ee | |
| 7d | 3-CF3C6H4 | 6g 68%, 86% ee | 7g 21%, 94% ee | |
| 8 | 2-MeOC6H4 | 6h 60%, 83% ee | 7h 24%, 93% ee | |
| 9 | 2-ClC6H4 | 6i 75%, 92% ee | 7i 20%, 85% ee | |
| 10 | 2-Naphthyl | 6j 96%, 87% ee | 7j —b | |
| 11 | 2-Pyridyl | 6k 75%, 82% ee | 7k 19%, 46% ee | |
| 12 | CH2CH2OBn | 6l 35%, 92% ee | 7l 24%, 80% ee | |
|
|
|||
| 13 | Allyl | Ph | 6m 89%, 86% ee | 7m 6%, 94% ee |
| 14 | 4-ClC6H4 | 6n 94%, 88% ee | 7n —b | |
|
|
|||
| 15 | Ph | Ph | 6o 68%, 94% ee | 7o —b |
| 16 | 2-Thienyl | 6p 50%, 97% ee | 7p —b | |
|
|
|||
| 17 | PMP | Ph | 6q 63%, 94% ee | 7q —b |
| 18 | 2-Thienyl | 6r 49%, 92% ee | 7r —b | |
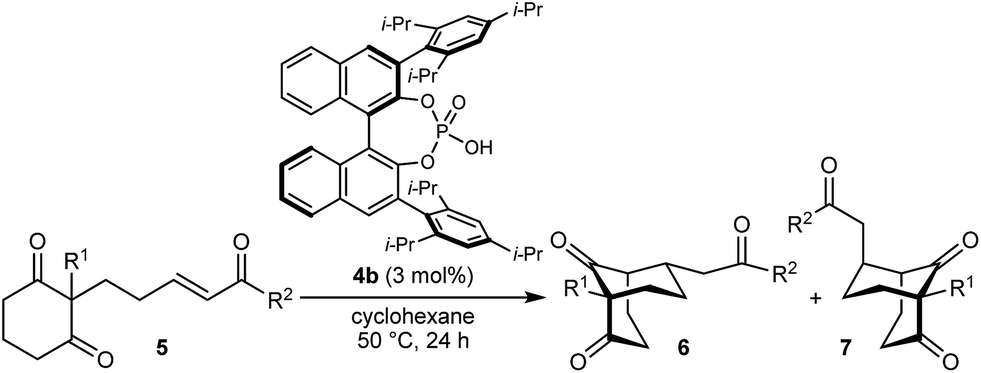
The process is not limited to α,β-unsaturated ketones as the Michael acceptor. For example, substrate 8, containing an α,β-unsaturated amide, successfully underwent cyclization to give bicyclo[3.3.1]nonane 9 in 52% yield and 77% ee, with the starting material being recovered in 40% yield (eqn (2)). Due to the lower reactivity of 8 compared with α,β-unsaturated ketones, a higher catalyst loading, temperature, and reaction time were required for reasonable results. Interestingly, the benzyl ester analogue of 8, in which the alkene is expected to be more electrophilic than in 8, was unreactive towards chiral phosphoric acid-catalyzed Michael cyclizations. This observation suggests that in addition to the electrophilicity of the electron-deficient alkene, the Lewis basicity of the oxygen atom of the α,β-unsaturated carbonyl (to facilitate binding of the chiral phosphoric acid) is also important for reactivity. Furthermore, the 2-alkenylbenzoxazole-containing substrate 10 also underwent cyclization in a good yield (eqn (3)) although the enantioselectivity of this reaction was modest (62% ee).18
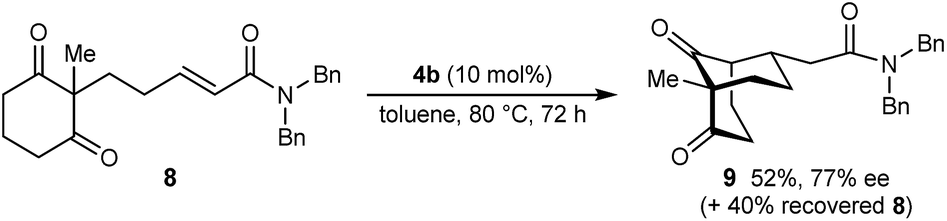 | (2) |
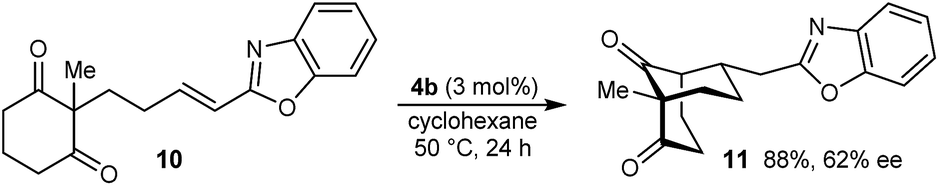 | (3) |
Scheme 2 presents a working hypothesis for the mode of action of the chiral phosphoric acid 4b, using substrate 1a for illustration. We propose that the catalyst promotes reversible keto-enol tautomerization of 1a,5 and is able to simultaneously bind the carbonyl group of the electrophilic enone and the hydroxyl group of the nucleophilic enol. The formation of the enantiomers 2a and ent-2a of the major product can be explained by chair-like conformations B and C, respectively, in which the enone occupies a pseudoequatorial position. The preferential formation of 2a is consistent with cyclization through conformation B being favored, in which the enol attacks the Si-face of the alkene.19 The formation of the two enantiomers 3a and ent-3a of the minor product can be explained by conformations D and E, where the enone occupies a pseudoaxial position. Again, attack of the Si-face of the alkene is favored (conformation D), leading to the preferential formation of 3a.19 This model is similar to one proposed by List and co-workers to explain the mode of enantioinduction in asymmetric chiral phosphoric acid-catalyzed Fischer indolizations.20
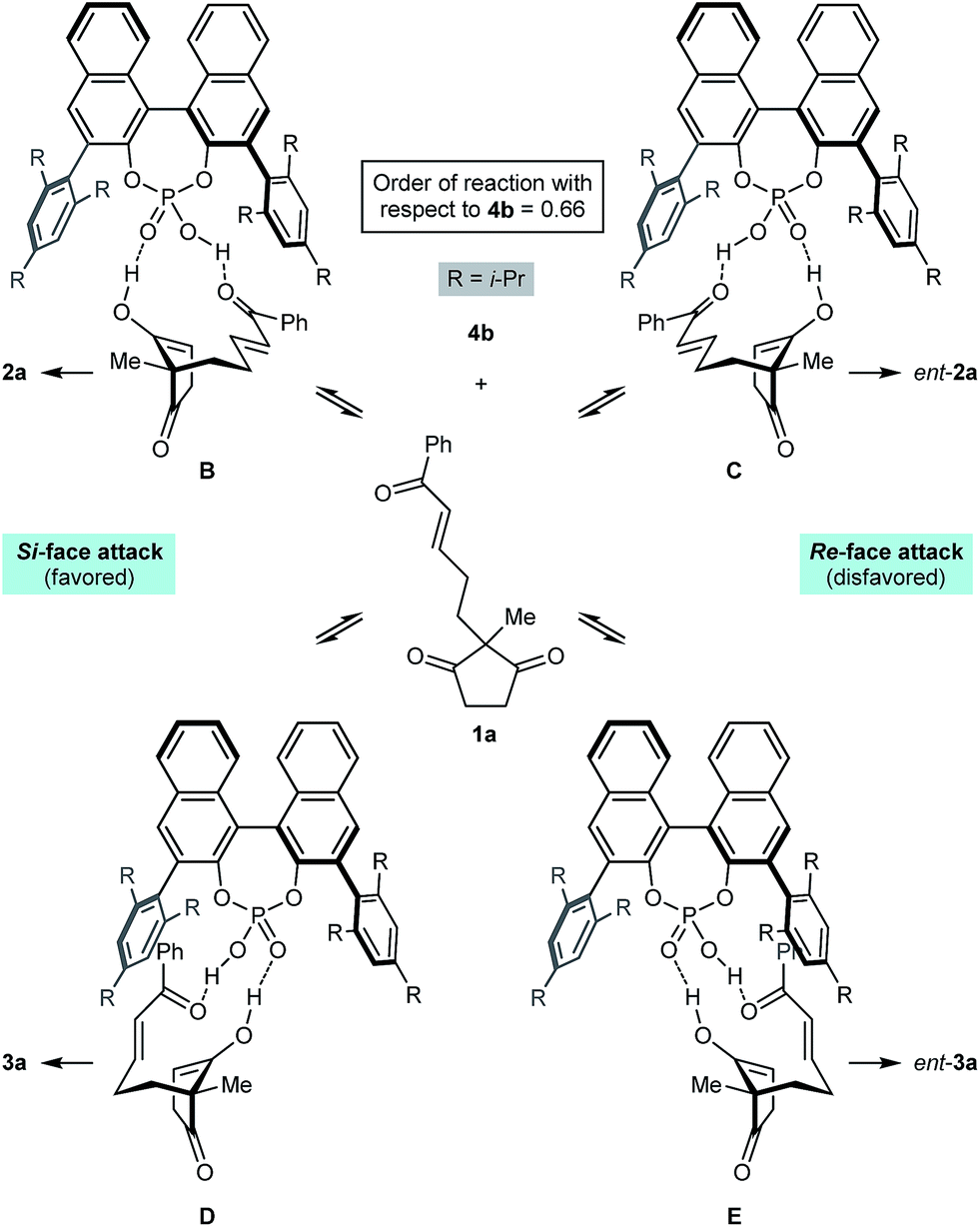 | ||
| Scheme 2 Proposed mode of action of catalyst 4b. | ||
Preliminary kinetic studies were also performed on the cyclization of 5d into 6d and 7d in toluene-d8 using different concentrations of catalyst 4b. From these experiments, the order of the reaction with respect to 4b was calculated to be 0.66.21 This non-integer value confirms the mechanism of the reaction is indeed complex, and may involve a series of equilibria as presented in Scheme 2. The complexity of the mechanism was further confirmed by a reaction in which the enantiomeric excess of 2j was measured during the course of the cyclization; the ee of 2j was not constant throughout, and increased from 70% ee after 1 h (15% conversion) to 90% ee after 24 h (75% conversion).21,22 Rationalization of these observations awaits the results of further studies.
In summary, the enantioselective synthesis of bicyclo[3.2.1]octanes and bicyclo[3.3.1]nonanes has been achieved by the chiral phosphoric acid-catalyzed Michael cyclizations of enone diones. These reactions involve the unusual enantioselective desymmetrization of 2,2-disubstituted cyclic 1,3-diketones, in which the bifunctional activation of the substrate by the catalyst is likely to be critical for success. This work further demonstrates the utility of chiral Brønsted acids in the enantioselective preparation of stereochemically complex structures, and investigation of these catalysts in other desymmetrization processes are likely to result in further advances in future. These studies, along with further mechanistic experiments, are topics for future study in our group.
Acknowledgements
We thank the EPSRC (Leadership Fellowship to H.W.L.), AstraZeneca, Pfizer, the University of Edinburgh, and the University of Nottingham for financial support. We thank Dr William Lewis (University of Nottingham) for X-ray crystallography, and Jorge Solana González for assistance in the preparation of substrates.Notes and references
- For reviews of bicyclo[3.n.1]alkanes, see: (a) M. Presset, Y. Coquerel and J. Rodriguez, Chem. Rev., 2013, 113, 525–595 CrossRef CAS PubMed; (b) M. Ruiz, P. Lopez-Alvarado, G. Giorgi and J. C. Menendez, Chem. Soc. Rev., 2011, 40, 3445–3454 RSC; (c) E. Butkus, Synlett, 2001, 1827–1835 CrossRef CAS PubMed; (d) M.-H. Filippini and J. Rodriguez, Chem. Rev., 1999, 99, 27–76 CrossRef CAS PubMed.
- For a review of the synthesis of bridge-containing compounds in general, see: W. Zhao, Chem. Rev., 2010, 110, 1706–1745 CrossRef CAS PubMed.
- M. Presset, Y. Coquerel and J. Rodriguez, ChemCatChem, 2012, 4, 172–174 CrossRef CAS PubMed.
- For examples, see: (a) N. Itagaki, M. Kimura, T. Sugahara and Y. Iwabuchi, Org. Lett., 2005, 7, 4185–4188 CrossRef CAS PubMed; (b) C. G. Bashore, M. G. Vetelino, M. C. Wirtz, P. R. Brooks, H. N. Frost, R. E. McDermott, D. C. Whritenour, J. A. Ragan, J. L. Rutherford, T. W. Makowski, S. J. Brenek and J. W. Coe, Org. Lett., 2006, 8, 5947–5950 CrossRef CAS PubMed; (c) C.-L. Cao, X.-L. Sun, Y.-B. Kang and Y. Tang, Org. Lett., 2007, 9, 4151–4154 CrossRef CAS PubMed; (d) M. Movassaghi and B. Chen, Angew. Chem., Int. Ed., 2007, 46, 565–568 CrossRef CAS PubMed; (e) M. L. Grachan, M. T. Tudge and E. N. Jacobsen, Angew. Chem., Int. Ed., 2008, 47, 1469–1472 CrossRef CAS PubMed; (f) B. M. Trost, P. J. McDougall, O. Hartmann and P. T. Wathen, J. Am. Chem. Soc., 2008, 130, 14960–14961 CrossRef CAS PubMed; (g) M. Rueping, A. Kuenkel, F. Tato and J. W. Bats, Angew. Chem., Int. Ed., 2009, 48, 3699–3702 CrossRef CAS PubMed; (h) C.-L. Cao, Y.-Y. Zhou, J. Zhou, X.-L. Sun, Y. Tang, Y.-X. Li, G.-Y. Li and J. Sun, Chem.–Eur. J., 2009, 15, 11384–11389 CrossRef CAS PubMed; (i) M. Rueping, A. Kuenkel and R. Fröhlich, Chem. –Eur. J., 2010, 16, 4173–4176 CrossRef CAS PubMed; (j) D. Ding, C.-G. Zhao, Q. Guo and H. Arman, Tetrahedron, 2010, 66, 4423–4427 CrossRef CAS PubMed; (k) C. Zhang, X.-H. Hu, Y.-H. Wang, Z. Zheng, J. Xu and X.-P. Hu, J. Am. Chem. Soc., 2012, 134, 9585–9588 CrossRef CAS PubMed; (l) C.-H. Wei, S. Mannathan and C.-H. Cheng, Angew. Chem., Int. Ed., 2012, 51, 10592–10595 CrossRef CAS PubMed; (m) W. Raimondi, M. d. M. Sanchez Duque, S. Goudedranche, A. Quintard, T. Constantieux, X. Bugaut, D. Bonne and J. Rodriguez, Synthesis, 2013, 45, 1659–1666 CrossRef CAS PubMed; (n) M. Tsakos, M. R. J. Elsegood and C. G. Kokotos, Chem. Commun., 2013, 49, 2219–2221 RSC; (o) A. Lefranc, L. Guénée, S. Goncalves-Contal and A. Alexakis, Synlett, 2014, 25, 2947–2952 CrossRef CAS PubMed; (p) A. Lefranc, L. Gremaud and A. Alexakis, Org. Lett., 2014, 16, 5242–5245 CrossRef CAS PubMed; (q) A. D. Gammack Yamagata, S. Datta, K. E. Jackson, L. Stegbauer, R. S. Paton and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 4899–4903 CrossRef CAS PubMed.
- For examples of transformations of unactivated ketones by chiral phosphoric acid-catalyzed enolization, see: (a) K. Mori, T. Katoh, T. Suzuki, T. Noji, M. Yamanaka and T. Akiyama, Angew. Chem., Int. Ed., 2009, 48, 9652–9654 CrossRef CAS PubMed; (b) G. Pousse, F. L. Cavelier, L. Humphreys, J. Rouden and J. Blanchet, Org. Lett., 2010, 12, 3582–3585 CrossRef CAS PubMed; (c) I. Felker, G. Pupo, P. Kraft and B. List, Angew. Chem., Int. Ed., 2015, 54, 1960–1964 CrossRef CAS PubMed; (d) X. Yang and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3205–3208 CrossRef CAS PubMed.
- (a) D. W. Low, G. Pattison, M. D. Wieczysty, G. H. Churchill and H. W. Lam, Org. Lett., 2012, 14, 2548–2551 CrossRef CAS PubMed; (b) A. R. Burns, J. Solana González and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 10827–10831 CrossRef CAS PubMed; (c) B. M. Partridge, J. Solana González and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 6523–6527 CrossRef CAS PubMed.
- For the racemic synthesis of compounds similar to 2a, see: (a) S. Danishefsky, G. Koppel and R. Levine, Tetrahedron Lett., 1968, 9, 2257–2260 CrossRef; (b) Y. Wang, A. Jaunet, P. Geoffroy and M. Miesch, Org. Lett., 2013, 15, 6198–6201 CrossRef CAS PubMed; (c) D.-H. Jhuo, B.-C. Hong, C.-W. Chang and G.-H. Lee, Org. Lett., 2014, 16, 2724–2727 CrossRef CAS PubMed.
- For selected reviews covering catalytic asymmetric Michael reactions, see: (a) C. Schneider and F. Abels, Org. Biomol. Chem., 2014, 12, 3531–3543 RSC; (b) M. M. Heravi, P. Hajiabbasi and H. Hamidi, Curr. Org. Chem., 2014, 18, 489–511 CrossRef CAS; (c) Y. Zhang and W. Wang, Catal. Sci. Technol., 2012, 2, 42–53 RSC; (d) J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 2007, 1279–1300 CrossRef PubMed; (e) S. C. Jha and N. N. Joshi, ARKIVOC, 2002, vii, 167–196 CrossRef.
- During the final preparation of this manuscript, the synthesis of 2-azabicyclo[3.3.1]nonanes by catalytic enantioselective desymmetrizing Michael cyclizations was reported see ref. 4q.
- For reviews of asymmetric deprotonations, including enolizations, see: (a) N. S. Simpkins and M. D. Weller, in Stereochemical Aspects of Organolithium Compounds, Verlag Helvetica Chimica Acta, 2010, pp. 1–52 Search PubMed; (b) K. W. Henderson and W. J. Kerr, Chem. –Eur. J., 2001, 7, 3430–3437 CrossRef CAS; (c) P. O'Brien, J. Chem. Soc., Perkin Trans. 1, 1998, 1439–1458 RSC; (d) P. J. Cox and N. S. Simpkins, Tetrahedron: Asymmetry, 1991, 2, 1–26 CrossRef CAS; (e) N. S. Simpkins, Chem. Soc. Rev., 1990, 19, 335–354 RSC.
- J. I. Seeman, Chem. Rev., 1983, 83, 83–134 CrossRef CAS.
- For seminal reports of the use of chiral phosphoric acids as catalysts in asymmetric synthesis, see: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999–1010 CrossRef CAS PubMed; (b) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (c) M. Terada, Chem. Commun., 2008, 4097–4112 RSC; (d) D. Kampen, C. M. Reisinger and B. List, in Asymmetric Organocatalysis, ed. B. List, 2009, pp. 395–456 Search PubMed; (e) M. Terada, Synthesis, 2010, 1929–1982 CrossRef CAS PubMed; (f) M. Terada, Bull. Chem. Soc. Jpn., 2010, 83, 101–119 CrossRef CAS; (g) A. Zamfir, S. Schenker, M. Freund and S. B. Tsogoeva, Org. Biomol. Chem., 2010, 8, 5262–5276 CAS; (h) M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539–4549 RSC; (i) M. Terada, Curr. Org. Chem., 2011, 15, 2227–2256 CrossRef CAS; (j) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS PubMed; (k) P. Nagorny, Z. Sun and G. A. Winschel, Synlett, 2013, 24, 661–665 CrossRef CAS PubMed; (l) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- For enantioselective desymmetrizations of dicarbonyl compounds by preferential nucleophilic addition to one carbonyl group, catalyzed by chiral phosphoric acids or their derivatives, see ref. 5a and: (a) K. Higuchi, S. Suzuki, R. Ueda, N. Oshima, E. Kobayashi, M. Tayu and T. Kawasaki, Org. Lett., 2015, 17, 154–157 CrossRef CAS PubMed; (b) J.-B. Gualtierotti, D. Pasche, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 9926–9930 CrossRef CAS PubMed; (c) J. Wilent and K. S. Petersen, J. Org. Chem., 2014, 79, 2303–2307 CrossRef CAS PubMed; (d) M. Yang, Y.-M. Zhao, S.-Y. Zhang, Y.-Q. Tu and F.-M. Zhang, Chem. –Asian J., 2011, 6, 1344–1347 CrossRef CAS PubMed; (e) V. N. Wakchaure and B. List, Angew. Chem., Int. Ed., 2010, 49, 4136–4139 CrossRef CAS PubMed.
- For examples of enantioselective Michael additions of ketones catalyzed (or co-catalyzed) by chiral phosphoric acids, see ref. 5b and: (a) T. Akiyama, T. Katoh and K. Mori, Angew. Chem., Int. Ed., 2009, 48, 4226–4228 CrossRef CAS PubMed; (b) M. Yamanaka, M. Hoshino, T. Katoh, K. Mori and T. Akiyama, Eur. J. Org. Chem., 2012, 4508–4514 CrossRef CAS PubMed; (c) L. Liu, D. Wu, X. Li, S. Wang, H. Li, J. Li and W. Wang, Chem. Commun., 2012, 48, 1692–1694 RSC.
- The relative and absolute stereochemistries of the products described herein were assigned by analogy to those of products 2d, 3a, 6e, and 6f, which were determined by X-ray crystallography. See the ESI.†.
- Although the substrate and phosphoric acid 4b are poorly soluble in cyclohexane at room temperature, the reaction mixtures become homogeneous upon heating to 50 °C.
- For catalytic enantioselective conjugate additions of carbon-centered nucleophiles to 2-alkenylazaarenes, see: (a) A. Baschieri, L. Bernardi, A. Ricci, S. Suresh and M. F. A. Adamo, Angew. Chem., 2009, 121, 9506–9509 CrossRef PubMed; (b) G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373–14375 CrossRef CAS PubMed; (c) A. Saxena and H. W. Lam, Chem. Sci., 2011, 2, 2326–2331 RSC; (d) T. Akiyama, T. Katoh and K. Mori, Angew. Chem., Int. Ed., 2009, 48, 4226–4228 CrossRef CAS PubMed; (e) A. A. Friedman, J. Panteleev, J. Tsoung, V. Huynh and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 9755–9758 CrossRef CAS PubMed; (f) I. D. Roy, A. R. Burns, G. Pattison, B. Michel, A. J. Parker and H. W. Lam, Chem. Commun., 2014, 50, 2865–2868 RSC; (g) D. Best and H. W. Lam, J. Org. Chem., 2014, 79, 831–845 CrossRef CAS PubMed.
- However, the precise manner in which the catalyst 4b favors Si-face attack is not readily explained at the present time.
- S. Müller, M. J. Webber and B. List, J. Am. Chem. Soc., 2011, 133, 18534–18537 CrossRef PubMed.
- See ESI for full details.†.
- This experiment was conducted in toluene rather than cyclohexane to ensure complete homogeneity of the reaction mixture.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data for new compounds, and crystallographic data for 2d, 3a, 6e, and 6f. CCDC 1045000–1045003. See DOI: 10.1039/c5sc00753d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |