
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddressing, amplifying and switching DNAzyme functions by electrochemically-triggered release of metal ions
Lina
Freage†
a,
Alexander
Trifonov†
a,
Ran
Tel-Vered
a,
Eyal
Golub
a,
Fuan
Wang
a,
John S.
McCaskill
b and
Itamar
Willner
*a
aInstitute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel. E-mail: willnea@vms.huji.ac.il
bBiomolecular Information Processing (BioMIP), Ruhr-Universität Bochum, Universitätsstr 150, Bochum, 44801, Germany
First published on 8th April 2015
Abstract
The design of artificial cells, which mimic the functions of native cells, is an ongoing scientific goal. The development of stimuli-responsive chemical systems that stimulate cascaded catalytic transformations, trigger chemical networks, and control vectorial branched transformations and dose-controlled processes, are the minimum requirements for mimicking cell functions. We have studied the electrochemical programmed release of ions from electrodes, which trigger selective DNAzyme-driven chemical reactions, cascaded reactions that self-assemble catalytic DNAzyme polymers, and the ON–OFF switching and dose-controlled operation of catalytic reactions. The addressable and potential-controlled release of Pb2+ or Ag+ ions into an electrolyte that includes a mixture of nucleic acids, results in the metal ion-guided selection of nucleic acids yielding the formation of specific DNAzymes, which stimulate orthogonal reactions or activate DNAzyme cascades.
The design of artificial cells is a major scientific challenge that has attracted substantial research efforts in the last two decades.1,2 Different approaches to construct simple, cell-like structures for specific applications have been reported.3–6 Different components have been integrated with cell-mimetic compartments, and the implementation of these constructs to drive complex transformations has been discussed.7,8 Although important scientific progress in developing building units of artificial cells has been reported, an operational man-made cell is still an unresolved goal. Different challenges exist in developing artificial cells. These include the fabrication of membrane-like compartments,9,10 the development of amplification feedback mechanisms and cascaded chemical transformations responding to environmental stimuli,11 and the replication of the cell configuration and its constituent parts.12,13 Tackling these issues would allow for the construction of complex chemical networks capable of controlling vectorial branched transformations, dose-controlled processes, oscillatory reactions etc. By studying the electrochemical properties of electrodes, and the triggering of specific chemical transformations by electrical stimuli to the extent that cell-like systems are duplicated, might provide a basis to construct “electronic cells”.14 That is, the addressable, potential-induced release of different ions, the local electrically-stimulated pH changes that control the local electrical properties at the electrodes, or the separation of molecular/biomolecular complexes, which regulate chemical transformations and catalytic cascades might provide important steps towards an electronic cell. Although substantial research efforts to develop “artificial cells” have been reported, limited advances have been made and the concept remains a scientific “holy grail”. Here we report the first step to develop an electronic (electrochemical) cell that highlights the electrical addressing of the electrodes, the release and uptake of metal ions from the electrodes, the subsequent control of catalytic nucleic acids (DNAzymes), and the activation of DNAzyme cascades. Specifically, the electrically-triggered, dose-controlled release of the ions allows for the regulation of secondary DNAzyme-catalyzed reactions.
Catalytic nucleic acids, DNAzymes, have attracted recent research efforts as catalytic labels for amplifying sensing events,15–19 as catalysts for the activation of DNA machines,20–22 and as building blocks for the assembly of nanostructures.23 Specifically, metal ion-dependent DNAzymes that stimulate the hydrolytic nicking of nucleic acids,24–26 and hemin/G-quadruplex horseradish peroxidase-mimicking DNAzymes were reported.27–29 In the present study, we electrically trigger the release of Pb2+ and Ag+ ions from electrode surfaces, thereby activating the secondary Pb2+-dependent DNAzyme30–32 and the hemin/G-quadruplex DNAzyme, respectively. We demonstrate the cyclic and reversible electrical “ON”/“OFF” activation and deactivation of the DNAzymes, and highlight the DNAzyme-driven operation of a catalytic cascade that synthesizes polymeric DNAzyme wires.
Results and discussion
The study is based on the electrochemical deposition of layers of Pb0 and/or Ag0 on Au supports. These layers act as metallic reservoirs that can be stripped off from the electrodes upon the application of specific bias potentials. Fig. 1, curves (a) and (b), depicts the linear sweep voltammograms (LSVs) corresponding to the stripping of the Pb2+ or the Ag+ ions from the Pb0 or Ag0 reservoirs, respectively. Fig. 1, curve (c), shows an LSV corresponding to the stripping of both Pb2+ and Ag+ from an electrode which contains the two metal reservoirs. The results imply that upon application of a potential higher than −0.6 V vs. the Ag quasi-reference electrode (QRE), Pb2+ ions are released from the Pb0-deposited surface, whereas application of a potential higher than 0.1 V vs. the Ag QRE oxidizes the Ag0 reservoir and releases Ag+ ions. Subjecting the electrode that includes the two metallic reservoirs to a potential higher than 0.1 V vs. the Ag QRE, results in the release of both metal ions from the electrode. Furthermore, the potential applied on the electrode determines the specific metal which is oxidized to the solution and the extent of the release process, while the amount of released metal ions can also be controlled by the time-interval of the applied potential step. Therefore, this potential-induced release of metal ions from the electrode can then be designed to electrochemically trigger interactions between these metal ions and nucleic acids solubilized in the electrolyte. Specifically, our study demonstrated that electrochemically-released Pb2+ ions triggered the operation of the Pb2+-dependent DNAzyme and that the electrochemical release of Ag+ ions cooperatively stabilized a DNA duplex through the formation of cytosine–Ag+–cytosine (C–Ag+–C) bridges. | ||
| Fig. 1 Linear sweep voltammograms (LSVs) corresponding to the electrochemical release of: (a) Pb2+ ions from a Pb0 reservoir associated with an electrode, (b) Ag+ ions from a Ag0 reservoir associated with an electrode, and (c) both Pb2+ and Ag+ ions from a Pb0/Ag0 reservoir deposited on an electrode. All measurements were performed in a HEPES buffer (0.05 M, pH = 7.0) containing NaCl, 50 mM. Scan rate: 100 mV s−1. | ||
Fig. 2(A) depicts the electrically-controlled activation of the Pb2+-dependent DNAzyme. Lead was deposited on a Au electrode and served as the source of Pb2+. The Pb0-functionalized electrode was immersed in an electrolyte solution that included the Pb2+-dependent DNAzyme sequence (1) and its fluorophore/quencher (ROX/BH2)-functionalized substrate, (2). Subjecting the electrode to a potential step from −0.6 V to −0.2 V vs. the Ag quasi-reference electrode (QRE) resulted in the stripping of the Pb2+ ions into the solution. The time interval of the applied potential pulse controlled the amount of released Pb2+, which activated the Pb2+-dependent DNAzyme, catalyzing the nicking of the substrate (2). The cleavage of (2) was transduced by the fluorescence of the fluorophore, arising from the separation of the fluorophore from the quencher. Fig. 2(B), curves (a) to (c), depicts the time-dependent fluorescence changes of the system subjected to potential steps of different time intervals. As the time interval of the potential step is increased, the time-dependent fluorescence changes, as a result of the cleavage of (2), are intensified, consistent with a higher amount of released Pb2+ ions. The fact that no fluorescence changes are observed, implies that the potential-induced release of Pb2+ ions is essential for the activation of the DNAzyme. Fig. 2(B), curve (d), shows the time-dependent fluorescence changes of the system without applying the potential to release the Pb2+ ions. Introduction of Na2S into the electrolyte solution results in the precipitation of the electrically-released Pb2+ ions in the form of PbS, leading to the blocking of the DNAzyme activity, as shown in Fig. 2(B), curve (e). Evidently, no fluorescence changes are observed in the absence of the metal ions. Fig. 2(B), top, shows the emission spectra recorded after the application of the oxidation potential pulse for different time intervals. These results confirm that the electrically-released Pb2+ ions activate the catalytic functions of the DNAzyme.
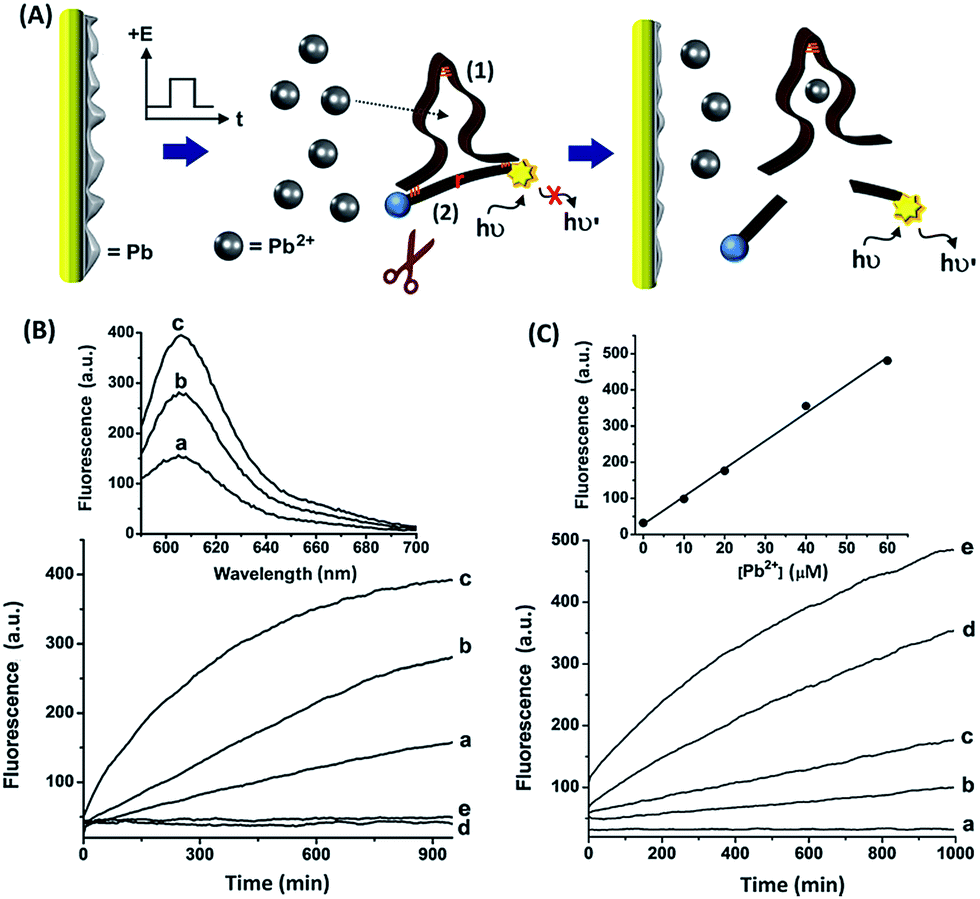 | ||
| Fig. 2 (A) Electrochemically-triggered activation of the Pb2+-dependent DNAzyme. (B) Time-dependent fluorescence changes, at λ = 590 nm, as a result of the Pb2+-stimulated, DNAzyme cleavage of (2) upon subjecting the electrode to potential pulses at −0.2 V vs. the Ag QRE for different time-intervals: (a) 1, (b) 2, and (c) 3 s. Curve (d) corresponds to the time-dependent fluorescence changes of the system in the absence of an applied potential, and curve (e) shows the time-dependent fluorescence changes of the system upon applying a potential of −0.2 V vs. the Ag QRE for 3 s with Na2S, 65 mM, added to the system. Top: fluorescence spectra of the systems described in curves (a)–(c) generated after 950 minutes. (C) Time-dependent fluorescence changes upon the manual activation of the Pb2+-dependent DNAzyme in the presence of variable Pb2+ concentrations: (a) 0, (b) 10, (c) 20, (d) 40, and (e) 60 μM Pb2+. Top: derived calibration curve corresponding to the fluorescence of the system generated at different concentrations of added Pb2+ ions after a fixed time interval of 1000 min. All measurements were performed in a HEPES buffer solution (0.05 M, pH = 7.0) containing NaCl, 50 mM, (1), 1 μM, and (2), 0.75 μM. | ||
We further examined the catalytic activity of the Pb2+-dependent DNAzyme towards the hydrolytic cleavage of (2) by manually adding Pb2+ ions to vary the Pb2+ concentration. Fig. 2(C) depicts the time-dependent fluorescence changes upon subjecting the mixture of (1) and (2) to solutions containing variable concentrations of Pb2+ ions. As the concentration of Pb2+ ions increases, the fluorescence changes are intensified, consistent with the higher activity of the Pb2+-dependent DNAzyme at elevated concentrations of Pb2+ ions. Fig. 2(C), top, depicts the calibration curve corresponding to the fluorescence intensities generated by the system after a fixed time interval of 1000 minutes, as a function of the concentration of the manually added Pb2+ ions. The derived calibration curve allows us to evaluate the concentrations of Pb2+ released upon applying the oxidation pulses of 1, 2, and 3 seconds to be 16, 33, and 50 μM, respectively. Coulometric analyses of the electrical release of Pb2+ during the three time interval pulses revealed the associated charges to be 1.0, 1.8, and 2.5 mC. Hence, the current efficiencies for the release of the Pb2+ ions corresponded to 62, 70, and 77%, respectively.
We next attempted to activate a different DNAzyme through the electrically-driven release of another ion, Ag+. This is exemplified in Fig. 3(A), which shows the electrically-triggered release of Ag+ ions and the activation of the hemin/G-quadruplex horseradish peroxidase-mimicking DNAzyme. The design of this system is based on the following elements: (i) Ag+ ions form cytosine–Ag+–cytosine complexes, and these cooperatively stabilize the formation of duplex nucleic acid structures,33 (ii) subunits of the G-quadruplex sequence can self-assemble in the presence of K+ to yield G-quadruplexes.34,35 The quadruplexes can be stabilized by cooperative duplex domains between the subunits. Accordingly, we designed two nucleic acids, (3) and (4), that included the G-quadruplex domains I and II, and the domains III and IV that include partial complementary sequences and two C–C mismatches, respectively. Under the experimental conditions, the strands (3) and (4) do not assemble into a stable G-quadruplex, and the formation of the hemin/G-quadruplex DNAzyme is prohibited. The electrically-triggered release of Ag+ ions from the electrode results in the formation of stable C–Ag+–C-bridged duplexes between domains III/IV of (3)/(4), resulting in the synergistic stabilization of the G-quadruplex. The incorporation of hemin into the G-quadruplex then yields the catalytically-active hemin/G-quadruplex DNAzyme. The activity of the DNAzyme is followed by the DNAzyme-catalyzed oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid), ABTS2−, by H2O2, and the formation of the colored product, ABTS˙− (λ = 415 nm). Fig. 3(B) depicts the time-dependent absorbance changes generated by the hemin/G-quadruplex DNAzyme, which is formed upon the electrically-triggered release of Ag+ ions by voltammetric pulses applied on the electrode for different time intervals. In these experiments, Ag0 was collected on a Au electrode and acted as a reservoir for Ag+ ions. The electrode was then subjected to a potential step from −0.6 V to 0.2 V vs. the Ag QRE, and the Ag+ ions were stripped off for different time intervals (100, 200 and 300 ms). The released Ag+ ions self-assembled the hemin/G-quadruplex DNAzyme structure, and its formation was probed by the catalytic oxidation of ABTS2− by H2O2. As the time interval of the applied stripping pulse increased, the DNAzyme-catalyzed oxidation of ABTS2− was enhanced, consistent with the higher content of the DNAzyme generated upon the triggered release of Ag+, shown in curves (a) to (c). Control experiments revealed that using potential pulses that did not strip off the Ag+ ions, or in the absence of an applied potential on the electrode, the formation of the hemin/G-quadruplex was prohibited, as shown by Fig. 3(B), curve (d). The observation of the minor formation of ABTS˙− is attributed to the inefficient H2O2-stimulated oxidation of ABTS2− by free hemin in the system. In a comparative assay, a series of fixed concentrations of Ag+ ions were added to the solution containing the two nucleic acids (3) and (4), and by monitoring the catalytic oxidation of ABTS2− by H2O2, the current efficiency corresponding to the electrochemical release of the Ag+ ions in Fig. 3(B) was estimated to be 76–80%.
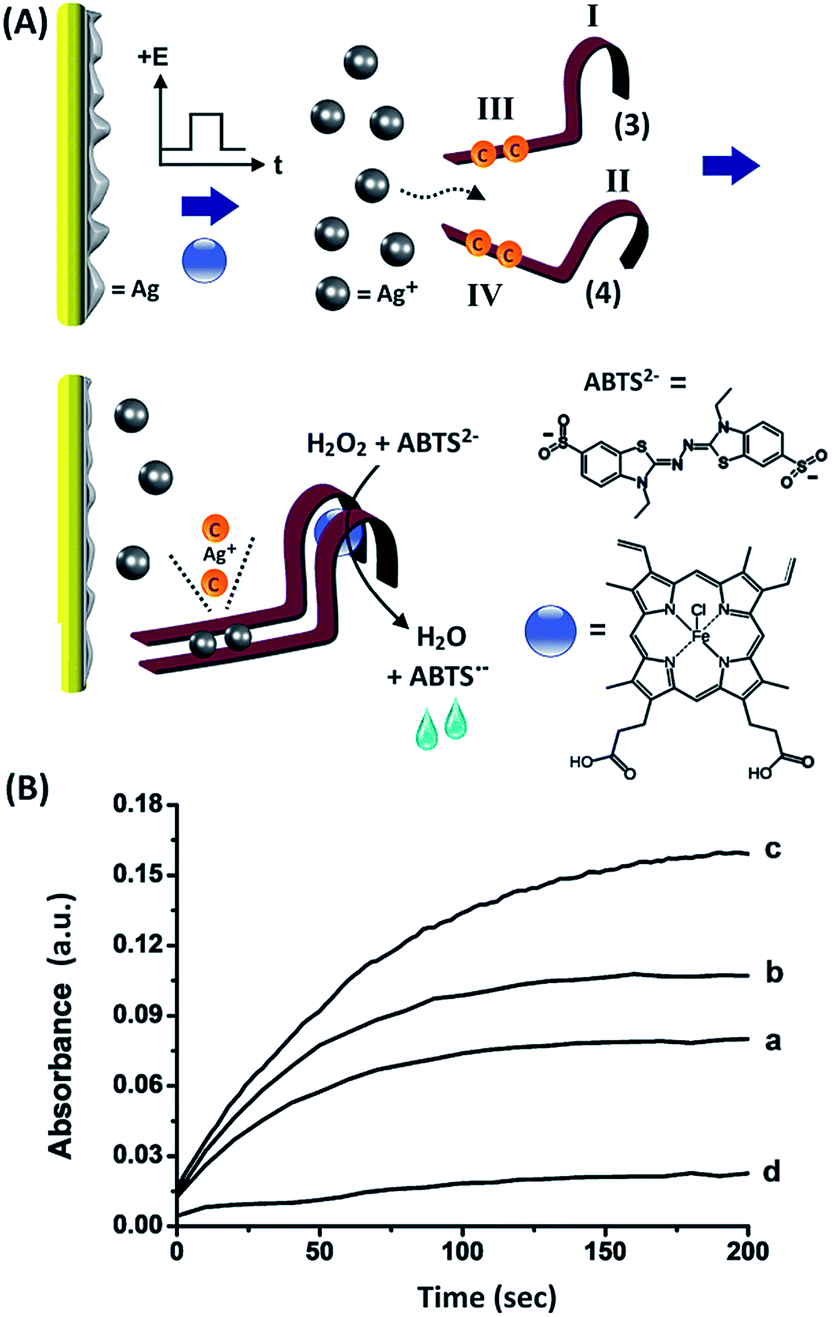 | ||
| Fig. 3 (A) Electrochemically-triggered release of Ag+ ions resulting in the activation of an HRP-mimicking DNAzyme by the Ag+ ion-induced assembly of a hemin/G-quadruplex structure. (B) Time-dependent absorbance changes, at λ = 415 nm, as a result of the DNAzyme-catalyzed oxidation of ABTS2− by H2O2. The oxidation of Ag was carried out by the application of potential pulses at E = +0.2 V vs. the Ag QRE for variable time intervals: (a) 100, (b) 200, and (c) 300 msec. Curve (d) corresponds to the absorbance changes recorded in the absence of an applied potential pulse. All measurements were performed in a HEPES buffer (0.05 M, pH = 7.0) containing (3), 1 μM, (4), 1 μM, ABTS2−, 2 mM, H2O2, 2 mM, and hemin 0.5 μM. | ||
The electrical release of Pb2+ or Ag+ ions, and the sequestered activation of DNAzyme-driven transformations reveal the possibility of addressing the electrochemistry of different electrodes and the ability to program catalytic transformations of mixtures of metal ion-controlled DNAzymes. Nonetheless, the fact that the ion release proceeds at different potentials suggests that a single conductive support that contains different metal-ion reservoirs could selectively release one or more ions, depending on the applied potential pulses, thus dictating the subsequent catalytic reactions. Accordingly, the circuit containing the two electrodes functionalized with Pb0 and Ag0 was shorted, so that the electrodes could be subjected to the same externally biased potentials. Fig. 4(A) depicts the catalytic properties of the system upon subjecting the two electrodes to an external potential pulse of −0.2 V vs. the Ag QRE (time interval 3 s), at which only Pb2+ is being released.
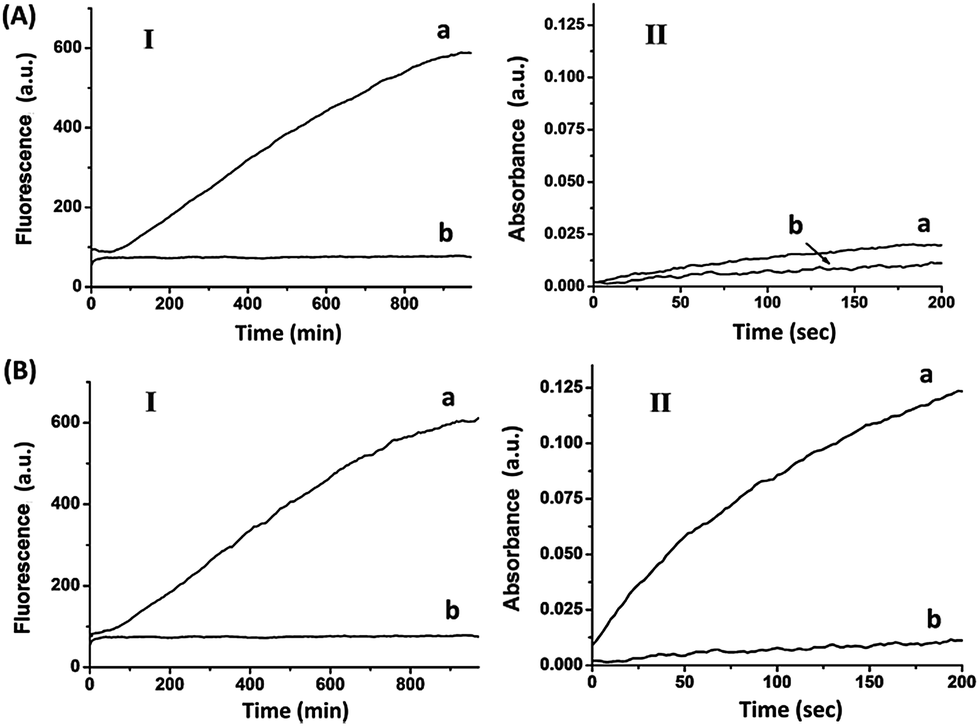 | ||
| Fig. 4 (A) Time-dependent fluorescence at λem = 590 nm (Panel I), and time-dependent absorbance at λ = 415 nm (Panel II), corresponding to the electrochemical release of Pb2+ ions from the short-circuited Pb- and Ag-deposited Au surfaces: (a) upon the application of a potential pulse at E = −0.2 V vs. the Ag QRE for 3 s, (b) in the absence of an applied potential. (B) Time-dependent fluorescence at λem = 590 nm (Panel I), and time-dependent absorbance at λ = 415 nm (Panel II), corresponding to the electrochemical release of Pb2+ and Ag+ ions from the short-circuited Pb- and Ag-deposited Au surfaces: (a) upon the application of a potential pulse at E = +0.2 V vs. the Ag QRE for 3 s, (b) in the absence of an applied potential. All measurements were performed in a HEPES buffer (0.05 M, pH = 7.0) containing (1), 1 μM, (2), 0.75 μM, (3), 1 μM, (4), 1 μM, ABTS2−, 2 mM, H2O2, 2 mM, and hemin 0.5 μM. | ||
The results indicate that under these conditions only the Pb2+-dependent DNAzyme is activated, Fig. 4(A), Panel I, curve (a), while the hemin/G-quadruplex DNAzyme is not formed, as shown by Panel II, curve (a). Fig. 4(B) shows the catalytic functions of the system upon the application of a potential pulse from −0.6 V to 0.2 V. Under these conditions, the two ions Pb2+ and Ag+ ions are released, leading to the activation of the Pb2+-dependent DNAzyme, Fig. 4(B), Panel I, curve (a), and of the hemin/G-quadruplex DNAzyme, Panel II, curve (a).
The systems discussed up to now have demonstrated the electrically-triggered release of metal ions. The reverse uptake of metal ions could, however, switch off the catalytic functions of the DNAzyme, thus introducing an additional means to control the catalytic functions of the cell. The reversible “ON–OFF” electrical switching of the Pb2+-dependent DNAzyme by the cyclic release/uptake of the Pb2+ ions is shown in Fig. 5. In these experiments, the Pb2+ ions were released from the Pb0 reservoir associated with the electrode, thus activating the Pb2+-dependent DNAzyme, shown as release point R1 in Fig. 5, curve (a). At the time marked with U1, the electrode was biased at −0.95 V vs. the Ag QRE (under stirring conditions). The uptake of Pb2+ by the electrode resulted in a decrease in the rate of the biocatalytic process, which was significantly but not completely blocked.
 | ||
| Fig. 5 Time-dependent fluorescence at λem = 590 nm, corresponding to the electrochemical release (R) of Pb2+ ions from the electrode and the activation of the Pb2+-dependent DNAzyme, or the uptake (U) of the Pb2+ ions from the DNAzyme-containing solution and their deposition onto the Au surface. Curve (a) corresponds to the intermittent release and uptake of Pb2+ ions to and from the solution through the repetitive application of oxidative stripping pulses at −0.2 V vs. the Ag QRE for 3 s at the times indicated by the arrows R1–4, and reductive pulses at −0.95 V vs. the Ag QRE for 300 s at the times indicated by the arrows U1–3. The measurements were performed under stirring conditions. Curve (b) corresponds to the time-dependent fluorescence changes recorded upon the application of a single oxidation step at −0.2 V vs. the Ag QRE for 3 s at the time indicated by the arrow R1. All measurements were performed in a HEPES buffer (0.05 M, pH = 7.0) containing NaCl, 50 mM, the Pb2+-dependent DNAzyme sequence, (1), 1 μM, and the ROX/BH2-functionalized substrate (2), 0.75 μM. | ||
The time intervals between ion release (R) and uptake (U) signals were then shortened, resulting in more complete blocking. At point R2, the system was re-subjected to the potential step at −0.2 V, releasing Pb2+ ions, which led to the reactivation of the catalytic process. A second reduction pulse was applied at U2, leading to a further blockage of the biocatalytic process. By the cyclic application of the potential steps to release (R) and uptake (U) the Pb2+ ions, the catalytic functions of the system were switched between the “ON”/“OFF” states, respectively. For comparison, Fig. 5, curve (b), shows the continuous catalytic function of the Pb2+-dependent DNAzyme upon the application of a single potential step at −0.2 V for 3 s. Furthermore, the electrically-triggered activation of the Pb2+-dependent DNAzyme was implemented to stimulate a catalytic cascade that synthesizes DNAzyme wires, as shown in Fig. 6(A). In this system, the Pb0-modified electrode is subjected to a mixture, consisting of the Pb2+-dependent DNAzyme sequence, (5), its substrate, (6), and two hairpins, Hα (7) and Hβ (8). The fluorophore/quencher-modified substrate of the Mg2+-dependent DNAzyme was also included in the mixture. The hairpins contained the Mg2+-dependent DNAzyme subunits I and II. The electrically-triggered activation of the Pb2+-dependent DNAzyme leads to the cleavage of (6) and the fragmented product, (9), is complementary to a domain of hairpin Hα. Opening of hairpin Hα drives the hybridization chain reaction (HCR)36 that results in the cross-opening of hairpins Hα and Hβ to yield the polymer wire.
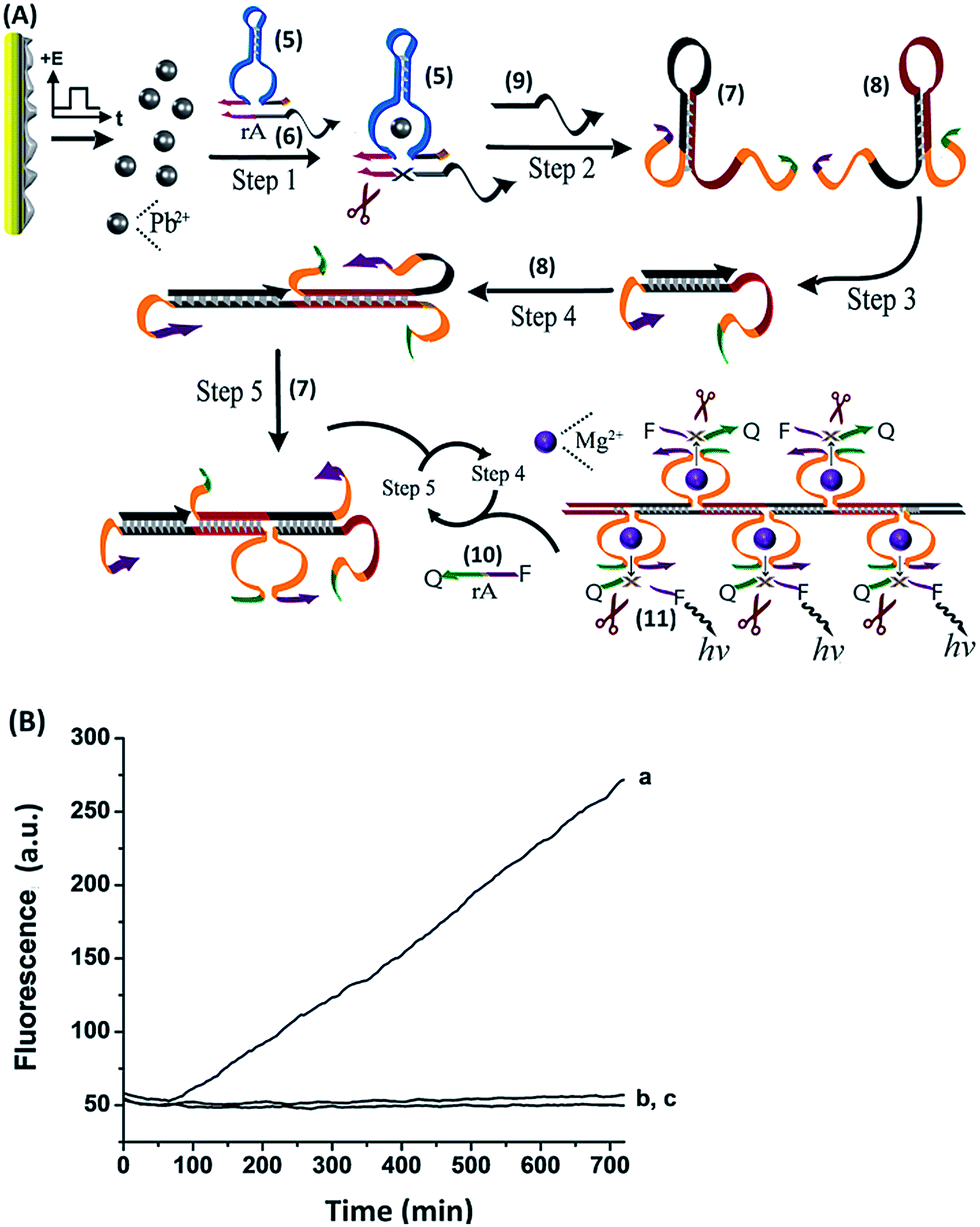 | ||
| Fig. 6 (A) The electrochemically-triggered activation of a catalytic cascade that synthesizes DNAzyme nanowires through the primary release of Pb2+ ions and the subsequent hybridization chain reaction. (B) Time-dependent fluorescence measurements at λem = 590 nm, corresponding to: (a) the electrochemical release of Pb2+ ions from a Pb-deposited Au surface upon the application of a potential pulse at E = −0.2 V vs. the Ag QRE for 3 s, (b) time-dependent fluorescence response of the system in the absence of an applied potential pulse to release Pb2+ ions, or (c) upon the application of the potential pulse in the absence of the substrate sequence (6). All systems included a HEPES buffer (0.1 M, pH = 7.0) containing NaCl, 50 mM, and the DNAs (5), 0.2 μM, (6), 0.05 μM, (7), 3 μM, (8), 3 μM, (10) 0.75 μM. | ||
The tethers I and II associated with the polymer wire self-assemble into Mg2+-dependent DNAzyme units37 that catalyze the cleavage of (10). The fluorescence of the fragmented product, (11), then provides the readout signal for the DNAzyme cascade. Fig. 6(B), curve (a) depicts the time-dependent fluorescence changes as a result of the operation of the DNAzyme cascade. Control experiments indicate that the Pb2+-dependent DNAzyme sequence, (5), and the substrate (6), do not activate the DNAzyme cascade in the absence of the electrically-triggered release of Pb2+ ions, as shown by curve (b). Similarly, exclusion of the substrate (6) from the system does not lead to the activation of the DNAzyme cascade and to the formation of the Mg2+-dependent DNAzymes upon the electrically-triggered release of Pb2+ ions, as shown by curve (c). These experiments imply that the primary electrically-triggered release of the Pb2+ ions, and the Pb2+ ion-dependent DNAzyme cleavage of (6), yield the product (9) that initiates the HCR process and the formation of the Mg2+-dependent DNAzyme. The electrically-driven activation of the DNAzyme cascade has important implications as it mimics, by electronic triggers, cellular processes, such as addressability, amplification, directed catalytic cascades, and branching of biocatalytic cascades.1–6
Methods
Chemicals and Instrumentation
Lead acetate, silver nitrate, hemin and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS2−) were purchased from Sigma.The DNA sequences applied in the study were:
(1) 5′-GTCATTTGAAGTAGCGCCGCCGTAACAGTCA-3′
(2) 5′-(ROX)-TGACTGTTrAGGAATGAC-(BH2)-3′
(3) 5′-TCTCTGTGGAGGG-3′
(4) 5′-ACACAGGGACGGG-3′
(5) 5′-GTCATTCCTGCTCCTGAAGTAGCGCCGCCGTTCAATTA-3′
(6) 5′-AAGACTTCTAATTGArGGAGCAGGAATGAC-3′
(7) 5′-GATATCAGCGATCTTCTAATTGAAAGTTATTAATCAATTAGAAGTCTTATGAAGCACCCATGTTACTCT-3′
(8) 5′-GATATCAGCGATCTTTTAATAACTTTCAATTAGCATAAGACTTCTAATTGAAAGCACCCATGTTACTCT-3′
(10) 5′-(FAM)-AGAGTATrAGGATATC-(BH1)-3′
An Autolab potentiostat (ECO Chemie, the Netherlands) driven by GPES software was used for the electrochemical measurements. A Ag wire (0.5 mm) and a Pt wire (0.5 mm) were used as the quasi-reference (QRE) and counter electrodes, respectively. The cell volume was 200 μL. UV/Visible spectroscopic measurements were performed using a Shimadzu UV-2401 PC spectrophotometer driven by UVProbe 2.33 software. Emission values were recorded using a Carry Eclipsed Fluorescence Spectrophotometer (Agilent Technologies).
Electrode preparation
A clean Au wire (0.5 mm diameter), was immersed in 1 M HClO4 containing lead acetate, 12 mM, and hydroquinone, 70 mM. In order to deposit a dense Pb0 layer on the Au surface, a potential pulse corresponding to E = −0.95 V vs. the Ag QRE was applied for 3 minutes under stirring conditions. Similarly, in order to prepare the Ag-modified Au surface, a solution containing silver nitrate, 10 mM in 1 M HNO3 was used. In this case, a potential pulse corresponding to E = −0.2 V vs. the Ag QRE was applied for 3 minutes under stirring conditions. The resulting metal-deposited electrodes were carefully washed using HNO3 and copious amounts of water. The release of the metal ions from the modified surfaces was performed using an in situ procedure, in which the target DNA sequences were presented in the electrochemical cells during the application of the oxidative potential pulses. The cells were assembled inside standard plastic cuvettes, which were subsequently used for measuring the fluorescence and absorbance spectra associated with the different DNA systems.Conclusions
To conclude, the present study has introduced the first steps for the development of an artificial “electronic cell”. It was demonstrated that the electrochemical release of ions was coupled to the subsequent activation of the catalytic functions of DNAzymes and DNAzyme cascades. The formation of the DNAzymes translated the electronic stimuli into chemical transformations and provided amplification of the electrochemical triggers. Also, the ON/OFF switching of the electronically-triggered DNAzyme functions demonstrated a means to electrically control the extent of the biocatalytic transformations. Such control could be linked to the electrochemical sensors of specific DNA or other cell concentrations to provide regulated “metabolic” feedback between the progress of artificial cell reactions and the initiation of further phases of the cell cycle.Acknowledgements
This research is supported by the EU FET Open MICREAgents Project # 318671.Notes and references
- A. Pohorille and D. Deamer, Trends Biotechnol., 2002, 20, 123–128 CrossRef CAS.
- G. Murtas, Mol. BioSyst., 2009, 5, 1292–1297 RSC.
- A. C. Forster and G. M. Church, Mol. Syst. Biol., 2006, 2, 45 CrossRef PubMed.
- G. Murtas, Origins Life Evol. Biospheres, 2007, 37, 419–422 CrossRef CAS PubMed.
- A. V. Pietrini and P. L. Luisi, ChemBioChem, 2004, 5, 1055–1062 CrossRef CAS PubMed.
- J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387–390 CrossRef CAS PubMed.
- P. L. Luisi, F. Ferri and P. Stano, Naturwissenschaften, 2006, 93, 1–13 CrossRef CAS PubMed.
- K. Ishikawa, K. Sato, Y. Shima, I. Urabe and T. Yomo, FEBS Lett., 2004, 576, 387–390 CrossRef CAS PubMed.
- G. Murtas, Y. Kuruma, P. Bianchini, A. Diaspro and P. L. Luisi, Biochem. Biophys. Res. Commun., 2007, 363, 12–17 CrossRef CAS PubMed.
- V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci U. S. A., 2004, 101, 17669–17674 CrossRef CAS PubMed.
- I. Vilotijevic and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 5250–5281 CrossRef CAS PubMed.
- P. Walde, R. Wick, M. Fresta, A. Mangone and P. L. Luisi, J. Am. Chem. Soc., 1994, 116, 11649–11654 CrossRef CAS.
- P. K. Schmidli, P. Schurtenberger and P. L. Luisi, J. Am. Chem. Soc., 1991, 113, 8127–8130 CrossRef CAS.
- P. F. Wagler, U. Tangen, T. Maeke and J. S. McCaskill, BioSystems, 2012, 109, 2–17 CrossRef CAS PubMed.
- Y. Xiao, V. Pavlov, T. Niazov, A. Dishon, M. Kolter and I. Willner, J. Am. Chem. Soc., 2004, 126, 7430–7431 CrossRef CAS PubMed.
- D. M. Kolpashchikov, J. Am. Chem. Soc., 2008, 130, 2934–2935 CrossRef CAS PubMed.
- F. Wang, J. Elbaz, C. Teller and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 295–299 CrossRef CAS PubMed.
- G. Pelossof, R. Tel-Vered and I. Willner, Anal. Chem., 2012, 84, 3703–3709 CrossRef CAS PubMed.
- T. Li, L. Shi, E. Wang and S. Dong, Chem.–Eur. J., 2009, 15, 1036–1042 CrossRef CAS PubMed.
- Y. Weizmann, M. K. Beissenhirtz, Z. Cheglakov, R. Nowaski, M. Kolter and I. Willner, Angew. Chem., Int. Ed., 2006, 45, 7384–7388 CrossRef CAS PubMed.
- Y. Tian, Y. He and C. Mao, ChemBioChem, 2006, 7, 1862–1864 CrossRef CAS PubMed.
- Z. Cheglakov, Y. Weizmann, B. Basnar and I. Willner, Org. Biomol. Chem., 2007, 5, 223–225 CAS.
- F. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149–17151 CrossRef CAS PubMed.
- B. Cuenoud and J. W. Szostak, Nature, 1995, 375, 611–614 CrossRef CAS PubMed.
- K. B. Chapman and J. W. Szostak, Chem. Biol., 1995, 2, 325–333 CrossRef CAS.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS.
- P. Travascio, Y. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef CAS.
- S. Nakayama and H. O. Sintim, J. Am. Chem. Soc., 2009, 131, 10320–10333 CrossRef CAS PubMed.
- E. Golub, R. Freeman and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 11710–11714 CrossRef CAS PubMed.
- A. K. Brown, J. Li, C. M. B. Pavot and Y. Lu, Biochemistry, 2003, 42, 7152–7161 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Anal. Chem., 2003, 75, 6666–6672 CrossRef CAS PubMed.
- H. K. Kim, J. Liu, J. Li, N. Nagraj, M. Li, C. M. B. Pavot and Y. Lu, J. Am. Chem. Soc., 2007, 129, 6896–6902 CrossRef CAS PubMed.
- C. K. Chiang, C. C. Huang, C. W. Liu and H. T. Chang, Anal. Chem., 2008, 80, 3716–3721 CrossRef CAS PubMed.
- D. M. Kolpashchikov, J. Am. Chem. Soc., 2008, 130, 2934–2935 CrossRef CAS PubMed.
- J. Elbaz, M. Moshe, B. Shlyahovsky and I. Willner, Chem.–Eur. J., 2009, 15, 3411–3418 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1995, 2, 655–660 CrossRef CAS.
Footnote |
| † Equally contributed to this study. |
| This journal is © The Royal Society of Chemistry 2015 |