
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxalyl amide assisted palladium-catalyzed synthesis of pyrrolidones via carbonylation of γ-C(sp3)–H bonds of aliphatic amine substrates†
Chao
Wang
,
Li
Zhang
,
Changpeng
Chen
,
Jian
Han
,
Yingming
Yao
* and
Yingsheng
Zhao
*
Key Laboratory of Organic Synthesis of Jiangsu Province College of Chemistry, Chemical Engineering and Materials Science Soochow University, Suzhou 215123, PR China. E-mail: yszhao@suda.edu.cn
First published on 19th May 2015
Abstract
The first Pd-catalyzed regioselective γ-carbonylation of oxalyl amide protected aliphatic amines with carbon monoxide leading to synthesis of pyrrolidones has been developed. Both γ-methyl and cyclopropyl methylene C–H bonds are well activated to obtain the corresponding pyrrolidones in moderate to excellent yields. The role of 3-(trifluoromethyl)benzoic acid as an additive is critical as it helps in stabilizing the palladium intermediate formed during the catalytic cycle. The reaction scope is extended to benzylamine and allyl amine derivatives, thereby affording the corresponding products in good to excellent yields.
Introduction
Recently, the development of practical methods for the construction of synthetically useful building blocks through direct C–H functionalization has emerged as an attractive and fruitful area in modern organic chemistry.1 Among these methods, the transition-metal-catalyzed carbonylation of aryl C(sp2)–H bonds using CO as a carbon atom source to form an easily transformable carbonyl group has experienced landmark development.2–5 The first example was in the 1950s when Murahashi reported the cobalt-catalysed direct C–H functionalization/carbonylation of arenes with CO.6 In 1980, Fujiwara et al. also described a protocol involving Pd(OAc)2-catalyzed direct carboxylation of some aromatic compounds with CO (15 atm) in an autoclave to give the corresponding carboxylic acids in poor to moderate yields.7 However, large excess of the arenes and lack of regioselectivity has hampered further application of this method in organic synthesis.In the last decades, the challenge of selectivity in carbonylation of C(sp2)–H bonds has been overcome by the employment of directing groups,8 or where regioselectivity could also be achieved based on the substrates own intrinsic reactivity, e.g. amines,4d,8d,9 carboxylics,10 phenols,11 and alcohols.12 In spite of these great developments in the carbonylation of arenes, there is still limited success in the direct functionalization of C(sp3)–H bonds with CO. In 2011, Chatani et al. described the Ru(0)-catalyzed regioselective carbonylation of unactivated C(sp3)–H bonds of aliphatic amides using bidentate directing groups (Scheme 1A). Similarly, Yu et al. reported the synthesis of succinimides via a Pd(II)-catalyzed β-carbonylation of aliphatic amide with CO (Scheme 1B).13 Recently, Gaunt et al. described a Pd-catalyzed carbonylation of secondary aliphatic amines at the β-position via a rarely reported four-membered palladacycle, leading to β-lactams in moderate to good yields (Scheme 1C).14 To the best of our knowledge, only these two examples of Pd-catalyzed carbonylation of C(sp3)–H bonds at specific positions have been reported. We speculated that during the catalytic cycle, Pd(II) might be easily reduced to Pd(0), thereby undergoing β-hydride elimination, and that there could be a competitive coordination with the palladium centre between CO and the specific C–H bond under CO atmosphere.15 For these reasons, the direct carbonylation of C(sp3)–H bonds remained a significant challenge.
 | ||
| Scheme 1 Carbonylation of C(sp3)–H Bonds. | ||
In 2005, Daugulis and co-workers reported the first use of picolinamide and aminoquinoline auxiliaries as bidentate directing groups in Pd-catalyzed regioselective functionalization of C(sp3)–H bonds.16 Later, several groups demonstrated that these groups have an immense ability for enabling a wide variety of C–H transformation.17 Inspired by these results, our group has developed another bidentate directing group, oxalyl amide, which could be easily synthesized from oxalyl chloride and diisopropylamine via an SN type reaction. We found that it could be used as a powerful directing group in enabling a variety of C–H transformations for both unactivated C(sp3)–H and C(sp2)–H bonds.18
In order to expand the reaction scope of oxalyl amide assisted C–H functionalization and explore its potential application towards transformation not achievable by other auxiliary-assisted C–H functionalization, we therefore proceeded to address the unreported γ-carbonylation of aliphatic amine substrates in a bid to synthesize important structural motifs of pyrrolidones.19 Herein, we report a novel protocol for the preparation of pyrrolidones by Pd(II)-catalyzed γ-carbonylation of oxalyl amide protected aliphatic amine substrates. Both γ-methyl and cyclopropyl methylene C–H bonds were well activated and gave the corresponding pyrrolidones in moderate to excellent yields. Furthermore, pyrrolidones could be transformed into synthetically useful γ-amino acids under acidic conditions,20 which offers a potential route towards synthesising unnatural amino acids (Scheme 1D). The substrate scope could also be further extended to oxalyl amide protected benzyl amine and allyl amine derivatives, affording the corresponding valuable synthons in good to excellent yields.
Results and discussion
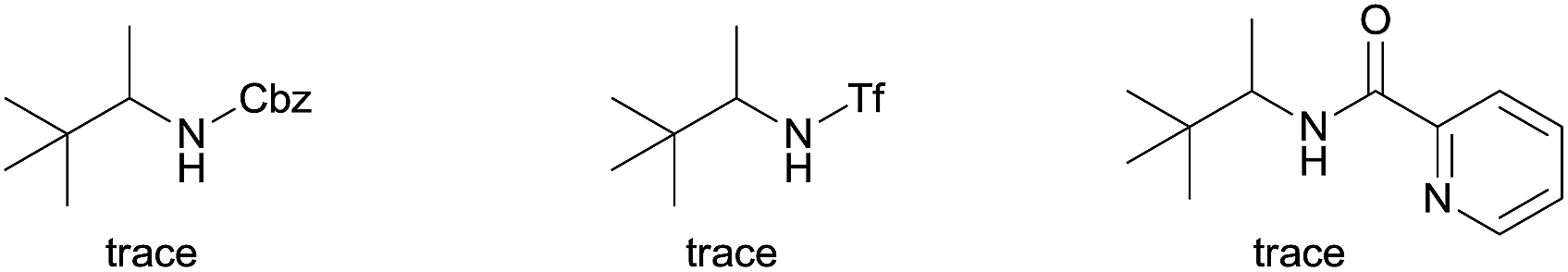
We started our investigation with the reaction of oxalyl amide protected 1a in the presence of Pd(OAc)2 catalyst and silver acetate as oxidant under atmospheric pressure of CO at 140 °C in mesitylene for 24 h. Gratifyingly, the desired pyrrolidone 2a was obtained in 22% yield. A variety of additives, such as AcOH, PivOH, (nBuO)2PO2H, Ac-Gly-OH, which are well known to improve the reactivity of a palladium center, were applied to this reaction,21 with all of them displaying different levels of positive effect on the product yield. Pivalic acid greatly aided the conversion of 1a, affording 2a in 57% yield, but benzoic acid had a better promoting effect. However, further screening revealed that 3-(trifluoromethyl)benzoic acid was the most effective additive, with an excellent conversion of 1a giving 85% yield of 2a. During optimization studies, reactions with various different additives proceeded cleanly and it was observed that Pd(II) was reduced to Pd(0) during the reaction thus rationalizing the low efficiency of some of these additives (Table 1, entries 2–8). Although the reason behind the promoting effect of 3-(trifluoromethyl)benzoic acid was not clear, one plausible explanation was that the palladium(0) stabilized by the electron-deficient arene during the catalytic cycle, could be further reoxidized to Pd(II). Meanwhile, it played the same role as pivalic acid during the C–H activation step.22 Other oxidants such as Cu(OAc)2 or BQ were not effective in this transformation. Notably, alternative directing groups or protecting groups, such as Cbz, triflamide and picolinamide were also tested under optimized conditions. However, none could give the corresponding carbonylated products in more than 5% yield analyzed by LC-MS.|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd(OAc)2 (mol%) | Oxidant (2.5 equiv.) | Additive (0.3 equiv.) | Conva (%) | Yielda,b (%) |
| a Reactions were carried out at 0.2 mmol scale under CO (1 atm), using mesitylene (0.3 mL) as the solvent; conversion and yield were based on GC using tridecane as the internal standard. b Isolated yield in parentheses. Ac = acetyl, Gly = glycine. | |||||
| 1 | 10 | AgOAc | 26 | 22 | |
| 2 | 10 | AgOAc | AcOH | 21 | 19 |
| 3 | 10 | AgOAc | PivOH | 60 | 57 |
| 4 | 10 | AgOAc | (nBuO)2PO2H | 66 | 62 |
| 5 | 10 | AgOAc | Ac-Gly-OH | 12 | 8 |
| 6 | 10 | AgOAc | PhCO2H | 76 | 75 |
| 7 | 10 | AgOAc | o-PhPhCO2H | 27 | 18 |
| 8 | 10 | AgOAc | m-CF3PhCO2H | 87 | 85(83) |
| 9 | 10 | Ag2CO3 | m-CF3PhCO2H | 21 | 20 |
| 10 | 10 | Cu(AOc)2 | m-CF3PhCO2H | 2 | 0 |
| 11 | 10 | BQ | m-CF3PhCO2H | 5 | 4 |
| 12 | 0 | AgOAc | m-CF3PhCO2H | 2 | 0 |
|
|||||
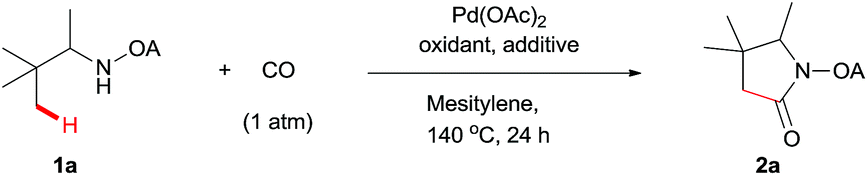
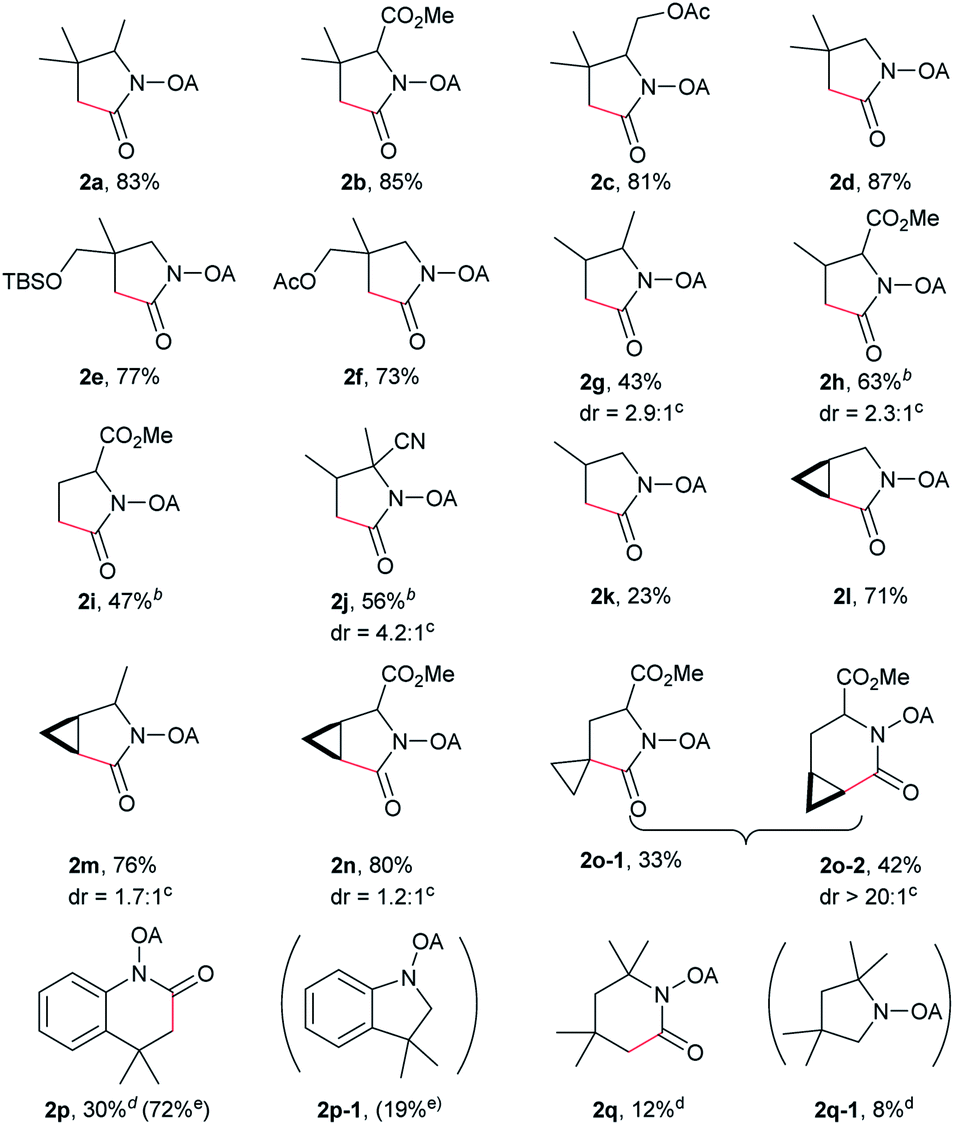
With the optimized conditions in hand, reactions involving a series of oxalyl amide protected propylamine derivatives were carried out. The carbonylation of substrates bearing two β-substituents proceeded smoothly to provide the corresponding pyrrolidines in good to excellent yields (Table 2, 2b–f). Ester derivatives were tolerated, giving the corresponding products in good yields (2b,c,f,h,i,n,o). Substrates 1g–k, bearing only one β methyl group, were less reactive and acceptable yields could only be obtained by slightly modifying the reaction conditions. Delightfully, substrate 1j, with an α-substituted cyano group, which is a highly valuable synthon in synthetic chemistry, also gave the desired carbonylated product 2j in 56% yield. The methylene C–H bonds in cyclopropane rings were all carbonylated giving good yields of products (2l–n). Interestingly, substrate 1o gave a mixture of five and six-membered carbonylated products in a total of 75% yield (2o), which could be easily separated by silica gel chromatography. Inspired by these result, substrate 1p was also tested, but a lower yield of product than expected was recovered, together with unreacted starting material. It was thought that the probable competitive carbonylation reaction happening at the aromatic ring was not detected by NMR and GC-MS.23 However, when one equivalent of Pd(OAc)2 was used, the δ-carbonylated product 2p was obtained in 72% yield, along with 19% yield of intramolecular aminated product 2p-1. Unfortunately, when the same reaction was attempted on substrate 1q, only 12% of the corresponding carbonylated product 2q could be isolated along with 8% of the product from intramolecular amination. Regrettably, the use of one equivalent of Pd(OAc)2 as in the case of substrate 1p, did nothing to improve the yield of carbonylation and intramolecular amination products. However, it can be inferred from these results that an unstable alkyl seven-membered palladacycle24 might have been formed during the catalytic cycle, which could open doors for the development of newer transformation. A series of other oxalyl amide protected amines (1r–u) were also tested, all failing to give acceptable yields.
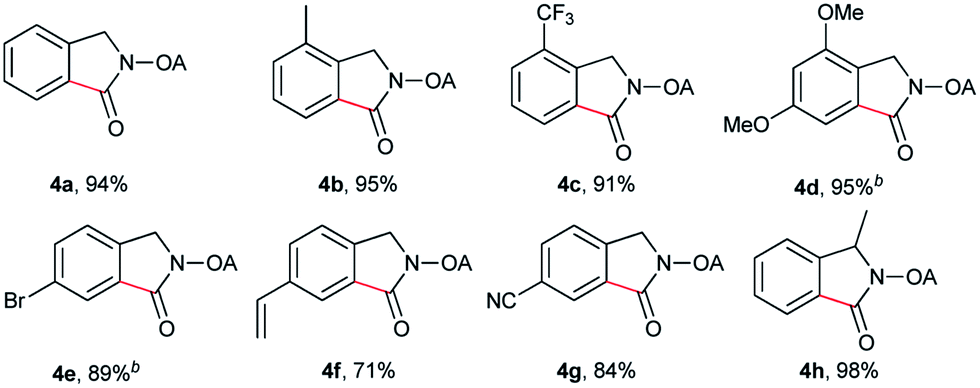
Despite the great development in palladium-catalyzed C(sp2)–H activation/carbonylation,25 there have been only few examples on the direct preparation of benzolactams from benzylamines substrates assisted by directing groups. Encouraged by the results obtained above, oxalyl amide protected benzylamines were also examined, and representative data were listed in Table 3. A wide variety of functional groups, such as CF3, OMe, Br, vinyl, cyano, were compatible with this protocol, affording the corresponding products in good to excellent yields. Both electron-donating and electron-withdrawing substituents on the phenyl ring had little or no effect on the yield of products. Interestingly, substrates 3d and 3e needed slightly higher temperature for complete transformation.
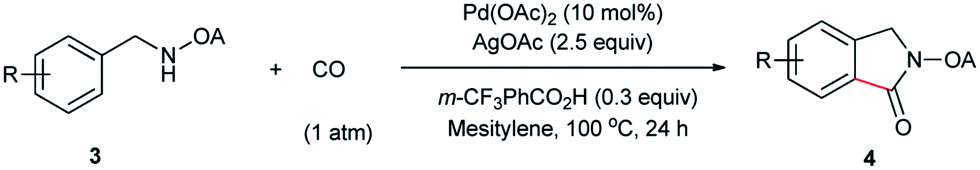
The scope of γ-carbonylation of oxalyl amide protected primary amine could be further extended to allylamine substrates. Substrates 5 and 7 smoothly reacted with CO to give the corresponding carbonylated products 6 and 8 in acceptable yield respectively, which could be used as synthons for a variety of further elaborations (Scheme 2).26
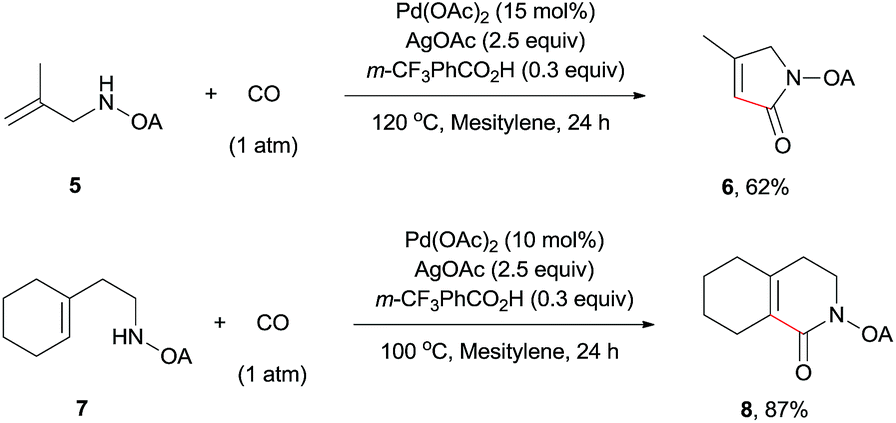 | ||
| Scheme 2 Carbonylation of allylamine reaction. | ||
To demonstrate the powerful use of this protocol, a gram scale reaction involving substrate 1a was carried out and the corresponding pyrrolidone was isolated in 72% yield (Scheme 3). The oxalyl amide directing group could be removed under basic conditions at 50 °C, thus underlining the potential utility of this newly developed synthetic method.
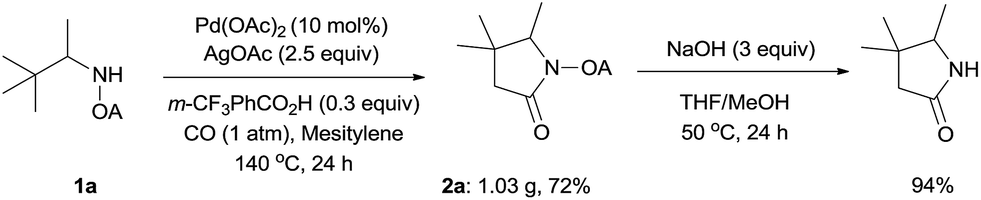 | ||
| Scheme 3 Scaling up and auxiliary removal. | ||
Conclusion
In conclusion, we have developed the first Pd-catalyzed γ-carbonylation of oxalyl amide protected aliphatic amine substrates by using oxalyl amide as a bidentate directing group with AgOAc, and ArCO2H under carbon monoxide atmosphere. This method is a novel strategy for the preparation of pyrrolidones. The substrate scope can be extended to benzylamine and allyl amine derivatives, giving products in good to excellent yields. The use of 3-(trifluoromethyl)benzoic acid is important for this transformation, and a detailed mechanistic study is now being undertaken in our laboratory.Acknowledgements
We gratefully acknowledge financial support from the Natural Science Foundation of Jiangsu Province of China (BK20130294), a start-up fund (Q410901212) from Soochow University and the Young National Natural Science Foundation of China (No. 2140213321402133) for support of this work. The PAPD and the Qing Lan Project are also gratefully acknowledged.Notes and references
- For recent reviews: (a) K. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826 CrossRef PubMed; (b) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (c) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (e) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (f) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (g) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (h) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (i) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2012, 46, 412 CrossRef PubMed; (j) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (k) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (l) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (m) J. L. Roizen, M. E. Harvey and J. D. Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed; (n) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- (a) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E.-I. Negishi, Wiley-Interscience, New York, 2002 Search PubMed; (b) R. Jazzar, J. Hitce, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS PubMed; (c) Y. Zhao, L. Jin and A. Lei, J. Am. Chem. Soc., 2008, 130, 9429 CrossRef CAS PubMed; (d) R. H. Munday, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 2754 CrossRef CAS PubMed; (e) C. F. J. Barnard, Organometallics, 2008, 27, 5402 CrossRef CAS; (f) A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed.
- For select examples of Pd-catalyzed carbonylation: (a) J. Schranck, M. Burhardt, C. Bornschein, H. Neumann, T. Skrydstrup and M. Beller, Chem.–Eur. J., 2014, 20, 9534 CrossRef CAS PubMed; (b) J. Chen, K. Natte, A. Spannenberg, H. Neumann, P. Langer, M. Beller and X.-F. Wu, Angew. Chem., Int. Ed., 2014, 53, 7579 CrossRef CAS PubMed; (c) L. Wang, Y. Wang, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 5657 CrossRef CAS PubMed; (d) P. Xie, Y. Xie, B. Qian, H. Zhou, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 9902 CrossRef CAS PubMed; (e) H. Zhang, R. Shi, P. Gan, C. Liu, A. Ding, Q. Wang and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 5204 CrossRef CAS PubMed; (f) H. Li, G.-X. Cai and Z.-J. Shi, Dalton Trans., 2010, 10442 RSC.
- (a) M. Tobisu, N. Chatani, T. Asaumi, K. Amako, Y. Ie, Y. Fukumoto and S. Murai, J. Am. Chem. Soc., 2000, 122, 12663 CrossRef CAS; (b) S. Inoue, H. Shiota, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2009, 131, 6898 CrossRef CAS PubMed; (c) N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070 CrossRef CAS PubMed; (d) Y. Du, T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 12074 RSC.
- (a) Z.-H. Guan, Z.-H. Ren, S. M. Spinella, S. Yu, Y.-M. Liang and X. Zhang, J. Am. Chem. Soc., 2009, 131, 729 CrossRef CAS PubMed; (b) I. S. noue, H. Shiota, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2009, 131, 6898 CrossRef PubMed.
- S. Murahashi, J. Am. Chem. Soc., 1955, 77, 6403 CrossRef CAS.
- (a) Y. Fujiwara, T. Kawauchi and H. Taniguchi, J. Chem. Soc., Chem. Commun., 1980, 220 RSC; (b) Y. Fujiwara, K. Takaki and Y. Taniguchi, Synlett, 1996, 591 CrossRef CAS PubMed.
- (a) W. Y. Yu, W. N. Sit, K. M. Lai, Z. Y. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2008, 130, 3304 CrossRef CAS PubMed; (b) R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686 CrossRef CAS PubMed; (c) V. Rajeshkumar, T.-H. Lee and S.-C. Chuang, Org. Lett., 2013, 15, 1468 CrossRef CAS PubMed; (d) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688 CrossRef CAS PubMed; (e) S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267 CrossRef CAS PubMed.
- (a) B. Haffemayer, M. Gulias and M. J. Gaunt, Chem. Sci., 2011, 2, 312 RSC; (b) B. Lopez, A. Rodriguez, D. Santos, J. Albert, X. Ariza, J. Garcia and J. Granell, Chem. Commun., 2011, 47, 1054 RSC.
- (a) R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082 CrossRef CAS PubMed; (b) B. R. Rosen, L. R. Simke, P. S. Thuy-Boun, D. D. Dixon, J.-Q. Yu and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 7317 CrossRef CAS PubMed.
- S. Luo, F.-X. Luo, X.-S. Zhang and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 10598 CrossRef CAS PubMed.
- Y. Lu, D. Leow, X. Wang, K. M. Engle and J.-Q. Yu, Chem. Sci., 2011, 2, 967 RSC.
- E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378 CrossRef CAS PubMed.
- A. McNally, B. Haffemayer, B. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129 CrossRef CAS PubMed.
- (a) T. Nishiguchi, K. Nakata, K. Takaki and Y. Fujiwara, Chem. Lett., 1992, 21, 1141 CrossRef; (b) K. Nakata, Y. Yamaoka, T. Miyata, Y. Taniguchi, K. Takaki and Y. Fujiwara, J. Organomet. Chem., 1994, 473, 32 CrossRef.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- For select references: (a) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed; (b) Y. Feng, Y. Wang, B. Landgraf, S. Liu and G. Chen, Org. Lett., 2010, 12, 3414 CrossRef CAS PubMed; (c) Y. Zhao and G. Chen, Org. Lett., 2011, 13, 4850 CrossRef CAS PubMed; (d) G. He, C. Lu, Y. Zhao, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2944 CrossRef CAS PubMed; (e) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124 CrossRef CAS PubMed; (f) Y. Ano, M. Tobisu and N. Chatani, Org. Lett., 2012, 14, 354 CrossRef CAS PubMed; (g) F.-R. Gou, X.-C. Wang, P.-F. Huo, H.-P. Bi, Z.-H. Guan and Y.-M. Liang, Org. Lett., 2009, 11, 5726 CrossRef CAS PubMed; (h) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed.
- (a) C. Wang, C. Chen, J. Zhang, J. Han, Q. Wang, K. Guo, P. Liu, M. Guan, Y. Yao and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9884 CrossRef CAS PubMed; (b) Q. Wang, J. Han, C. Wang, J. Zhang, Z. Huang, D. Shi and Y. Zhao, Chem. Sci., 2014, 5, 4962 RSC; (c) J. Han, P. Liu, C. Wang, Q. Wang, J. Zhang, Y. Zhao, D. Shi and Y. Zhao, Org. Lett., 2014, 16, 5682 CrossRef CAS PubMed.
- (a) G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS PubMed; (b) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed; (c) D. E. A. Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766 CrossRef CAS PubMed; (d) D. Q. Tan, K. S. Martin, J. C. Fettinger and J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6781 CrossRef CAS PubMed.
- (a) S. Rousseaux, B. Liegault and K. Fagnou, Chem. Sci., 2012, 3, 244 RSC; (b) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 12842 CrossRef CAS PubMed; (c) J.-X. Yan, H. Li, X.-W. Liu, J.-L. Shi, X. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 4945 CrossRef CAS PubMed.
- (a) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed; (b) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed; (c) S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124 CrossRef CAS PubMed.
- (a) M. Lafrance, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 14570 CrossRef CAS PubMed; (b) S. Rousseaux, M. Davi, J. Sofack-Kreutzer, C. Pierre, C. E. Kefalidis, E. Clot, K. Fagnou and O. Baudoin, J. Am. Chem. Soc., 2010, 132, 10706 CrossRef CAS PubMed.
- (a) W. Li, Z. Duan, X. Zhang, H. Zhang, M. Wang, R. Jiang, H. Zeng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 1893 CrossRef CAS PubMed; (b) W. Li, Z. Duan, R. Jiang and A. Lei, Org. Lett., 2015, 17, 1397 CrossRef CAS PubMed.
- (a) K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342 CrossRef CAS PubMed; (b) K. Orito, M. Miyazawa, T. Nakamura, A. Horibata, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita, T. Yamazaki and M. Tokuda, J. Org. Chem., 2006, 71, 5951 CrossRef CAS PubMed; (c) J. Albert, X. Ariza, T. Calvet, M. Font-Bardia, J. Garcia, J. Granell, A. Lamela, A. Lopez, M. Martinez, L. Ortega, A. Rodriguez and D. Santos, Organometallics, 2013, 32, 649 CrossRef CAS.
- (a) M. Otsuka, T. Masuda, A. Haupt, M. Ohno, T. Shiraki, Y. Sugiura and K. Maed, J. Am. Chem. Soc., 1990, 112, 838 CrossRef CAS; (b) J. S. Clark, P. B. Hodgson, M. D. Goldsmith and L. J. Street, J. Chem. Soc., Perkin Trans. 1, 2001, 3312 CAS.
- Y. Hoshimoto, T. Ohata, Y. Sasaoka, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2014, 136, 15877 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for all new compounds. CCDC 1048649. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00519a |
| This journal is © The Royal Society of Chemistry 2015 |