
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect in vivo imaging of ferrous iron dyshomeostasis in ageing Caenorhabditis elegans†
Simon A.
James
abc,
Blaine R.
Roberts
c,
Dominic J.
Hare
 cde,
Martin D.
de Jonge
a,
Ian E.
Birchall
c,
Nicole L.
Jenkins
c,
Robert A.
Cherny
c,
Ashley I.
Bush
c and
Gawain
McColl
*c
cde,
Martin D.
de Jonge
a,
Ian E.
Birchall
c,
Nicole L.
Jenkins
c,
Robert A.
Cherny
c,
Ashley I.
Bush
c and
Gawain
McColl
*c
aAustralian Synchrotron, Clayton, Victoria, Australia
bCommonwealth Scientific and Industrial Research Organisation, Clayton, Victoria, Australia
cThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Kenneth Myer Building, 30 Royal Parade, Parkville, Victoria, Australia 3052. E-mail: gawain.mccoll@florey.edu.au; ashley.bush@florey.edu.au; Tel: +61 3 9035 6608
dElemental Bio-imaging Facility, University of Technology Sydney, Broadway, New South Wales, Australia
eExposure Biology Laboratory, Lautenberg Environment Health Sciences Laboratory, Department of Preventive Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA
First published on 3rd March 2015
Abstract
Iron is essential for eukaryotic biochemistry. Systematic trafficking and storage is required to maintain supply of iron while preventing it from catalysing unwanted reactions, particularly the generation of oxidising reactive species. Iron dyshomeostasis has been implicated in major age-associated diseases including cancers, neurodegeneration and heart disease. Here, we employ population-level X-ray fluorescence imaging and native-metalloproteomic analysis to determine that altered iron coordination and distribution is a pathological imperative of ageing in the nematode, Caenorhabditis elegans. Our approach provides a method to simultaneously study iron metabolism across different scales of biological organisation, from populations to cells. Here we report how and where iron homeostasis is lost during C. elegans ageing, and its relationship to the age-related elevation of damaging reactive oxygen species. We find that wild types utilise ferritin to sustain longevity, buffering against exogenous iron and showing rapid ageing if ferritin is ablated. After reproduction, escape of iron from safe-storage in ferritin raised cellular Fe2+ load in the ageing C. elegans, and increased generation of reactive species. These findings support the hypothesis that iron-mediated processes drive senescence. We propose that loss of iron homeostasis may be a fundamental and inescapable consequence of ageing that could represent a critical target for therapeutic strategies to improve health outcomes in ageing.
Introduction
The near-universal requirement of iron in life is due to its ability to mediate single-electron chemistry under physiological conditions, from ATP production to oxygen transport.1 The unique chemical properties of iron facilitate innumerable metabolic processes, suggesting that iron dyshomeostasis may be an upstream event preceding biological senescence.Age is the greatest risk factor for a number of diseases including cancer, heart disease and neurodegeneration. The associated impairments to iron metabolism and increased iron-mediated oxidative stress have been implicated in disease pathogenesis.1 The precise mechanism through which iron imparts cytotoxicity depends on the chemical state in which the metal exists within the cell. Biological iron is predominantly either ferrous (Fe2+) or ferric (Fe3+), with the majority of bioavailable iron tightly bound by dedicated enzymes and storage proteins, and a small proportion likely existing in dynamic equilibrium as low molecular weight complexes. The normally desirable propensity of iron to participate in single-electron redox chemistry is non-specific; unchecked interaction of Fe2+ with biological oxygen species can generate powerful oxidants and thus become cytotoxic. The reactivity of iron is determined by local bonding environment. Biological ligands typically prevent iron from engaging in side reactions particularly the indiscriminate reduction of endogenous cellular peroxides (i.e. the Fenton reaction) to generate the damaging OH˙ radical.
Iron accumulation appears to be a common feature of ageing organisms.2–4 In the human brain, for instance, both iron and proteins associated with iron metabolism are elevated in aged individuals, particularly in regions most susceptible to damage in diseases including Alzheimer's and Parkinson's.5,6 Stable isotope tracing in rats has identified an imbalance favouring iron influx over efflux, leading to an approximately 30% increase in brain iron as the animals aged.7 In concert with this gradual yet significant elevation in iron levels, a changing redox environment favouring unrestrained oxidative activity of Fe2+ is an intriguing candidate that might explain both age-related disease and general senescence. For example, ceruloplasmin-deficient mice that lack the capacity to oxidise Fe2+ to Fe3+ display marked age-dependent neurodegeneration.8
Direct appraisal of iron redox chemistry in vivo is an analytical challenge. Post mortem artefacts arising from tissue handling, fixation and cryoprotection are all potential sources of experimental error, including metal redistribution and altered cellular redox state.9 Achieving mechanistic insight into the specific redox chemistry associated with iron in an ageing organism requires the capacity to obtain snapshots of true biophysical conditions with minimal disturbance to normal Fe2+/Fe3+ balance. When applied to this problem the results of any analytical technology are only as relevant as the degree to which the native redox state of the cell is maintained. For instance, fluorescent probes offer one method to assess native redox state.10 Hard X-ray microprobes and mass spectrometry also offer an opportunity to probe biological microstructures at high resolution,11 and are compatible with hydrated specimens.12 Synchrotron-based X-ray fluorescence microspectroscopy allows direct quantitative appraisal of the abundance of inorganic species, as well as a means to assess coordination environment within intact specimens at micrometer length scales. Combined with the ability to image live, hydrated biological samples, this unique approach is a step closer to permitting accurate measurement of the true in vivo chemical environment in which iron exists in the ageing cell.
The nematode Caenorhabditis elegans provides an excellent model to explore the bioinorganic chemistry of ageing in an entire multicellular organism.13,14 Like higher-order counterparts, C. elegans deposit fat, accumulate lipofuscin, develop sarcopenia and suffer neurodegeneration with age. Genetic modulation of ageing has been well characterised in this animal,15 however, the biochemical processes and underlying molecular mechanisms driving these events are uncertain.
We have previously identified that C. elegans express high concentrations of iron in intestinal cells,16,17 which house the majority of metabolic processes in this animal. To expand our understanding of the chemistry of ageing we here imaged iron coordination chemistry within live C. elegans, quantitated age-related iron accumulation, and assessed biochemical iron alterations in this genetic model of longevity. A complementary native metalloproteomic analysis identified senescent changes in endogenous iron ligands. We propose that systematic failure of iron metabolism is a major contributor to the ageing process, which has implications for understanding the risk for several age-related diseases.
Results
Iron accumulation and redistribution during ageing
We have previously shown that C. elegans adults exhibit a highly compartmentalised bioinorganic architecture,16,17 including high iron concentrations in the metabolically active intestinal cells. Using synchrotron-based X-ray fluorescence microscopy (XFM; ∼2 μm resolution) we quantified the spatial distribution of iron in young and aged intact wild-type adults (Fig. 1a). This revealed a 77% increase in mean total iron from young (4 days post egg lay, 62.5 pg iron per individual) to post-reproductive animals (12-day old, 110.4 pg iron per individual; two-tailed unpaired Student's t-test, p < 0.001, Fig. 1b), with marked intracellular accumulation in the intestinal cells (Fig. 1a). Measurement of iron using inductively coupled plasma-mass spectrometry (ICP-MS) in aged C. elegans cohorts (n = 100 adults per aliquot) closely corresponded and confirmed a significant increase in total iron (ESI Fig. 1a†). For instance, post-reproductive senescent 10-day old adults had a 66% increase in mean iron compared to 6-day old adults (two-tailed unpaired Student's t-test, p < 0.001). Iron also increased with age when normalised against dry weight, indicating that the iron elevation was not due to increased body mass (ESI Fig. 1b†). | ||
| Fig. 1 Elevation and spatial redistribution of iron in ageing C. elegans. (a) Representative X-ray fluorescence micrographs of wild type adult C. elegans (4- and 12-day old). Inelastic scatter of incident photons (Compton scatter) provides anatomical visualisation (greyscale), and intestinal cells are highlighted by calcium (yellow). Iron (32-colour scale) was elevated throughout aged animals (scale bar = 100 μm). (b) Quantification of iron content in individual animals by XFM at intervals across lifespan. For all tested genotypes, 12- and 15-day old worms contained significantly more iron than 4-day old individuals (two-tailed unpaired Student's t-test; ** p < 0.01 and *** p < 0.001; mean ± SEM). Long-lived daf-2 mutants accumulated significantly less iron than age-matched wild type and daf-16(−);daf-2(−) (p < 0.001). (c) DAB-enhanced Perls' staining for non-heme iron (brown) in young (4-day, left) and aged (12-day, right) wild type C. elegans. Top row: Head sections. Middle row: Mid-body sections. Bottom row: Posterior sections. Insets show (i) intestinal cell nuclei free of iron in young C. elegans (filled triangles); (ii) discrete vesicular deposits of iron close to the intestinal lumen in young adults; (iii) iron in inclusions within the head of aged worms (filled arrowhead); (iv) dispersed and intranuclear iron (open triangles) within the intestine in aged individuals (scale bar = 100 μm). | ||
Lifespan can be genetically modulated in C. elegans, which allows us to determine if iron accumulation scales with ageing rate. Insulin-like signaling (ILS) is a conserved pathway that has been shown to increase lifespan in nematodes, flies, and mammals.18 Mutation in daf-2, encoding an insulin/IGF-1 receptor ortholog,19 doubles C. elegans life span but requires the activity of daf-16,20 which encodes a FOXO family transcription factor.21 Mutation of daf-16 suppresses the longevity gains of daf-2 mutants. When examined by XFM, the age-dependent rise in iron was markedly suppressed in the long-lived daf-2 mutants. daf-16;daf-2 double mutants mirrored the marked age-dependent rise in iron observed in wild type (Fig. 1b; ESI Fig. 2†). When aged to 30 days (their approximate median lifespan), daf-2 mutants also showed an approximate two-fold increase in total iron (two-tailed unpaired Student's t-test; p < 0.01), consistent with iron elevation heralding death even where the rate of ageing is slowed (ESI Fig. 3†).
To examine the presence of potentially harmful non-heme iron, we used histological Perls' staining, visually enhanced with 3,3′-diaminobenzidine (DAB).22 Consistent with the XFM data, we found an age-dependent accumulation of non-heme iron in intestinal cells, progressing from discrete vesicular to dispersed distribution (Fig. 1c). Aged individuals also exhibited iron staining within intestinal cell nuclei, and dense inclusions in the head. Adult C. elegans are post-mitotic and lose intestinal cell nuclei during ageing by an unknown process,23 which our histological findings indicate might be caused by toxic accumulation of nuclear non-heme iron. In addition, we observed conspicuous iron accumulation in the germ nuclei of post-reproductive adults (ESI Fig. 4†).
We hypothesised that age-dependent elevation of iron is a significant source of harmful redox activity if the accumulation is not coordinated in a redox-silenced manner. Both XFM and Perls staining revealed significant changes in iron concentration, cellular distribution and chemical state. Consequently, we assessed the contribution of elevated iron to age-related oxidative stress.
Accumulated iron contributes to oxidative stress
The relationship between reactive iron elevation and oxidative damage within post-mitotic intestinal cells during ageing was examined in living C. elegans. The calcium sensor calcein exhibits decreased fluorescence in the presence of chemically available iron. Consistent with an accumulation of calcein-accessible iron, in vivo fluorescence was suppressed in aged adults (Fig. 2a). This suppression could not be explained by a decrease in total calcium levels (Fig. 1a and ESI Fig. 5†).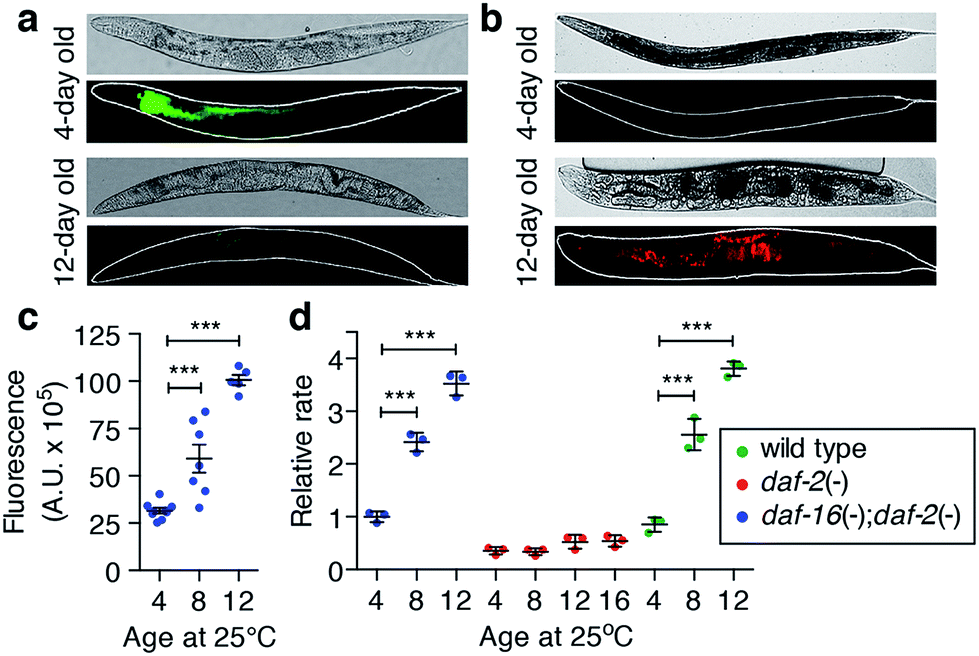 | ||
| Fig. 2 Elevated iron in C. elegans promotes oxidative stress in vivo. (a) Live imaging of exchangeable iron using calcein-AM fluorescence in (top) young (4-day) and (bottom) aged (12-day) C. elegans. Fluorescence is quenched by chemically accessible iron in the aged intestine. Bright field image (above) and fluorescence image (below) with an outline in white. (b) In vivo oxidative activity was detected by dichlorofluoroscein (DCF) fluorescence and increased in the intestine of old (bottom) compared to young (top) C. elegans. (c) Quantification of in vivo reactive species by DCF fluorescence. In the ageing worm, fluorescence significantly increased with age relative to 4-day old adults (mean ± SEM, n = 9, 7 and 5 individuals worms respectively; two-tailed unpaired Student's t-test, *** p < 0.001). (d) Ex vivo ROS generation is a product of iron accumulation. Long-lived daf-2 mutants lacked the increase in levels of reactive species with age (measured by DCF fluorescence; relative to 4-day old adults) compared to wild type and daf-16;daf-2 mutants (mean ± SEM, n = 4; two-tailed unpaired Student's t-test, *** p < 0.001). | ||
Increased accessible iron appeared concurrently with elevated formation of reactive species in vivo, as measured by oxidation of 2′,7′-dichlorodihydrofluorescein (DCFH) to dichlorofluorescein (DCF; Fig. 2b). DCF fluorescence was markedly increased in the intestine compared to young adults (two-tailed unpaired Student's t-test, p < 0.001; Fig. 2c), consistent with a pro-oxidant chemical environment within the aged C. elegans intestine.
To confirm whether age-dependent iron elevation is a catalytic source of the in vivo oxidative stress, we measured DCFH oxidation in the soluble fraction of lysed C. elegans. This ex vivo oxidative activity from wild type C. elegans increased throughout lifespan (two-tailed unpaired Student's t-test, p < 0.001; Fig. 2d), matching the observed steady-state increase in oxidation in vivo (Fig. 2b). The increased rate of ex vivo DCF fluorescence is consistent with an increased pool of catalytically reactive iron because it was abolished by the redox-silencing chelator diethylenetriamine penta-acetic acid (DTPA) (ESI Fig. 6a†). Paralleling iron levels, pro-oxidant chemistry was significantly attenuated in the long-lived daf-2 mutants at each of the ages studied, remaining below the reference baseline rate (4-day old wild type), even by 16 days of age (Fig. 2d, ESI Fig. 6b†). As with its impact on iron levels with ageing, loss of daf-16 function increased oxidative activity in the abbreviated lifespan of the daf-16;daf-2 double mutants.
Normally, careful chaperoning of the metal by protein ligands prevents unregulated iron activity. Elevated oxidative stress arising from an increased pool of accessible iron, typical of many age-related disease states, implicates a failure of containment is a feature of the ageing process.
Ageing impairs iron buffering
The cellular reservoir of exchangeable iron is normally stored by ferritin, which oxidises ferrous (Fe2+) iron to the more chemically stable hydrous ferric (Fe3+) form, protecting the cell from unwanted incidental redox reactions. Ferritin proteins are highly conserved and typically organise as a 24-mer capable of storing up to ∼4500 atoms of iron, although are rarely saturated in vivo.24To determine whether the age-related tandem elevation of iron and oxidative activity were caused by loss of ferritin sequestration of iron we used native size-exclusion chromatography with on-line inductively coupled plasma-mass spectrometry (SEC-ICP-MS) to examine the soluble iron-binding protein profile of wild type C. elegans homogenates. Soluble protein-bound iron resolved into three major peaks (Fig. 3a). Peak #2 represented most (∼84%) of the total soluble iron in 4-day-old adults. Protein purification and analysis by mass spectrometry identified Peak #2 as ferritin, FTN-2, (ESI Fig. 7†). Soluble iron redistributed from ferritin towards high molecular weight (HMW, >1 MDa; Peak #1), and low molecular weight (LMW)-iron species (<30 kDa; Peak #3; Fig. 3b). In total, by 13 days of age, the amount of iron associated with ferritin was reduced by approximately 50%, despite a near doubling of total iron levels (Fig. 1b and c). Integration of all peaks indicated that total soluble iron did not change with age, rather, the increased burden of iron we observed was driven by elevations in the insoluble fraction (two-tailed unpaired Student's t-test, p < 0.001, ESI Fig. 8†).
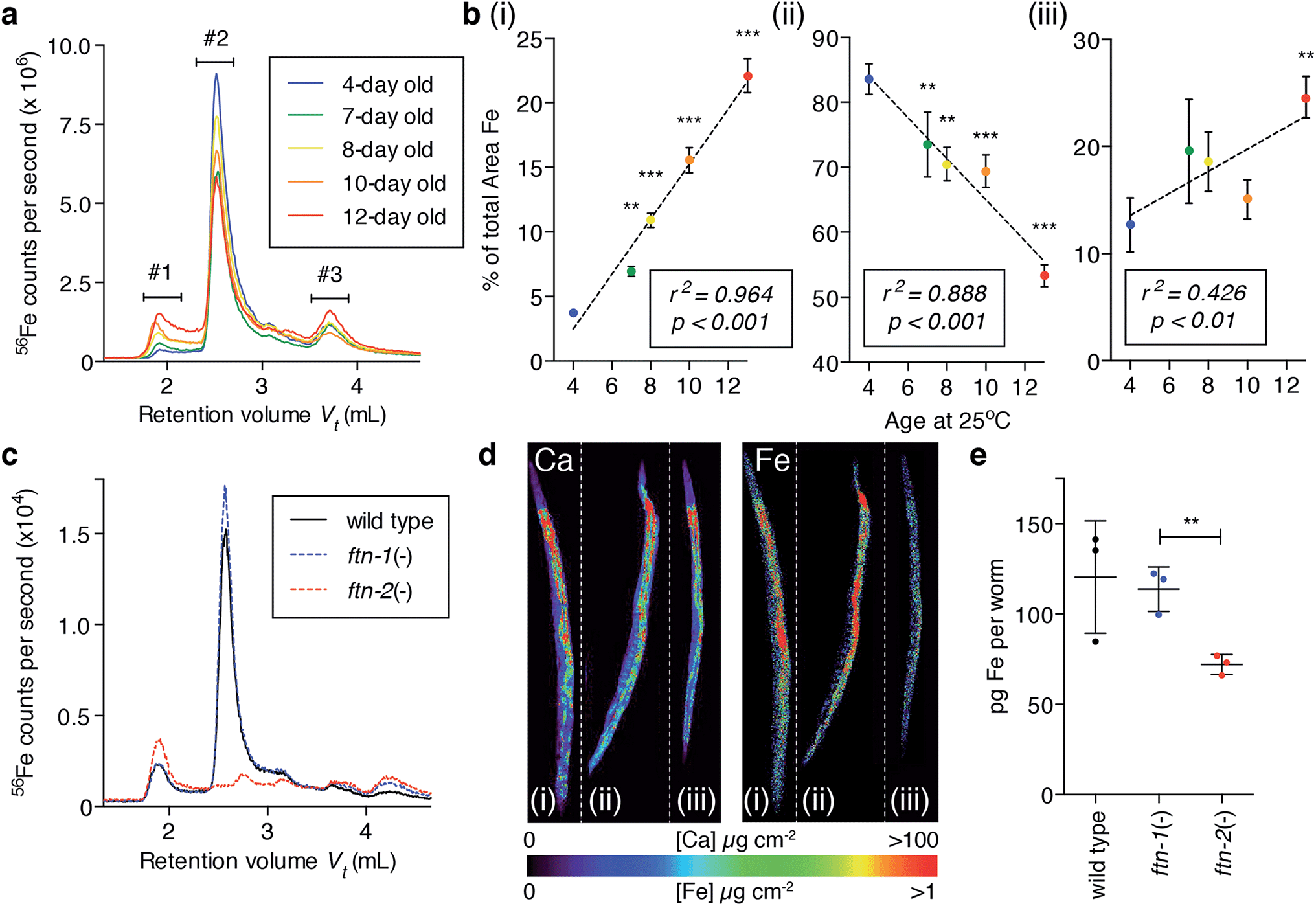 | ||
| Fig. 3 Iron storage capacity of ferritin is compromised during ageing. (a) Native soluble iron-binding species separated and detected by size exclusion chromatography-inductively coupled plasma-mass spectrometry (SEC-ICP-MS). In ageing C. elegans, greater levels of iron are associated with high molecular weight (Peak #1) and low molecular weight (Peak #3) species, whilst ferritin iron is decreased (Peak #2). (b) Integration of these three major chromatographic peaks across age (mean ± SEM, n = 3, one-way ANOVA with Dunnett's post hoc test, ** p < 0.01, *** p < 0.001). Linear regression analysis showed correlation between iron levels in each peak and age. (c) Loss of ftn-2 (null) ablated soluble ferritin-associated iron in young adults. In contrast, ftn-1 nulls did not differ from wild type, retaining the same iron-binding protein profile. (d) Representative X-ray fluorescence micrographs of wild type (i), ftn-1 null (ii) and ftn-2 null (iii) young adults confirmed loss of ftn-2 and resulted in a specific decrease in total iron with unaltered calcium levels. (e) Quantification of iron per adult revealed ftn-2 (ferritin) accounts for approximately 40% total iron in young C. elegans (mean ± SEM, n = 3, two-tailed unpaired Student's t-test, ** p < 0.01). | ||
The C. elegans genome encodes two heavy-chain ferritin orthologs, ftn-1 and ftn-2.14 FTN-1 (predicted MW = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 524.8 Da) expression is induced in the intestine by high iron exposure, while FTN-2 (experimentally observed MW = 19502.2 Da; ESI Fig. 7f†) has constitutive as well as iron-responsive expression in intestinal cells.25,26 Mass spectrometry detected no peptides of FTN-1 within Peak #2. Therefore, ageing is associated with the escape of now-chemically available iron from previously redox-protected storage in FTN-2 (Fig. 3b) to become species that can facilitate oxidation (Fig. 2).
524.8 Da) expression is induced in the intestine by high iron exposure, while FTN-2 (experimentally observed MW = 19502.2 Da; ESI Fig. 7f†) has constitutive as well as iron-responsive expression in intestinal cells.25,26 Mass spectrometry detected no peptides of FTN-1 within Peak #2. Therefore, ageing is associated with the escape of now-chemically available iron from previously redox-protected storage in FTN-2 (Fig. 3b) to become species that can facilitate oxidation (Fig. 2).
Genetic ablation of FTN-2 abolished Peak #2 (ferritin-bound iron) and increased iron associated with Peak #1, whereas loss of FTN-1 had only minor effects compared to wild type (Fig. 3c), likely due to its low constitutive expression.26 Populations of whole wild type and null mutants for ftn-1 and ftn-2 were imaged using XFM (Fig. 3d), which revealed that ftn-2 nulls had ∼46% less total iron compared to wild type (Fig. 3e). Total iron in ftn-1 nulls was not altered. Young ftn-1;ftn-2 null animals had no detectable histological non-heme iron (ESI Fig. 9†), demonstrating that ferritin accounts for the bulk of Perls staining in young animals (Fig. 1c-i & ii).
Total iron load does not explain senescence
Iron markedly increases and spreads in the ageing animal (Fig. 1), but this must represent iron that fails to be incorporated into ferritin as FTN-2 loading of iron decreases with age (Fig. 3a and b), consistent with a concurrent decline in ftn-2 expression.27 We tested whether total iron burden is responsible for age-related oxidative stress is an insufficient explanation for the biochemical causes of senescence. We exposed young wild type and ftn-2 null animals to high levels of exogenous iron and found no detrimental effect on wild type survival (Fig. 4a), or oxidative stress (Fig. 4b), despite inducing a clear elevation in total iron levels, which were largely stored in Peak #2 FTN-2 (Fig. 4c). FTN-2 is responsible for preventing iron-mediated senescence because ftn-2 null animals failed to appropriately buffer iron and displayed a further curtailed lifespan with an expected concomitant increase in oxidation (Fig. 4a–c).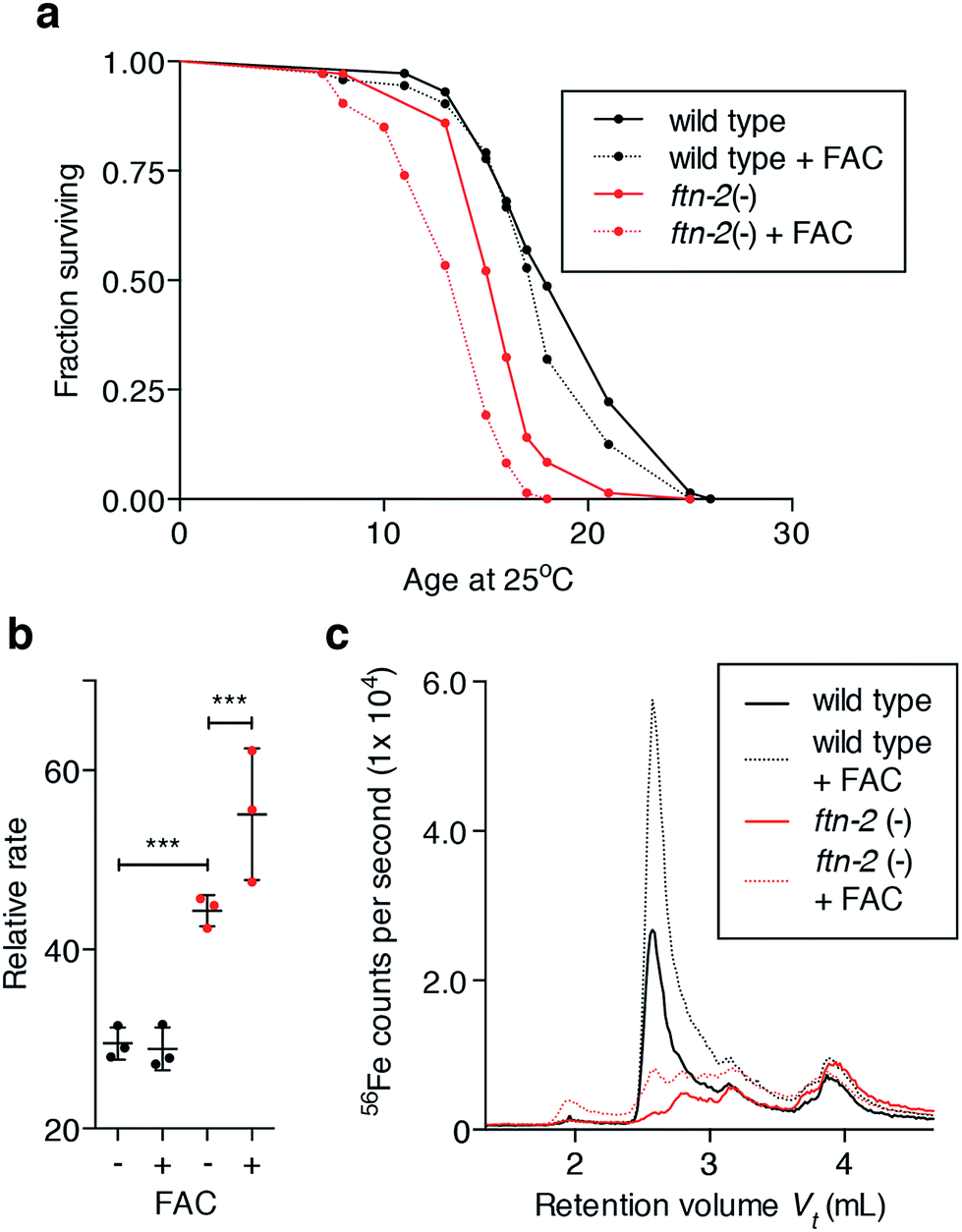 | ||
| Fig. 4 Elevated iron alone does not decrease lifespan (a) exposure of young adult wild type C. elegans to high iron (as ferric ammonium citrate, FAC; 5 mg mL−1) did not affect lifespan. In contrast, ftn-2 null mutants (16 day median lifespan) were significantly short-lived relative to wild type (18 day median lifespan; log-rank test, p < 0.001). The median lifespan of ftn-2 nulls was further reduced by exposure to FAC (15 day median lifespan; log-rank test, p < 0.001). (b) Ex vivo reactive species (as indicated by DCF fluorescence rate) generation was unaltered in lysates from 5-day old wild type exposed to high iron (5 mg mL−1 FAC) for 48 h. Fluorescence rate from ftn-2 nulls raised on FAC was elevated relative to wild type (two-tailed unpaired Student's t-test, *** p < 0.001) and further elevated following iron exposure (two-tailed unpaired Student's t-test, p < 0.001). (c) Consistent with previously observed effects, wild type buffer high iron exposure (FAC) in ferritin, as shown by SEC-ICP-MS, whereas ftn-2 nulls were incapable. | ||
Considering the primary role of ferritin is to store and buffer potentially cytotoxic iron species, our data suggested an underlying mechanism of wild type ageing as being loss of ferritin function and/or cellular iron homeostasis, with resultant increases in pro-oxidant reactive iron (Fig. 2 & 3). Therefore, we sought to investigate how the aggregate coordination environment responsible for maintaining the cellular Fe2+/Fe3+ balance changes within age.
Corrupted buffering shifts iron oxidation state in vivo
X-ray absorption near-edge spectroscopy (XANES) reports on the electronic structure and organisation of nearest neighbors around the absorbing atom. Ideally, XANES assessment of metal coordination in vivo requires procedures that avoid inducing chemical disruption, (e.g. artefacts that arise from fixation). To visualise the coordination chemistry of iron in vivo, we stepped beyond single point projection-based XANES and performed fluorescence-XANES imaging on intact hydrated C. elegans. Serial scanning of young and aged wild type, and young ftn-2 null animals (2–4 individuals per group) across an incident energy range spanning the Fe K-edge (7112 eV) produced a hyperspectral dataset (incident energy, x position, y position, X-ray fluorescence intensity) where each pixel is associated with an iron XANES spectrum.As the relevant ligand set is difficult to define a priori we did not seek to localise individual iron species, rather we gained an overview of iron metabolism by identifying aggregate modes of iron coordination associated with physiological structures (Fig. 5 and ESI Fig. 11a–c†). For biological systems the aggregate iron coordination environment is composed of numerous iron complexes (including ferritin, Fe–S and heme proteins, etc.) with distinct spatial distributions and present at a wide range of concentrations. We used the approach of Lerotic et al.28 to identify the spatial distribution of unique coordination states. Principal component analysis was used to decompose the XANES spectra using k-means clustering (ESI Fig. 10†) into distinct aggregate iron coordination states, which we used to segment the XANES image into 5–6 regions of interest (ROI).
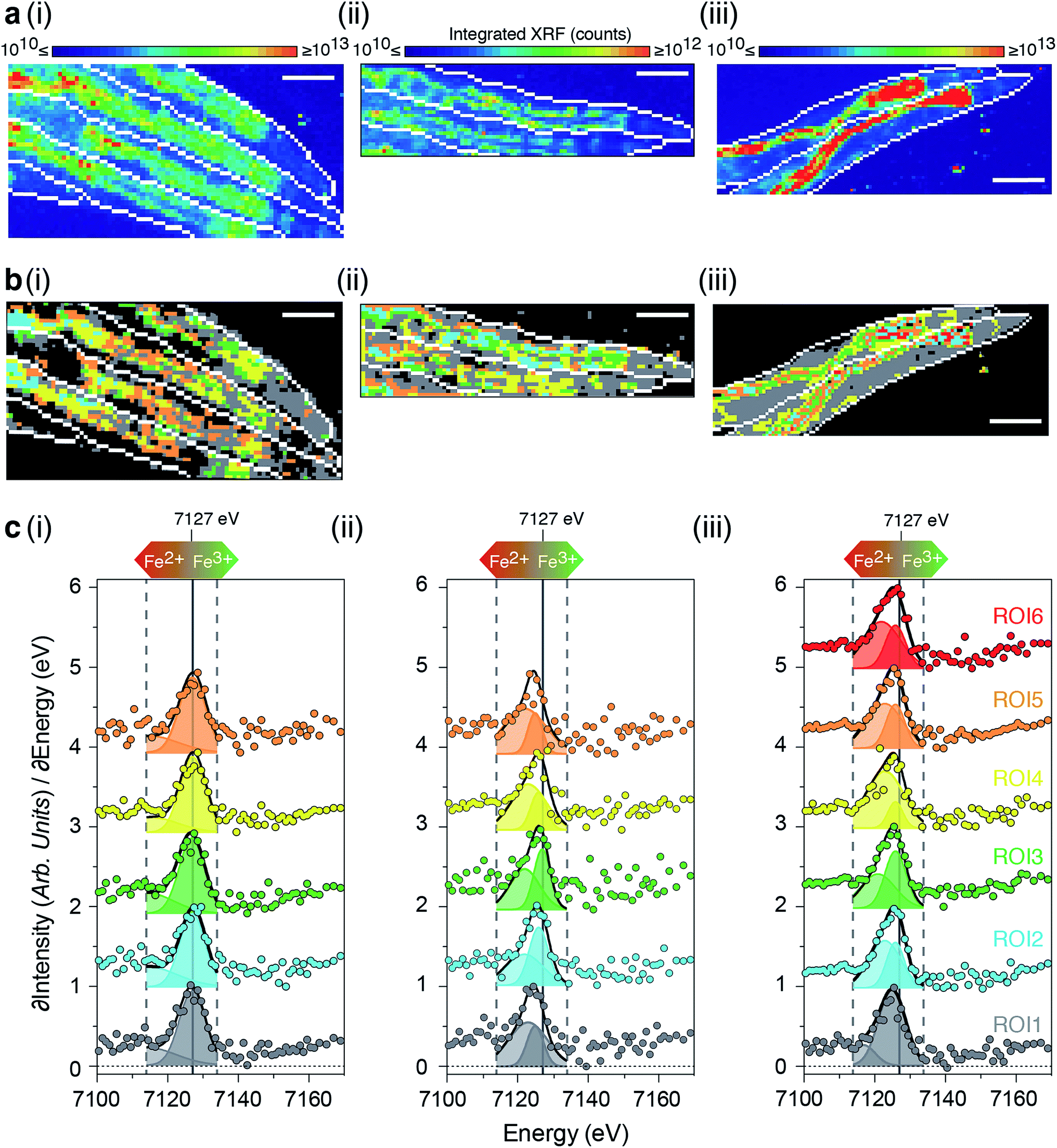 | ||
| Fig. 5 Ageing disturbs iron redox balance (a) integrated intensity of 71 X-ray fluorescence micrographs spanning the iron K-edge (7100 to 7170 eV; dashed grey lines) for the anterior portion of hydrated (i; n = 4) 5-day old wild type, (ii; n = 2) 5-day old ftn-2;ftn-1 nulls; and (iii; n = 2) 12-day old wild type adults. (b) Spatial distribution of ROIs defined by grouping pixels with similar spectra using k-means clustering. ROI-1 corresponds predominately to non-intestinal tissues, whilst ROI-2 through 5 are localized to the intestine. Scale bar = 100 μm. (c) Derivative iron K-edge spectra (∂XRF/∂Energy) for each ROI. Derivative spectra were modeled as the sum of two Gaussians between 7114 and 7134 eV (solid black lines) and the peak area associated with the 1s → 4s (light shaded curves) and 1s → 4p (dark shaded curves) transitions used to assess the Fe2+/Fe3+ balance of each ROI. Impaired ferritin function in aged wild type and ftn-2;ftn-1 null adults animals resulted in a proportional elevation of Fe2+. | ||
Consistent with our earlier results, we observed decreased total iron in ftn-2 nulls and a marked increase in aged wild types (Fig. 5a) with distinct ROIs corresponding to areas of similar iron metabolism. In all animals ROI1 (grey) was localised to tissues outside the intestine, while all other ROIs primarily defined intestinal structures (Fig. 5b and c).
Both the 1s → 3d pre-edge peak and edge position (defined as the peak in the first derivative spectra) shift to higher energy with oxidation state, independent of coordination geometry29 and can be used to assess the relative proportion of Fe2+ and Fe3+. Both of these features revealed changes in the dominant iron oxidation state in young ftn-2 nulls and aged wild type animals. Compared to young wild types all ftn-2 null ROI K-edges were shifted to lower energies (wild type having a K-edge at approximately 7127 eV). This is consistent with a biochemical shift towards increased Fe2+ content within these animals that are unable to safely store iron in ferritin as Fe3+. Aged wild types also displayed a similar shift of ROI K-edge indicative of elevated Fe2+ content. These data are supported by a concomitant shift in the energy of iron pre-edge feature (ESI Fig. 11†). A unique mode of Fe coordination was identified in the aged animals (ROI6 red; Fig. 5b and c), representing distinct foci of aggregate iron coordination localised to the intestine.
The relative intensity of the 1s → 4s (7114–7123 eV) and 1s → 4p (7124–7134 eV) transitions provides a means for quantifying the Fe2+:Fe3+ ratio.29,30 Fitting the contribution of these electronic transitions to the structure of the first derivative we determined the relative abundance of Fe2+ within each ROI. This analysis showed both the aged wild type and ftn null had significantly more Fe2+ content than the young wild type (Fig. 5 & ESI Fig. 11†).
Discussion
Accumulation of iron with age may be a universal feature of multicellular organisms, from invertebrates to humans.2–5,31 Although iron is an essential redox-active element for eukaryotes, it can induce uncontrolled oxidative chemistry if it is not appropriately regulated. The human brain appears particularly vulnerable to improper iron handling, probably because its exceptionally high metabolic output calls for constitutively high iron concentrations.We have recapitulated the biochemical features of the ageing brain in the well-characterised animal model C. elegans. Using several advanced analytical approaches to study bioinorganic chemistry we identified iron dyshomeostasis as an imperative feature of C. elegans senescence, and we have characterised a possible mechanism for this. Our data indicate that the C. elegans intestine is particularly susceptible to senescent iron changes. The C. elegans intestine is the primary site responsible for digestion, macromolecule storage, endocrine function, detoxification of exogenous toxins, and host-pathogen interactions. The intestine comprises approximately one third of the somatic mass of C. elegans, has the greatest relative concentrations (>10 μM) of iron,17 and is the predominant site of ferritin expression and function. Collapse of iron homeostasis is marked in the intestine of aged C. elegans. The ILS pathway acts on intestinal DAF-16 transcription factor activity to modulate ageing,32 and therefore loss of normal intestinal cell function could mirror the accelerated ageing effects of daf-16 mutation. Other lifespan-affecting transcription factors such as SKN-1 (a nuclear factor E2-related factor family member), also act in the intestinal cells to alter ageing rates.33 Intestinal cell nuclei are progressively lost as C. elegans age,23 and the resultant cumulative loss of intestinal function is likely to further compound disruption of iron homeostasis. We observed nuclear iron accumulation (Fig. 1f) within the intestine that is likely to contribute to demise of these nuclei via elevated iron-mediated free radical generation. Assays designed to directly appraise H2O2 have also suggested nuclear accumulation of this damaging oxidant.34 Intra-nuclear aggregates of iron-rich ferritin have also been observed in mammalian models of toxic iron overload.35,36
The age-dependent rise in iron is markedly delayed in long-lived daf-2 mutants. However, at their median lifespan, daf-2 mutants still show a significant increase in total iron (ESI Fig. 3†), demonstrating that iron elevation invariably heralds death even where the rate of ageing is slowed. The suppression of iron accumulation accounts for the comparatively low levels of reactive species generated by the daf-2 mutants versus age-matched wild type (Fig. 2d). Loss of daf-16 by mutation reverses the longevity effects of the daf-2 mutant and not only restores but also exaggerates the age-dependent rise in iron (Fig. 1b). DAF-16 need be expressed only in the intestine to slow ageing in C. elegans.32 This is consistent with uncontrolled iron elevation and altered coordination in the metabolically critical intestinal cells being a primary contributor to oxidative load in aged C. elegans.
The formation of non-ferritin iron collections in HMW soluble- and insoluble forms (Fig. 3; ESI Fig. 8†) precedes death, and is reminiscent of iron-filled intracellular inclusions, lipofuscin, neuromelanin and hemosiderin, which all feature in the pathologies of ageing and age-related disease, but have questionable or unclear roles in cytotoxicity. We hypothesise that the underlying mechanism of ageing in wild type C. elegans is the escape of iron from safe storage in ferritin (FTN-2), where it emerges as redox-active Fe2+ species that redistribute primarily to soluble HMW and LMW-iron, as well as insoluble precipitates, where it catalyses toxic free radical generation. HMW-iron may represent misfolded, mis-metallated proteinaceous material transitioning towards an insoluble aggregate typical of ageing pathology.37 The withdrawal of soluble iron from the cytoplasm into these aggregates could explain why the rise in total somatic iron is not sensed by the transcriptional mechanisms that should upregulate the expression of ferritin14 and other iron-regulatory elements.
The C. elegans genome encodes two ferritin genes; ftn-1 and ftn-2, however we have determined that only ftn-2 contributes to iron storage under basal conditions. Although understanding the specific function of ftn-1 will require further investigation, our data are consistent with previous reports showing the impact of ftn-1 is negligible with respect to lifespan under normal conditions.38
Conclusion
Extreme levels of iron can be toxic,14,31,38 however, our results indicate that the age-related accumulation of iron observed in C. elegans is, by itself, insufficient to cause senescence. Rather than being determined by simple accumulation, we find that normal ageing is characterised by a loss of ferritin function and/or cellular iron homeostasis, with resultant increases in reactive iron and oxidative stress. With ferrous iron a catalyst for radical generation, the clear shift from Fe3+ to Fe2+ in both aged and ferritin null worms in vivo provides a mechanism to drive senescence. These observations have broader implications for understanding how cellular iron dyshomeostasis may contribute to major age-related human diseases. Strategies derived to delay or mitigate the risks associated with ageing may consider the inevitability of contributions of late-life iron.Methods and materials
Strains
N2 (wild-type), TJ1060: spe-9(hc88); fer-15(b26), CB1370: daf-2(e1370), and DR1309: daf-16(m26); daf-2(e1370), RB2603; ftn-1(ok3625) and RB668; ftn-2(ok404) were obtained from the Caenorhabditis Genetics Center. As the ftn-1(ok3625) deletion allele has not been mapped, we sequenced the genomic DNA across the putative deletion site. A 495 bp deletion was identified that removes all of exon 2 and most of exon 3 (ESI Fig. 12†), resulting in a premature stop codon (UGA), so that the likely product is truncated to 20 amino acids (out of the predicted 170). The ftn-2(ok404) is a previously-characterised null allele.25 RB2603; ftn-1(ok3625) and RB668; ftn-2(ok404) were each backcrossed four times to wild type prior to further analysis. New strains reported in this study are listed in ESI Table 1.† All strains were maintained at 20 °C on standard nematode growth media (NGM)39 and aged at 25 °C as required, with the exception of the fertility mutant TJ1060 which was maintained at 16 °C.DNA primers 5′-ATGTGTCTCAGATTTCCGCC-3′ and 5′-GAACCCTTTCGTTGCCAATA-3′ were used to track the ftn-1(ok3625) deletion, and 5′-CCAGCCACAAACTACCATCA-′3 and 5′-AAAGAAGATGCGATGAGCAGA-′3 for the ftn-2(ok404) deletion respectively during back crossing to wild type and crossing to generate the double null strains GMC005 and GMC007.
X-ray fluorescence microscopy (XFM)
Aged C. elegans were prepared as previously described17 on Si3N4 windows (Silson) for analysis at the XFM beamline at the Australian Synchrotron and the experimental setup has been described previously.16 Briefly, the distribution of metals was mapped using a beam of 12.7 keV X-rays focused to 2 μm (full-width at half-maximum) using a Kirkpatrick–Baez mirror pair.40 The X-ray energy was chosen to induce K-shell ionisation of elements with atomic numbers below 34, while also separating the elastic and inelastic scatter from fluorescence lines of lighter elements. Specimens were continuously scanned through X-ray focus and the real-time event-mode processing approach used in the Maia detector system,41 installed at the Australian Synchrotron XFM beamline, allowed flexible selection of effective per pixel dwell times, though 8 ms was typical. Resulting elemental maps ranged up to 50000 pixels in size and the total acquisition time varied around 7 minutes per specimen. Two single-element foils of known areal density, Mn and Pt (Micromatter, Canada), were scanned during the experiment as references for determination of elemental areal density.42 Deconvolution of the X-ray fluorescence spectra was performed using the GeoPIXE v7.0f (CSIRO, Australia) and calibrated using the metal foil measurements, with corrections made for self-absorption in the sample, absorption in air, and the efficiency response of the detector.
Comparison of total iron quantitated from whole worms was examined via one-way ANOVAs with a Dunnett's post-hoc test. Within each genotype aged cohorts were compared to young (4 days of age) adults.
Histology
C. elegans were washed in S-basal,39 fixed overnight in 10% (v/v) neutral buffered formalin (NBF) at 4 °C, embedded in 2% (w/v) agar in phosphate buffered saline (PBS) blocks and then fixed again in 10% NBF overnight. Following processing of the agar blocks into paraffin, 5 μm sections were prepared, dewaxed and stained with DAB-enhanced modified Perl's Prussian blue following a standard protocol.43 Samples were counter stained with Harris haematoxylin solution (Amber Scientific).Live imaging of iron
C. elegans cultures were aged as indicated, washed in S-basal,39 then co-cultured in S-basal containing 1 × 108 cells OP50 (E. coli) and 0.05 μg mL−1 calcein-AM (Invitrogen) for 1 h and then in S-basal with 1 × 108 cells OP50 for 1 h. Samples were then mounted for epi-fluorescence microscopy using standard techniques. Calcein fluoresces in the presence of calcium ions in solution, but this fluorescence is quenched by ionic iron. Calcein has a slight selectivity for Fe2+ over Fe3+. Other divalent metal ions, Cu2+, Ni2+ and Co2+ can also quench calcein fluorescence, however, these were found to be present at ≥2 orders of magnitude lower than Fe in the intestine and therefore not considered to be able to interfere with the signal.ROS detection
2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) at 10 μM was used as a fluorescent probe for reactive oxygen species (ROS) detection in vivo using standard protocols.44 DCFH-DA enters and accumulates in the intestinal cells, where it is oxidised by several ROS (including hydroxyl radicals) to form the fluorophore DCF.45 Analysis of fluorescence rate increase was performed on samples sonicated in 1× TBS, pH 7.4 and recovered as the supernatant from a 100000g centrifugation at 4 °C. Total protein concentration was determined by a Nanodrop spectrophotometer (Thermo Scientific). Ex vivo ROS detection were made by bringing lysate supernatants (50 μg total protein) to a reaction volume of 200 μL with 200 mM ammonium acetate pH 7.5, 100 μM DCFH-DA (Sigma-Aldrich, made from a 10 mM stock in acetonitrile), and 400 μM ascorbate, in black 96-well micro titer plates. Fluorescence in 8 replicate wells was quantitated (Ex: 485 nm, Em: 535 nm using a 495 nm cut-off) by a FlexStation (Molecular Devices) plate reader, using 30 reads every minute for 1 h at medium PMT setting. Values from lysate-free (negative) controls were subtracted and the fluorescence data was then baseline corrected. A linear regression fitted from 20–40 minutes and the slopes were normalised against 4-day old wild type and plotted (Prism v5.0d, Graphpad Software).
Fluorescence microscopy
Animals were mounted on a glass slide with 2% (w/v) agar pad containing 2% NaN3 under a glass cover slip and examined using an Olympus BX40 epifluorescence microscope equipped with SPOT RTKE cooled color CCD camera (Diagnostic Instruments, MI). The GFP fusion protein was visualised by using an Olympus U-MWG filter set (Ex: bandpass 530 ± 20 nm, Em: longpass 590 nm), and the images imported into SPOT software (Diagnostic Instruments). ImageJ v1.45s (NIH, USA) was used for image preparation and overlays.Bulk iron quantitation
Total iron was measured using a 7700 Series (Agilent) inductively coupled plasma mass spectrometry (ICP-MS) as previously reported.46 Samples consisted of 100 adults per replicate for different aged cohorts as indicated.Purification of C. elegans ferritin
10 g of mixed stage wild type cultured on 8P media47 at 20 °C were frozen as pellets in liq-N2. Pellets were crushed in a liq-N2 chilled mortar and pestle, then added to 20 mL PBS pH 7.4 with EDTA-free proteinase inhibitors (Roche). The lysate was further disrupted with 20 strokes of an ice-cooled Dounce homogeniser (in 50 mL). The extract was centrifuged at 3300g at 4 °C and the supernatant (∼30 mL) dialyzed overnight in 18 MΩ pure H2O (Millipore) at 4 °C using pleated dialysis tubing with a 10 kDa molecular weight cut-off (Thermo Scientific). 20 mL of dialysate was then diluted to 60 mL with pH 3–10 ampholytes and iso-electrically focused via a Rotofor II (Bio-Rad), as per manufacturer's protocols to 2500 Vh. Fractions were collected and iron content measured by graphite furnace atomic absorption spectroscopy (AAS). A standard method was used with ashing and atomisation temperatures of 700 °C and 2300 °C respectively, and with linear absorbance to a concentration of 100 μg L−1. The three highest contiguous iron-containing fractions were then pooled and re-focused via the Rotofor II as above. Fractions containing iron were identified by AAS. To the six contiguous iron containing fractions NaCl was added to a final concentration of 150 mM and then size-excluded via FPLC (Bio-Rad) using a Superdex 200 10/300 GL column and PBS buffer at 0.6 mL min−1. Fractions were collected and iron was measured by AAS. Fractions 9 and 10 (F9/10) were identified, pooled and concentrated to 300 μL via vacuum centrifugation (SpeedVac, Savant). Aliquots were then frozen at −80 °C until required for further analysis.Electrophoretic analysis
Samples were suspended in 1× Laemmli sample buffer (with 10 mM TCEP, 6 M urea, and 2% SDS), boiled for 10 min, and analyzed by SDS-PAGE (NuPage 4–12% Bis-Tris, Invitrogen). Samples were prepared in parallel were either stained with Oriole (Bio-Rad) in preparation for mass spectrometry or immunoblotted using a 1:1000 dilution of polyclonal anti-horse spleen ferritin antibody produced in rabbit (Sigma-Aldrich), and imaged via standard chemiluminescence.
Mass spectrometry
Matrix-assisted laser desorption/ionisation-mass spectrometry (MALDI-MS) using an UltrafleXtreme (Bruker Daltronics), a two-layer sample preparation method48 and α-cyano-4-hydroxycinnamic acid as the MALDI matrix, was used to determine the protein parent mass of the purified C. elegans F9/10 as ferritin. For further analysis the purified C. elegans ferritin was digested with LysC (Roche) in 4 M urea in a ratio of 50:1 protein to LysC overnight at 37 °C. Digested material was then desalted and concentrated through a C4 ZipTip (Millipore) for MALDI-MS/MS analysis (as above) to generate a peptide mass fingerprint. The peptide masses were searched using MASCOT (Matrix Science).
DNA sequencing
The ftn-1 ORF amplicon was amplified using the following nested primers: outer-forward 5′-ATGTGTCTCAGATTTCCGCC, inner-forward 5′-GGTTGAACCTTTTTAGGAACTGC, inner-reverse 5′-ACAGTCCCGGACACGTAATC and outer-reverse 5′-GAACCCTTTCGTTGCCAATA. Sequencing was performed using the inner primer pair at the Applied Genetic Diagnostic facility (Department of Pathology, University of Melbourne) using ABI3130xl capillary genetic analyzers and BDV3.1 terminators. Three independent ampicons, from both wild type and ftn-1(ok3625), were sequence on both complementary DNA strands.Size exclusion chromatography-inductively-coupled plasma mass spectrometry (SEC-ICP-MS)
Samples were homogenised in TBS (pH 8.0) with added proteinase inhibitors (EDTA-free, Roche), then clarified by a 15 min centrifugation at 175000g, 4 °C. The protein concentration of the supernatant was determined via UV absorbance (Nanodrop, ThermoScientific) and equivalent protein amounts were size-excluded using a Bio SEC-5 (4.6 × 300 mm, 5 μm, Agilent) column with 200 mM ammonium nitrate (trace analysis grade, Sigma) pH 8.0 buffer the flow rate was 0.4 mL min−1 at 30 °C. The eluant from the column was directly connected to the ICP-MS for elemental detection as previously described.49,50
Lifespan analysis
The effects of genetic ablation of ftn-2 and ftn-1 on wild type was measured using established protocols.51,52 For the life span effects of elevated exogenous iron we used ferric ammonium citrate (FAC) supplemented NGM. Due to the semi defined chemical formulae of FAC, xFe·yNH4C6H7O8, we determined the iron content of our batch of FAC (9.60 ± 0.61% by weight; n = 3), NGA (246.24 ng g−1 of media, n = 10) and the NGA + 5 mg mL−1 FAC high iron media (1.172 mg g−1 of media, n = 10) by ICP-MS. For the media measurements the media was aliquoted into ∼500 mg aliquots, weighed and then freeze dried, digested in 50 μL 60% (w/v) HNO3 + 50 μL 30% (w/v) H2O2 at 60 °C for 5 min, diluted to 1 mL and analysed by ICP-MS. All life span assays were conducted at 25 °C following adulthood.Spatially resolved fluorescence X-ray absorption near edge structure (XANES) spectroscopy
Hydrated, anesthetised C. elegans samples were prepared for XFM as previously described.16 A series of 100 XFM maps were collected using energies 7282–7042 eV (energy steps: 7282–7242 in 20 eV increments; 7222–7197 in 5 eV increments; 7192–7164 in 2 eV increments; 7162–7093 in 1 eV increments; 7092–7042 in 10 eV increments). To reduce the applied dose and limit radiation damage the beam was defocused to 3 μm and specimens scanned with a 7 μm vertical step. Data was binned every 7 μm in the horizontal to produce 7 × 7 μm pixel with an effective dwell time of ∼27.34 ms per pixel.As above, deconvolution of the Maia data was performed using GeoPIXE v7.1f (CSIRO, Australia). The X-ray photons from each pixel were related to calculated-model fluorescence X-ray yield for an assumed specimen composition and thickness as previously described.16 To minimise the effects of sample movement subsequent analysis was limited to the information rich iron K-edge (7100–7170 eV, consisting of 71 micrographs). The distribution of diffusible calcium remained unchanged throughout the data collection and was used to correct small positional discrepancies between micrographs. Cross correlation as implemented in MANTiS v2.06 was used to align the 71 iron micrographs into the final XANES stack. For each group the maximum pixel displacement through all series was <1 pixel (ESI Fig. 13†). Pixels were grouped by spectral similarity using MANTiS v2.06 (ref. 53) into distinct regions of interest. The associated XANES spectra for each region (ESI Fig. 10†) were normalised to the intensity of the white line and the K-edge structure analysed. Edge position was determined by fitting a single Gaussian to the first derivative spectra between 7114–7134 eV. The relative intensities of the 1s → 4s and 1s → 4p electronic transitions within the structure of the first derivative was used to indicate the Fe2+:Fe3+ ratio.29,30
Statistical tests
Kaplan–Maier survival curves were generated and compared via non-parametric log rank tests and t-tests performed using Prism v5.0d (Graphpad Software). Comparisons between the goodness of fit for nested models, i.e. Gaussian (X ∼ N(μ,σ2)) vs. a sum of Gaussians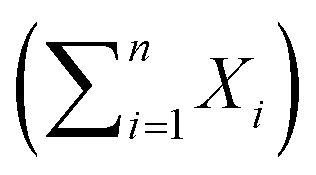 were assessed using the Akaike information criterion and the exact sum of squares F-test. The dependency and co-variance between parameters of candidate models were monitored and models containing redundant parameters were rejected.
were assessed using the Akaike information criterion and the exact sum of squares F-test. The dependency and co-variance between parameters of candidate models were monitored and models containing redundant parameters were rejected.
Acknowledgements
We thank Brian Stevens (Florey Institute) for technical assistance, the X-ray Fluorescence Microscopy and X-ray Absorption Spectroscopy beamlines (Australian Synchrotron), and the Caenorhabditis Genetics Center funded by the US National Institutes of Health National Center for Research Resources for providing several strains. The Australian Research Council (ARC) and the Victorian Government's Operational Infrastructure Support Program supported this research.Notes and references
- K. J. Barnham and A. I. Bush, Chem. Soc. Rev., 2014, 43, 6727–6749 RSC.
- H. R. Massie, V. R. Aiello and T. R. Williams, Mech. Ageing Dev., 1993, 67, 227–237 CrossRef CAS.
- C. I. Cook and B. P. Yu, Mech. Ageing Dev., 1998, 102, 1–13 CrossRef CAS.
- B. Bilgic, A. Pfefferbaum, T. Rohlfing, E. V. Sullivan and E. Adalsteinsson, NeuroImage, 2012, 59, 2625–2635 CrossRef CAS PubMed.
- L. Zecca, M. B. Youdim, P. Riederer, J. R. Connor and R. R. Crichton, Nat. Rev. Neurosci., 2004, 5, 863–873 CrossRef CAS PubMed.
- R. J. Ward, F. A. Zucca, J. H. Duyn and R. R. Crichton, Lancet Neurol., 2014, 13, 1045–1060 CrossRef CAS.
- J. H. Chen, N. Singh, H. Tay and T. Walczyk, Metallomics, 2014, 6, 1417–1426 RSC.
- S. Y. Jeong and S. David, J. Neurosci., 2006, 26, 9810–9819 CrossRef CAS PubMed.
- D. J. Hare, M. Gerlach and P. Riederer, J. Neural Transm., 2012, 119, 1515–1521 CrossRef CAS PubMed.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- A. Hong-Hermesdorf, M. Miethke, S. D. Gallaher, J. Kropat, S. C. Dodani, J. Chan, D. Barupala, D. W. Domaille, D. I. Shirasaki, J. A. Loo, P. K. Weber, J. Pett-Ridge, T. L. Stemmler, C. J. Chang and S. S. Merchant, Nat. Chem. Biol., 2014, 10, 1034–1042 CrossRef CAS PubMed.
- G. Schneider, P. Guttmann, S. Heim, S. Rehbein, F. Mueller, K. Nagashima, J. B. Heymann, W. G. Muller and J. G. McNally, Nat. Methods, 2010, 7, 985–987 CrossRef CAS PubMed.
- P. M. Chege and G. McColl, Front. Aging Neurosci., 2014, 6, 89 Search PubMed.
- B. L. Gourley, S. B. Parker, B. J. Jones, K. B. Zumbrennen and E. A. Leibold, J. Biol. Chem., 2003, 278, 3227–3234 CrossRef CAS PubMed.
- C. J. Kenyon, Nature, 2010, 464, 504–512 CrossRef CAS PubMed.
- S. A. James, M. D. de Jonge, D. L. Howard, A. I. Bush, D. Paterson and G. McColl, Metallomics, 2013, 5, 627–635 RSC.
- G. McColl, S. A. James, S. C. Mayo, D. L. Howard, C. G. Ryan, R. Kirkham, G. F. Moorhead, D. Paterson, M. D. de Jonge and A. I. Bush, PLoS One, 2012, 7, e32685 CAS.
- M. Tatar, A. Bartke and A. Antebi, Science, 2003, 299, 1346–1351 CrossRef CAS PubMed.
- K. D. Kimura, H. A. Tissenbaum, Y. Liu and G. Ruvkun, Science, 1997, 277, 942–946 CrossRef CAS.
- C. Kenyon, J. Chang, E. Gensch, A. Rudner and R. Tabtiang, Nature, 1993, 366, 461–464 CrossRef CAS PubMed.
- S. Ogg, S. Paradis, S. Gottlieb, G. I. Patterson, L. Lee, H. A. Tissenbaum and G. Ruvkun, Nature, 1997, 389, 994–999 CrossRef CAS PubMed.
- R. Meguro, Y. Asano, H. Iwatsuki and K. Shoumura, Histochem. Cell Biol., 2003, 120, 73–82 CrossRef CAS PubMed.
- M. D. McGee, D. Weber, N. Day, C. Vitelli, D. Crippen, L. A. Herndon, D. H. Hall and S. Melov, Aging Cell, 2011, 10, 699–710 CrossRef CAS PubMed.
- P. Arosio, R. Ingrassia and P. Cavadini, Biochim. Biophys. Acta, 2009, 1790, 589–599 CrossRef CAS PubMed.
- Y. I. Kim, J. H. Cho, O. J. Yoo and J. Ahnn, J. Mol. Biol., 2004, 342, 421–433 CrossRef CAS PubMed.
- U. Cha'on, N. Valmas, P. J. Collins, P. E. Reilly, B. D. Hammock and P. R. Ebert, Toxicol. Sci., 2007, 96, 194–201 CrossRef PubMed.
- Y. V. Budovskaya, K. Wu, L. K. Southworth, M. Jiang, P. Tedesco, T. E. Johnson and S. K. Kim, Cell, 2008, 134, 291–303 CrossRef CAS PubMed.
- M. Lerotic, C. Jacobsen, T. Schäfer and S. Vogt, Ultramicroscopy, 2004, 100, 35–57 CrossRef CAS PubMed.
- A. J. Berry, H. S. O'Neill, K. D. Jayasuriya, S. J. Campbell and G. J. Foran, Am. Mineral., 2003, 88, 967–977 CAS.
- A. J. Berry, L. V. Danyushevsky, H. S. C. O'Neill, M. Newville and S. R. Sutton, Nature, 2008, 455, 960–963 CrossRef CAS.
- I. M. Klang, B. Schilling, D. J. Sorensen, A. K. Sahu, P. Kapahi, J. K. Andersen, P. Swoboda, D. W. Killilea, B. W. Gibson and G. J. Lithgow, Aging, 2014, 6, 975–991 Search PubMed.
- N. Libina, J. R. Berman and C. Kenyon, Cell, 2003, 115, 489–502 CrossRef CAS.
- J. M. Tullet, M. Hertweck, J. H. An, J. Baker, J. Y. Hwang, S. Liu, R. P. Oliveira, R. Baumeister and T. K. Blackwell, Cell, 2008, 132, 1025–1038 CrossRef CAS PubMed.
- B. C. Dickinson, Y. Tang, Z. Chang and C. J. Chang, Chem. Biol., 2011, 18, 943–948 CrossRef CAS PubMed.
- A. G. Smith, P. Carthew, J. E. Francis, R. E. Edwards and D. Dinsdale, Hepatology, 1990, 12, 1399–1405 CrossRef CAS.
- A. Kondo, J. Deguchi and S. Okada, Virchows Arch., 1998, 433, 543–548 CrossRef CAS.
- P. Reis-Rodrigues, G. Czerwieniec, T. W. Peters, U. S. Evani, S. Alavez, E. A. Gaman, M. Vantipalli, S. D. Mooney, B. W. Gibson, G. J. Lithgow and R. E. Hughes, Aging Cell, 2012, 11, 120–127 CrossRef CAS PubMed.
- S. Valentini, F. Cabreiro, D. Ackerman, M. M. Alam, M. B. A. Kunze, C. W. M. Kay and D. Gems, Mech. Ageing Dev., 2012, 133, 282–290 CrossRef CAS PubMed.
- W. Wood, The Nematode Caenorhabditis elegans, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA, 1988 Search PubMed.
- D. Paterson, M. D. de Jonge, D. L. Howard, W. Lewis, J. McKinlay, A. Starritt, M. Kusel, C. G. Ryan, R. Kirkham, G. Moorhead and D. P. Siddons, 10th International Conference on X-Ray Microscopy, 2011, vol. 1365, pp. 219–222 Search PubMed.
- C. G. Ryan, D. P. Siddons, R. Kirkham, P. A. Dunn, A. Kuczewski, G. Moorhead, G. De Geronimo, D. J. Paterson, M. D. de Jonge, R. M. Hough, M. J. Lintern, D. L. Howard, P. Kappen and J. Cleverley, X-Ray Optics and Microanalysis, Proceedings, 2010, vol. 1221, pp. 9–17 Search PubMed.
- C. G. Ryan, Int. J. Imag. Syst. Tech., 2000, 11, 219–230 CrossRef.
- J. A. Duce, A. Tsatsanis, M. A. Cater, S. A. James, E. Robb, K. Wikhe, S. L. Leong, K. Perez, T. Johanssen, M. A. Greenough, H. H. Cho, D. Galatis, R. D. Moir, C. L. Masters, C. McLean, R. E. Tanzi, R. Cappai, K. J. Barnham, G. D. Ciccotosto, J. T. Rogers and A. I. Bush, Cell, 2010, 142, 857–867 CrossRef CAS PubMed.
- K. Ye, C. B. Ji, X. W. Lu, Y. H. Ni, C. L. Gao, X. H. Chen, Y. P. Zhao, G. X. Gu and X. R. Guo, J. Radiat. Res., 2010, 51, 473–479 CrossRef CAS.
- A. Gomes, E. Fernandes and J. L. F. C. Lima, J. Biochem. Biophys. Methods, 2005, 65, 45–80 CrossRef CAS PubMed.
- D. Kaur, F. Yantiri, S. Rajagopalan, J. Kumar, J. Q. Mo, R. Boonplueang, V. Viswanath, R. Jacobs, L. Yang, M. F. Beal, D. DiMonte, I. Volitaskis, L. Ellerby, R. A. Cherny, A. I. Bush and J. K. Andersen, Neuron, 2003, 37, 899–909 CrossRef CAS.
- L. Bianchi and M. Driscoll, in WormBook, 2006, DOI:10.1895/wormbook.1.122.1.
- Y. Dai, R. M. Whittal and L. Li, Anal. Chem., 1999, 71, 1087–1091 CrossRef CAS.
- D. J. Hare, A. Grubman, T. M. Ryan, A. Lothian, J. R. Liddell, R. Grimm, T. Matsuda, P. A. Doble, R. A. Cherny, A. I. Bush, A. R. White, C. L. Masters and B. R. Roberts, Metallomics, 2013, 5, 1656–1663 RSC.
- D. J. Hare, J. D. Doecke, N. G. Faux, A. Rembach, I. Volitakis, C. J. Fowler, R. Grimm, P. A. Doble, R. A. Cherny, C. L. Masters, A. I. Bush and B. R. Roberts, ACS Chem. Neurosci., 2015 DOI:10.1021/cn5003557.
- G. McColl, D. W. Killilea, A. E. Hubbard, M. C. Vantipalli, S. Melov and G. J. Lithgow, J. Biol. Chem., 2008, 283, 350–357 CrossRef CAS PubMed.
- G. McColl, A. N. Rogers, S. Alavez, A. E. Hubbard, S. Melov, C. D. Link, A. I. Bush, P. Kapahi and G. J. Lithgow, Cell Metab., 2010, 12, 260–272 CrossRef CAS PubMed.
- M. Lerotic, R. Mak, S. Wirick, F. Meirer and C. Jacobsen, J. Synchrotron Radiat., 2014, 21, 1206–1212 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc00233h |
| This journal is © The Royal Society of Chemistry 2015 |