
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
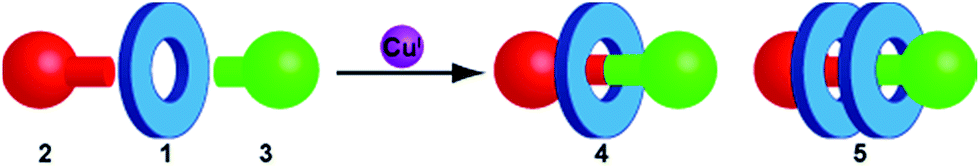
Competitive formation of homocircuit [3]rotaxanes in synthetically useful yields in the bipyridine-mediated active template CuAAC reaction†
Edward A.
Neal
a and
Stephen M.
Goldup
*b
aSchool of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London, E1 4NS, UK
bDepartment of Chemistry, University of Southampton, Highfield, Southampton, Hampshire SO17 1BJ, UK. E-mail: s.goldup@soton.ac.uk
First published on 3rd February 2015
Abstract
We recently identified competitive formation of doubly interlocked [3]rotaxanes as the origin of the non-linear variation in yield of [2]rotaxane with macrocycle size in the bipyridine-mediated AT-CuAAC reaction. Selection of reaction conditions gave [2]rotaxanes in essentially quantitative yield in all cases and hard to access doubly threaded [3]rotaxanes in up to 50% yield in a single, four component coupling. Based on the effect of macrocycle structure on the reaction outcome we propose a detailed mechanism of [3]rotaxane formation.
Introduction
Since Sauvage's first demonstration of a CuI template,1 the explosion of high yielding “passive template” methods for the synthesis of interlocked molecules has facilitated the synthesis of ever more complicated structures and their investigation in areas as diverse molecular electronics, drug delivery, sensors, materials, and as molecular machines.2 In 2006, Leigh and co-workers introduced the “active template” concept,3 in which the metal ion plays an active role in the final covalent bond-forming reaction that captures the interlocked structure, the first example of which was the active template Cu-mediated azide–alkyne cycloaddition4 reaction (AT-CuAAC) for the synthesis of [2]rotaxanes.5 This approach has since been applied to ever more complex interlocked molecules6 and molecular machines,7 as well as being extended to other bond-forming reactions.8During investigations into the effect of macrocycle size in the AT-CuAAC reaction, we recently reported a small bipyridine macrocycle that allows access to functionalised [2]rotaxanes in excellent yield, further increasing the utility of this powerful methodology.9 These results have now been extended to the synthesis of a water-stable CuI–triazolide,10 and the first scalable synthesis of mechanically chiral rotaxanes.11 However, at the time of our first report, the origin of the effect of macrocycle size was unclear, particularly as the yield of [2]rotaxane varied non-linearly with ring size.
Here we report that not only can the yield of [2]rotaxane for all macrocycle sizes be increased to >95% but that optimised conditions can produce novel [3]rotaxanes, challenging targets in their own right, in synthetically useful yields, uniting four components in a single step. Furthermore, comparison of the product distribution of macrocycles with and without a key benzylic ether unit allow us to propose detailed structures for the reactive intermediates leading to [3]rotaxane formation.
Results and discussion
We reinvestigated the AT-CuAAC reaction between macrocycles 1, alkyne 2 and azide 3 to determine the origin of lower yields of [2]rotaxanes 4 with macrocycles 1a–c (Scheme 1). Surprisingly, we found that although 1a produced [2]rotaxane 4a in just 48% yield, no 1a was recovered; typically the balance of macrocycle not incorporated into the rotaxane is recovered unchanged in AT reactions. | ||
| Scheme 1 Effect of bipyridine macrocycle size on product distribution in the AT-CuAAC reaction. a1 equiv. each 1, 2, 3, 0.90 equiv. [Cu(MeCN)4]·PF6, 10 mM conc. of 1, CH2Cl2, 80 °C, 72 h. bDetermined by 1H NMR analysis of the crude mixture. | ||
To our further surprise, on raising the polarity of the eluent during flash chromatography, a new interlocked product was unexpectedly isolated and subsequently identified as [3]rotaxane 5a. Similarly, in the case of macrocycles 1b and 1c no non-interlocked macrocycle was recovered, with all of the starting materials accounted for by the formation of novel [3]rotaxanes 5b and 5c respectively. Only in the case of macrocycle 1d was no [3]rotaxane observed; indeed 4d is the only species observed in the 1H NMR spectrum of the crude reaction product (Fig. 1e). With pure [3]rotaxanes 5a–c in hand, 1H NMR analysis of the crude reaction mixture revealed that this unexpected product accounted fully for the balance of the macrocycle added (Fig. 1). Thus, contrary to our original conclusion, the AT-CuAAC reaction with bipyridine macrocycles is essentially 100% efficient in the formation of the mechanical bond regardless of the size of the macrocycle, but the product distribution between [2]- and [3]rotaxane varies dramatically with macrocycle size.
 | ||
| Fig. 1 Partial 1H NMR (400 MHz, CDCl3, 300 K) of (a) pure [3]rotaxane 5a; (b)–(e) the crude product mixtures obtained from the reactions of macrocycles 1a, 1b, 1c, and 1d respectively (conditions as shown in Scheme 1). Selected signals are assigned with labelling as shown in Scheme 1. Signals corresponding to [3]rotaxanes 5 are denoted by primes (note: although the two macrocycles in 5 are chemically non-equivalent their signals are poorly separated and they are given a single label here for clarity). | ||
Although Leigh and co-workers previously observed the formation of [3]rotaxanes during the AT-CuAAC reaction of monodentate pyridine macrocycles, this was thought not be possible in the case of bidentate macrocycles.5b Furthermore, the yield of the [3]rotaxane product, based on the synthetically expensive macrocycle component, was extremely low (<7%; 33% with respect to the azide and alkyne components).12 Thus, having identified competitive [3]rotaxane formation in significant yield with a single equivalent of all reaction components as the origin of the variation in yield of [2]rotaxane with macrocycle size, we set out to investigate the effect of conditions and substrate on the product distribution, with a view to further optimising this efficient, versatile and reliable reaction.
Effect of reaction conditions on the product distribution
We selected the AT-CuAAC reaction of macrocycle 1a with half threads 2 and 3 to investigate the effect of conditions on the product distribution. The reactions were assessed by 1H NMR analysis of the crude reaction mixture after aqueous work up, as the signals for macrocycle, [2]- and [3]rotaxane, particularly those of the thread subunit (Hk of 5 and Hd and Hk of 6) are clearly separated (Fig. 1).We began our study by investigating the effect of reaction stoichiometry on the product distribution (Table 1), as this had previously been shown to have a large effect in the case of pyridine macrocycles, where high macrocycle–Cu ratios led to increased yields of [3]rotaxane.5b Unexpectedly, in the case of macrocycle 1adecreasing the equivalents of CuI from 0.9 (entry 1) led to an increase in the proportion of [2]rotaxane 4a formed (entries 2–4) and vice versa (entry 5). Variation of the number of equivalents of the half threads (entries 6–8), led to small changes in the ratio of [2]- and [3]rotaxane formed, although a larger change was observed when both were used in excess (entry 8). Conducting the standard reaction at higher concentration (entry 9) did not significantly affect the reaction outcome.
| Entry | CuI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1a:2:3 :1a:2:3 |
t/h | Conv. 1ac |
4a:5ac |
|---|---|---|---|---|
| a 80 °C in CH2Cl2, 10 mM conc. of 1a. b 50 mM concentration of 1a. c Determined by 1H NMR analysis of the crude mixture. | ||||
| 1a | 0.90:1:1:1 |
6 | >99% | 65:35 |
| 2a | 0.50:1:1:1 |
51 | >99% | 85:15 |
| 3a | 0.18:1:1:1 |
290 | 92% | 97:3 |
| 4b | 0.90:5:5:5 |
6 | >99% | 97:3 |
| 5a | 0.96:1:1:1 |
6 | >99% | 62:38 |
| 6a | 0.90:1:5:1 |
6 | >99% | 63:37 |
| 7a | 0.90:1:1:5 |
6 | >99% | 65:35 |
| 8a | 0.90:1:5:5 |
6 | >99% | 76:24 |
| 9b | 4.50:5:5:5 |
6 | >99% | 68:32 |
The AT-CuAAC reaction tolerates a wide range of solvents with excellent conversion of 1a to interlocked products observed in all solvents investigated (Table 2). The slightly reduced selectivity for interlocked products in the case of the strongly coordinating solvent NMP (entry 7) can be attributed to a small proportion of the catalytically competent CuI remaining bound to solvent rather than 1a. However, the product distribution shows a strong solvent dependency with, in general, less coordinating solvents (entries 1–4) favouring [3]rotaxane relative to more coordinating solvents (entries 5–7). In this regard PhMe (entry 4) is an outlier, being relatively non-coordinating but also producing less [3]rotaxane than CH2Cl2, EtOAc or CHCl3 (entries 1–3). However, it should be noted that the solubility of the [1a·Cu]·PF6 complex is poor in PhMe.
The reaction outcome shows a large dependence on temperature with a maximum in [3]rotaxane formation being observed between 60 and 100 °C (Table 3, entries 1–4). Reducing the temperature below 60 °C led to increased quantities of [2]rotaxane formation (entries 5–7). Although lowering the reaction temperature also decreased the reaction rate significantly this could be overcome by addition of base (NiPr2Et), which we have previously shown dramatically enhances the rate of the AT-CuAAC reaction.10 The addition of base does not alter the product distribution (entry 8).
| Entry | T/°C | t/h | Conv. 1ab |
4a:5ab |
|---|---|---|---|---|
| a 1 equiv. each 1, 2, 3, 0.90 equiv. [Cu(MeCN)4]·PF6, 10 mM conc. of 1a, CH2Cl2. b Determined by 1H NMR analysis of the crude mixture. | ||||
| 1 | 100 | 6 | >99% | 66:34 |
| 2 | 80 | 6 | >99% | 67:33 |
| 3 | 70 | 8 | >99% | 65:35 |
| 4 | 60 | 26 | 98% | 67:33 |
| 5 | 50 | 54 | 96% | 70:30 |
| 6 | 40 | 116 | 89% | 81:19 |
| 7 | 30 | 116 | 80% | 86:14 |
| 8 | 30 | 3 | >99% | 86:14 |
In summary, the formation of [3]rotaxane is favoured by (i) a macrocycle:CuI ratio close to unity; (ii) less coordinating solvents; and (iii) higher reaction temperatures. Combining these results allowed us to design new reaction conditions, A and B, that maximised the formation [2]rotaxane and [3]rotaxane respectively (Table 4).13 Using conditions A, all [2]rotaxanes 4 were isolated in excellent yield (>94%) with a reaction time of 6 h at rt, a significant improvement over our previously published conditions (Table 4, entries 1–4). Alternatively, adopting conditions B allowed the formation of [3]rotaxanes 5 in good yield; 53% and 65% for macrocycles 1a and 1b respectively (49% and 50% isolated yields; entries 5–6). However, even under these optimised conditions, 5d was not observed (entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | Macrocycle |
4:5c |
Isolated product | Yield |
| a 1 equiv. 1, 1.2 equiv. 2 and 3, 50 mM conc. of 1. b 1 equiv. 1, 1.2 equiv. 2 and 3, 10 mM conc. of 1. c Determined by 1H NMR analysis of the crude mixture. d Isolated yield. e 20 h. f 2 h. | ||||
| Conditions A: THF, 0.10 equiv. Cu I , N i Pr 2 Et, rt, 6 h | ||||
| 1 | 1a | 99:1 |
4a | 99c% (95%)d |
| 2 | 1b | 99:1 |
4b | 99c% (94%)d |
| 3 | 1c | >99:<1 |
4c | >99c% (99%)d |
| 4e | 1d | >99:<1 |
4d | >99c% (99%)d |
| Conditions B: CH 2 Cl 2 , 0.96 equiv. Cu I , 100 °C (μW), 20 min | ||||
| 5 | 1a | 64:36 |
5a | 53c% (49%)d |
| 6 | 1b | 52:48 |
5b | 65c% (50%)d |
| 7 | 1c | 87:13 |
5c | 23c% (19%)d |
| 8f | 1d | >99:<1 |
4d | >99c% (quant.d) |
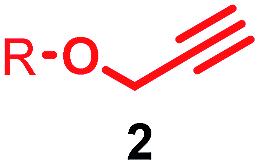
The effect of thread structure on [3]rotaxane formation
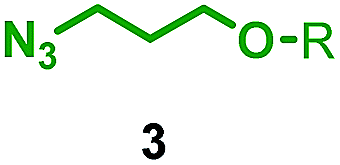
Having identified conditions that favour [3]rotaxane formation, and given that doubly threaded [3]rotaxanes are challenging synthetic targets in their own right,14 we turned our attention to the effect of substrate structure on the reaction outcome with a view to determining the generality of the procedure. Pleasingly, elongation of the linker between the azide and stopper moieties (Table 5, entry 1) did not lead to a significant change in the product distribution compared with the reaction between alkyne 2 and azide 3 suggesting that there is relative freedom in the choice of the azide component. Conversely, lengthening the spacer between the tritylphenoxy head group and the acetylene moiety from a single methylene to a (CH2)4 (entry 2) or (CH2)9 (entry 3) chain revealed that longer spacers lead to decreased quantities of [3]rotaxane. Thus, it appears that the structure of the azide component is largely irrelevant to the reaction outcome whereas the acetylene component plays a key role in [3]rotaxane formation, with longer linkers between the alkyne and stopper units, counter-intuitively, favouring less sterically demanding [2]rotaxane. In keeping with these results, when azide 6 was employed with alkyne 7, the ratio of interlocked products was largely unchanged from the reaction with the same acetylene and shorter azide 3 (entry 4). Finally, no [3]rotaxane product was formed in the reaction between 1d, azide 6 and alkyne 7, affording complete conversion to [2]rotaxane, despite the potential of the longer thread length to better accommodate a second macrocyclic component (entry 5).| Entry | Macrocycle | Alkyne | Azide | [2]:[3] rotaxaneb |
Yield of [3]rotaxane |
|---|---|---|---|---|---|
| a 1 equiv. macrocycle, 1.2 equiv. alkyne and azide, 0.96 equiv. [Cu(MeCN)4]·PF6, 10 mM of macrocycle, 100 °C (μW), 2 h. R = 4-(4tBuC6H4)3CC6H4–. b Determined by 1H NMR analysis of the crude reaction mixture. c Isolated yield. See ESI for further information. | |||||
| 1 | 1a |
|
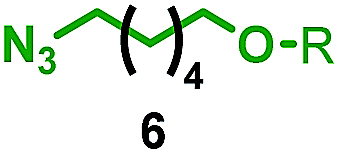
|
65:35 |
52b% (48%)c |
| 2 | 1a |

|

|
71:29 |
45b% (32%)c |
| 3 | 1a |
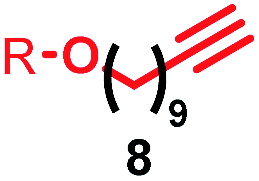
|
|
76:24 |
39b% (34%)c |
| 4 | 1a |
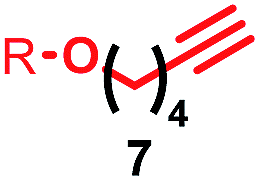
|

|
76:24 |
39b% (35%)c |
| 5 | 1d |
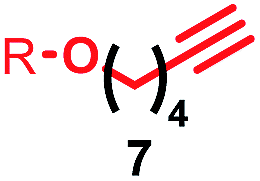
|

|
>99:<1 |
— |
The effect of macrocycle structure – a mechanistic clue
Having established that [3]rotaxane formation is relatively general with respect to the thread structure, we returned to the structure of the macrocycle in an effort to understand the origin on the doubly interlocked product. Working on the hypothesis that the benzylic ether oxygens in macrocycles 1 may play a role in assembling the key reactive intermediate, and that this is the interaction that coordinating solvents serve to disrupt, we investigated the AT-CuAAC reactions of macrocycles 9 in which this moiety was replaced with a simple methylene. Sure enough, macrocycles 9 give rise to [2]rotaxanes 10 in excellent yield with only traces (<2%) of the corresponding [3]rotaxane observed, even under conditions previously found to favour [3]rotaxane formation with macrocycles 1 (Scheme 2). The reactions of macrocycle 9 were slower than macrocycle 1, requiring a higher concentration to reach completion in a comparable time. No significant variation in product distribution with macrocycle size was observed; all [2]rotaxanes 10 were isolated in excellent yield (>95%). This observation, combined with the effect of coordinating solvents on the product distribution for macrocycles 1 strongly supports the proposal that the benzylic ether units help assemble the key reactive species en route to the doubly interlocked product.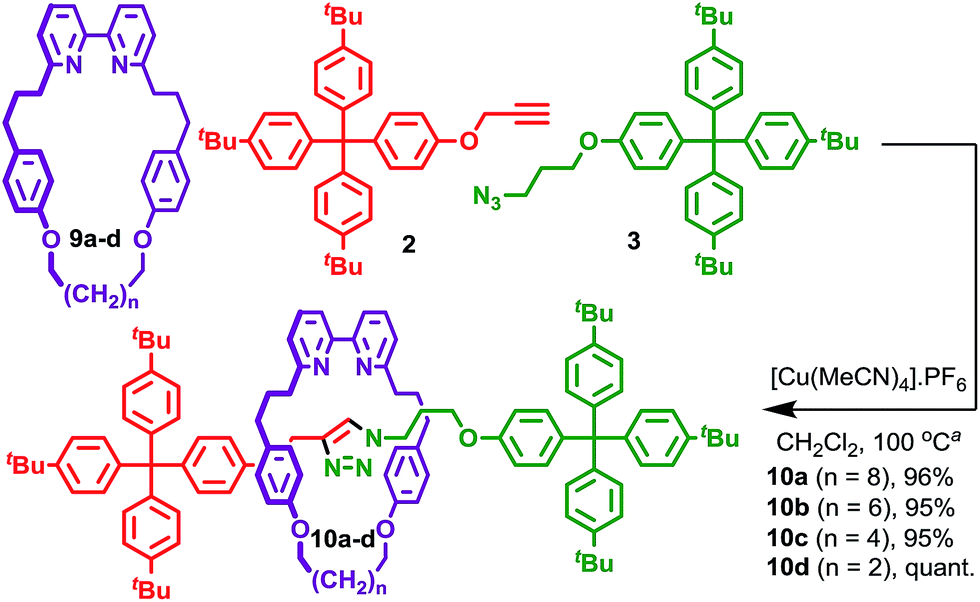 | ||
| Scheme 2 Selective synthesis of [2]rotaxanes 10 with macrocycles 9. a1 equiv. 9, 1.2 equiv. each 2 and 3, 0.96 equiv. [Cu(MeCN)4]·PF6, 50 mM conc. of 9, CH2Cl2 100 °C (μW), 15 min. The ratio of [2]:[3]rotaxane was >98:<2 in all cases. | ||
A mechanistic model of [3]rotaxane formation
The surprisingly large effect of the benzylic ether oxygens on the outcome of the AT-CuAAC reaction suggests that they play a key role in stabilising the intermediate leading to [3]rotaxane formation. By analogy to Leigh's proposed pathway in the case of monopyridine macrocycles,5b and in keeping with previous work on the mechanism of the CuAAC reaction,15 this leads us tentatively to propose a mechanism for [3]rotaxane formation (Scheme 3). Initial π-coordination of the acetylene by one equivalent of macrocycle–Cu complex leads to threaded intermediate I, lowering the pKa of the acetylenic C–H. Subsequent deprotonation and trapping with a second equivalent of macrocycle-bound Cu leads to key intermediate II that is stabilised by reciprocal Cu–O interactions bridging the two macrocycles. Finally, coordination of the azide component through the cavity of the σ-bound macrocycle, displacing the interaction between the σ-bound Cu and the benzylic ether unit of the π-bound macrocycle leads to doubly threaded intermediate III that collapses via a 1,3-dipolar cycloaddition to give the observed [3]rotaxane product. A simple computational model was constructed (Fig. 2), demonstrating the shape of intermediate II and the open face of the σ-bound macrocycle through which the azide can approach.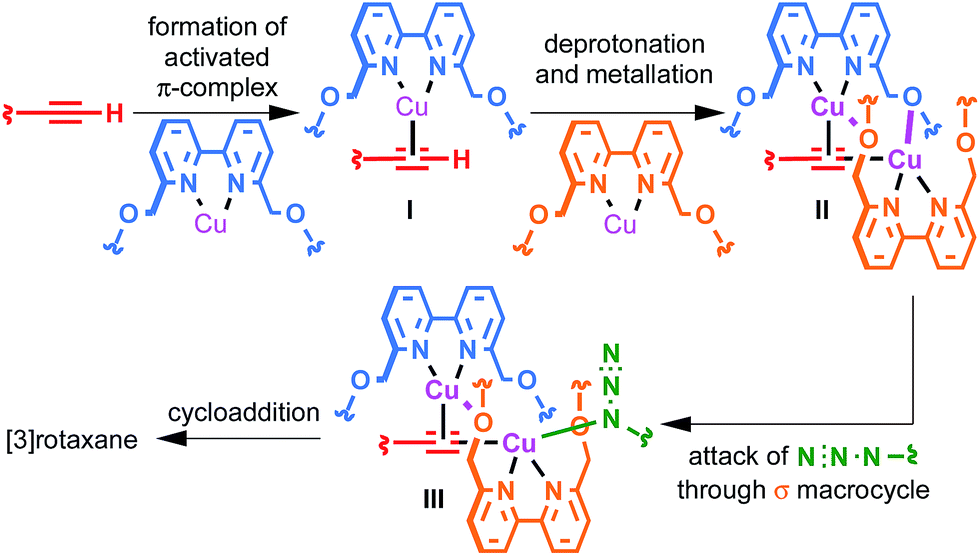 | ||
| Scheme 3 Proposed mechanism of [3]rotaxane formation. | ||
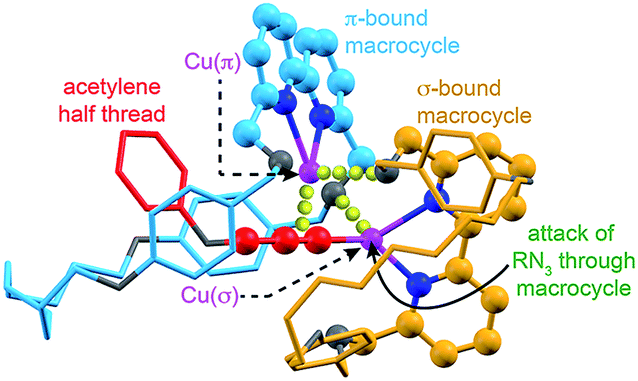 | ||
| Fig. 2 Simple computational model (MM2)16 of proposed intermediate II indicating the attack of the azide component through the cavity of the σ–bound macrocycle. O, N and Cu atoms shown in grey, dark blue, and pink respectively. Macrocycles coloured light blue (π-bound) and orange (σ-bound) for clarity. Bonding interactions between Cu(π) and the acetylenic triple bond; and O of the σ-bound macrocycle and Cu(σ) and the O of the π-bound macrocycle are represented by yellow spheres. The acetylene component has been truncated for clarity. | ||
This mechanistic scheme accounts for many of our observations: (i) the effect of coordinating solvents is explained by their tendency to competitively disrupt the key Cu–O interaction, destabilising II; (ii) the failure of smaller macrocycles to produce significant quantities of [3]rotaxane is accounted for by increased steric interactions in I that prevent formation of a threaded π-complex in the case of the smallest macrocycles employed; (iii) the slight reduction in [3]rotaxane yield with the largest macrocycle 1a compared with 1b may be due to the ability of larger macrocycles to adopt an unthreaded “side saddle” coordination mode in intermediate II leading to [2]rotaxane; (iv) similarly, the counterintuitive tendency of less sterically hindered acetylenes to produce more [2]rotaxane is rationalised by their added flexibility, facilitating the side saddle coordination of the π-bound macrocycle; (v) the insensitivity of the reaction to the structure of the azide component arises naturally from this model as the doubly threaded structure is assembled prior to azide coordination.
The mechanistic origin of the [2]rotaxane product is less clear. Leigh and co-workers previously proposed that, in the case of pyridine macrocycles, [2]rotaxane is formed by intermediates in which π-activation of the acetylene is achieved by a Cu centre not coordinated by a macrocyclic ligand, and the addition of excess macrocycle leading to enhanced yields of both [2]- and [3]rotaxanes was provided as evidence.5b However, in the case of bipyridine macrocycles the opposite trend was observed: high Cu–macrocycle loadings led to enhanced [3]rotaxane formation. This suggests that, in keeping with the stronger chelating nature of the bipyridine ligand, such “free” Cu is not involved in the reaction.
Although [2]rotaxane could potentially arise by side saddle coordination of the macrocycle in intermediate II, based on the effect of Cu loading it seems likely that a mono-metallic pathway is also in operation. This is supported by two simple experiments under strongly basic conditions (Scheme 3). Lithiation of acetylene 2 followed by trapping with [1b·Cu]·PF6 and then addition of azide 3 led almost exclusively to the expected [2]rotaxane, suggesting that the σ-bound macrocycle–Cu–acetylide complex formed quantitatively by transmetallation from the lithium acetylide is unable to rearrange to give a bimetallic intermediate such as II and, therefore, can only progress directly to [2]rotaxane. Conversely, repeating the same reaction with an additional equivalent [1b·Cu]·PF6 added along with azide 3 (Scheme 4b) led to partial recovery of [3]rotaxane formation, presumably as additional macrocycle–CuI complex allows the system to rearrange via ligand transfer to produce intermediates of the form of II.
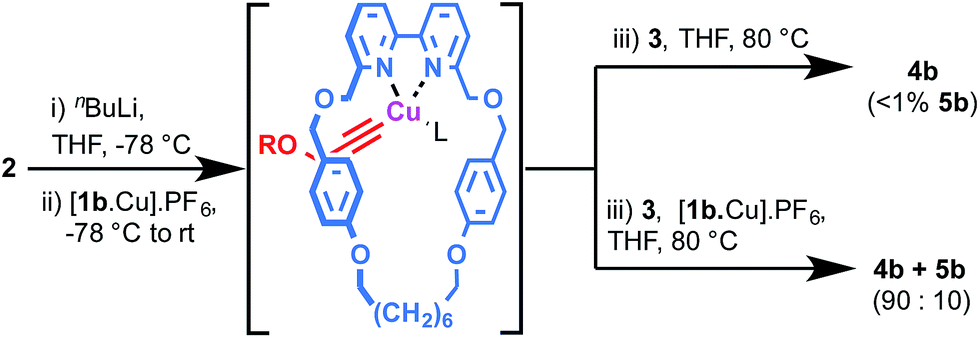 | ||
| Scheme 4 Effect of alkyne lithiation on rotaxane product distribution (see ESI† for full details). In both cases quantitative (>99%) conversion of 2 and 3 to interlocked products was observed. R = (4-tBuC6H4)3CC6H4–. L = THF or MeCN. | ||
Conclusions
In conclusion, we have demonstrated that, through optimisation of the reaction conditions, [2]rotaxanes can be produced in essentially quantitative yields in the AT-CuAAC reaction with all macrocycles investigated. Furthermore, by performing the reaction at higher temperature and Cu loading with larger macrocycles 1a and 1b, we have demonstrated the synthesis of seven novel doubly interlocked [3]rotaxanes in up to 65% yield (50% isolated yield), through a convergent four component coupling, the first time that such complex structures have been obtained in high yield in an active template reaction. Finally, we have presented a relatively detailed structure for the key reactive intermediate leading to [3]rotaxanes in the bipyridine-mediated AT-CuAAC reaction that provides an explanation for the effect of macrocycle and thread structure on the product distribution between [2]- and [3]rotaxane.Acknowledgements
The authors thank Hicham Lahlali for preliminary studies, the EPSRC National Mass Spectrometry Service for high resolution mass analysis and the Royal Society for funding. SMG is a Royal Society Research Fellow.Notes and references
- (a) C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS; (b) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1521–1529 RSC.
- For recent reviews on the synthesis, properties and applications of mechanically interlocked molecules see: (a) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed; (b) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; (c) G. Barin, R. S. Forgan and J. F. Stoddart, Proc. R. Soc. A, 2012, 468, 2849–2880 CrossRef CAS PubMed; (d) E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC; (e) N. H. Evans and P. D. Beer, Chem. Soc. Rev., 2014, 43, 4658–4683 RSC.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. a. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- (a) V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed; (b) V. Aucagne, J. Berna, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed.
- (a) S. M. Goldup, D. A. Leigh, T. Long, P. R. McGonigal, M. D. Symes and J. Wu, J. Am. Chem. Soc., 2009, 131, 15924–15929 CrossRef CAS PubMed; (b) S. M. Goldup, D. A. Leigh, P. R. McGonigal, V. E. Ronaldson and A. M. Z. Slawin, J. Am. Chem. Soc., 2010, 132, 315–320 CrossRef CAS PubMed; (c) P. E. Barran, H. L. Cole, S. M. Goldup, D. A. Leigh, P. R. McGonigal, M. D. Symes, J. Wu and M. Zengerle, Angew. Chem., Int. Ed., 2011, 50, 12280–12284 CrossRef CAS PubMed; (d) H. M. Cheng, D. A. Leigh, F. Maffei, P. R. McGonigal, A. M. Z. Slawin and J. Wu, J. Am. Chem. Soc., 2011, 133, 12298–12303 CrossRef CAS PubMed; (e) M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901 RSC; (f) L. D. Movsisyan, D. V. Kondratuk, M. Franz, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, Org. Lett., 2012, 14, 3424–3426 CrossRef CAS PubMed; (g) N. Weisbach, Z. Baranová, S. Gauthier, J. H. Reibenspies and J. A. Gladysz, Chem. Commun., 2012, 48, 7562–7564 RSC; (h) R. Hayashi, K. Wakatsuki, R. Yamasaki, Y. Mutoh, T. Kasama and S. Saito, Chem. Commun., 2014, 50, 204–206 RSC; (i) Y. Yamashita, Y. Mutoh, R. Yamasaki, T. Kasama and S. Saito, Chem.–Eur. J., 2014, 21, 2139–2145 CrossRef PubMed; (j) A. Noor, S. C. Moratti and J. D. Crowley, Chem. Sci., 2014, 5, 4283–4290 RSC.
- (a) B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed; (b) G. De Bo, S. Kuschel, D. A. Leigh, B. Lewandowski, M. Papmeyer and J. W. Ward, J. Am. Chem. Soc., 2014, 136, 5811–5814 CrossRef CAS PubMed.
- (a) S. Saito, E. Takahashi and K. Nakazono, Org. Lett., 2006, 8, 5133–5136 CrossRef CAS PubMed; (b) J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, A.-L. Lee and D. A. Leigh, Angew. Chem., Int. Ed., 2007, 46, 5709–5713 CrossRef PubMed; (c) J. D. Crowley, K. D. Hänni, A. Lee and D. A. Leigh, J. Am. Chem. Soc., 2007, 129, 12092–12093 CrossRef CAS PubMed; (d) J. Berná, S. M. Goldup, A.-L. Lee, D. A. Leigh, M. D. Symes, G. Teobaldi and F. Zerbetto, Angew. Chem., Int. Ed., 2008, 47, 4392–4396 CrossRef PubMed; (e) J. D. Crowley, K. D. Hänni, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem. Soc., 2010, 132, 5309–5314 CrossRef CAS PubMed; (f) J. D. Crowley, S. M. Goldup, N. D. Gowans, D. A. Leigh, V. E. Ronaldson and A. M. Z. Slawin, J. Am. Chem. Soc., 2010, 132, 6243–6248 CrossRef CAS PubMed; (g) S. M. Goldup, D. A. Leigh, R. T. McBurney, P. R. McGonigal and A. Plant, Chem. Sci., 2010, 1, 383–386 RSC; (h) K. Ugajin, E. Takahashi, R. Yamasaki, Y. Mutoh, T. Kasama and S. Saito, Org. Lett., 2013, 15, 2684–2687 CrossRef CAS PubMed.
- H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 CrossRef CAS PubMed For a recent study on the effect of macrocycle size in the pyridine-mediated AT-CuAAC reaction see: A. Noor, W. K. C. Lo, S. C. Moratti and J. D. Crowley, Chem. Commun., 2014, 50, 7044–7047 RSC.
- J. Winn, A. Pinczewska and S. M. Goldup, J. Am. Chem. Soc., 2013, 135, 13318–13321 CrossRef CAS PubMed.
- R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817–4820 CrossRef CAS PubMed.
- Leigh6b,d and others6i have previously reported the synthesis of [3]rotaxanes comprised of two threads and one macrocycle in high yield using the active template approach. For selected examples of the synthesis of these compounds using passive template approaches see: (a) E. J. F. Klotz, T. D. W. Claridge and H. L. Anderson, J. Am. Chem. Soc., 2006, 128, 15374–15375 CrossRef CAS PubMed; (b) A. I. Prikhod'ko and J.-P. Sauvage, J. Am. Chem. Soc., 2009, 131, 6794–6807 CrossRef PubMed; (c) C.-F. Lee, D. a. Leigh, R. G. Pritchard, D. Schultz, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Nature, 2009, 458, 314–318 CrossRef CAS PubMed.
- The use of microwave heating allowed the reaction time to be reduced compared with conventional heating. The product distribution was not affected.
- For selected examples of the syntheses and applications of homocircuit [3]rotaxanes see: (a) J.-S. Marois, K. Cantin, A. Desmarais and J.-F. Morin, Org. Lett., 2008, 10, 33–36 CrossRef CAS PubMed; (b) J.-P. Collin, J. Frey, V. Heitz, J.-P. Sauvage, C. Tock and L. Allouche, J. Am. Chem. Soc., 2009, 131, 5609–5620 CrossRef CAS PubMed; (c) A. Joosten, Y. Trolez, J. Collin, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 2012, 134, 1802–1809 CrossRef CAS PubMed; (d) C.-F. Lee, D. A. Leigh, R. G. Pritchard, D. Schultz, S. J. Teat, G. a. Timco and R. E. P. Winpenny, Nature, 2009, 458, 314–318 CrossRef CAS PubMed; (e) N. H. Evans, C. J. Serpell and P. D. Beer, Chem. Commun., 2011, 47, 8775–8777 RSC; (f) C. J. Campbell, D. A. Leigh, I. J. Vitorica-Yrezabal and S. L. Woltering, Angew. Chem., Int. Ed., 2014, 126, 13991–13994 CrossRef; (g) L. S. Witus, K. J. Hartlieb, Y. Wang, A. Prokofjevs, M. Frasconi, J. C. Barnes, E. J. Dale, A. C. Fahrenbach and J. F. Stoddart, Org. Biomol. Chem., 2014, 12, 6089–6093 RSC; (h) E. A Wilson, N. A. Vermeulen, P. R. McGonigal, A.-J. Avestro, A. A. Sarjeant, C. L. Stern and J. F. Stoddart, Chem. Commun., 2014, 50, 9665–9668 RSC.
- For selected articles on the mechanism of the CuAAC reaction invoking binuclear intermediates see: (a) V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS PubMed; (b) B. F. Straub, Chem. Commun., 2007, 3868–3870 RSC; (c) M. Ahlquist and V. V. Fokin, Organometallics, 2007, 26, 4389–4391 CrossRef CAS; (d) V. O. Rodionov, S. I. Presolski, D. D. Díaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705–12712 CrossRef CAS PubMed; (e) J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018–8021 CrossRef CAS PubMed; (f) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed; (g) S. Lal, H. S. Rzepa and S. Díez-González, ACS Catal., 2014, 4, 2274–2287 CrossRef CAS.
- A simple model (mechanics, MM2) of the minimum energy conformation of intermediate II was calculated using the Spartan molecular modelling program (Spartan'10, Wavefunction Inc.).
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation of all novel compounds. See DOI: 10.1039/c4sc03999h |
| This journal is © The Royal Society of Chemistry 2015 |