
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the correlation between metal induced aggregation and cellular uptake/subcellular localization of Znsalen: an overlooked rule for design of luminescent metal probes†
Juan
Tang
a,
Yuan-Bo
Cai
a,
Jing
Jing
b and
Jun-Long
Zhang
*a
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: zhangjunlong@pku.edu.cn; Fax: +86-10-62767034
bSchool of Chemistry, Beijing Institute of Technology, Beijing 100081, P. R. China
First published on 21st January 2015
Abstract
Unravelling the unique effects of metal coordination on biological behaviours is of importance to design metal based therapeutic and diagnostic agents. In this work, we chose luminescent Znsalen (ZnL1) as a case study to demonstrate that metal induced aggregation arising from the intermolecular Zn⋯O interaction influences its cellular uptake and subcellular localization. Comparative studies with the free bases (L1 and L2) show that ZnL1 undergoes cellular uptake through caveolae-mediated endocytosis and internalizes in endosomal/lysosomal compartments, in contrast to the localization of L1 and L2 in the mitochondria. Further studies of photophysical properties, TEM imaging and DLS analysis suggest that ZnL1 tends to form large sized fibrous structures in aqueous media. To investigate the relationship between ZnL1 aggregation and the biological behaviour, we used pyridine to tune the “aggregation-to-deaggregation” transition and found that, in the presence of pyridine, ZnL1 could localize in the mitochondria and internalize into cells through the passive diffusion pathway. Such distinctive biological behaviours resulting from the different Znsalen species clearly point out the importance of metal induced aggregation or metal speciation analysis in designing metal complexes as biological probes.
Introduction
Luminescent metal complexes have emerged as an important class of imaging agents, because of their photophysical properties such as high luminescence, good photostability, large Stokes shifts and long life-times.1–10 Moreover, metal coordination gives luminescent metal complexes greater flexibility and diversity in structures, compared to organic fluorophores.6,7,11–13 Thus, deciphering the structural rules governing their biological behaviours is of importance to explore the features of luminescent metal complexes in molecular imaging. Despite the tremendous progress which has been made in understanding the relationships between structural information arising from coordination, such as lipophilicity, oxidation state, charge state and coordination mode, and biological behaviours, much less attention has been given to the physical state or association of luminescent metal complexes in aqueous media.3,7,9,14 For metal complexes, the intermolecular metal–ligand or metal–metal interactions reinforce the aggregation process, which may influence the mobility of the metal complex, and therefore constrain the pathways of exposure to the cell and affect the subcellular distribution. Thus, the chemical and physical form of a metal complex in aqueous solution might be an important factor in designing luminescent imaging agents. To address this issue, we herein chose the Znsalen complex (salen = N,N′-bis(salicylidene)ethylenediamine) as a case study to demonstrate that metal induced aggregation arising from intermolecular Zn⋯O interaction indeed influences its cellular uptake and subcellular localization.Znsalens were selected because Zn2+ ions possess high Lewis acidity within a planar geometry that allows for extra coordination of a Lewis base ligand or coordinating solvent, or in their absence, self-assembly through an intermolecular Zn⋯O axial coordination to the phenolic group of another unit.15–22 Such speciation of Znsalens or salophens is accompanied by changes in morphology and “switched off/on” fluorescence corresponding to the “aggregation-to-deaggregation” transition.15,21,23–25 This forms a chemical basis to design optical sensors to detect anions, biological alkaloids, and even metal cations.23,26–30 Extending this prominent feature to living cell imaging, Znsalens could be used as low cytotoxicity agents, and display high fluorescence intensity in specific organelles, in which the environment is more hydrophobic than in cytosol or cell culture media.31–34 Since Znsalen tends to aggregate in aqueous media, it is feasible to investigate whether and how Znsalen aggregation affects its biological behaviours such as cellular uptake and subcellular location. This would be important to further design luminescent Znsalen complexes as bioprobes,35,36 combined with the insights gained from the coordination chemistry of Znsalens.
To demonstrate the effect of Zn coordination on biological behaviours, we performed comparative studies of cell imaging experiments between a water-soluble Znsalen, ZnL1, which is conjugated with the mitochondria-targeting triphenylphosphonium cation (TPP), and its free base congeners L1 and L2 (Scheme 1a).37–42 Distinctive subcellular distribution between ZnL1 (lysosomal/endosomal compartments) and the free bases L1 and L2 (mitochondria) indicated the significant biological effect arising from Zn coordination. Using L1 and L2 as controls, we excluded the effects of factors such as lipophilicity and charge state on biological behaviours. Then, we hypothesized that the intermolecular Zn⋯O interaction driving ZnL1 aggregation plays an important role in its distinctive biological behaviours, according to the photophysical properties and morphology of ZnL1 and L1 in aqueous media. To confirm this hypothesis, we used pyridine as an extra ligand to dissociate the aggregates by breaking the intermolecular Zn⋯O interaction, and found that ZnL1 undergoes the passive diffusion pathway and mainly localizes in mitochondria (Scheme 1b). Thus, these results point out the importance of metal induced aggregation in determining biological behaviours, which is potentially useful to the design of luminescent metal probes with organelle specificity.
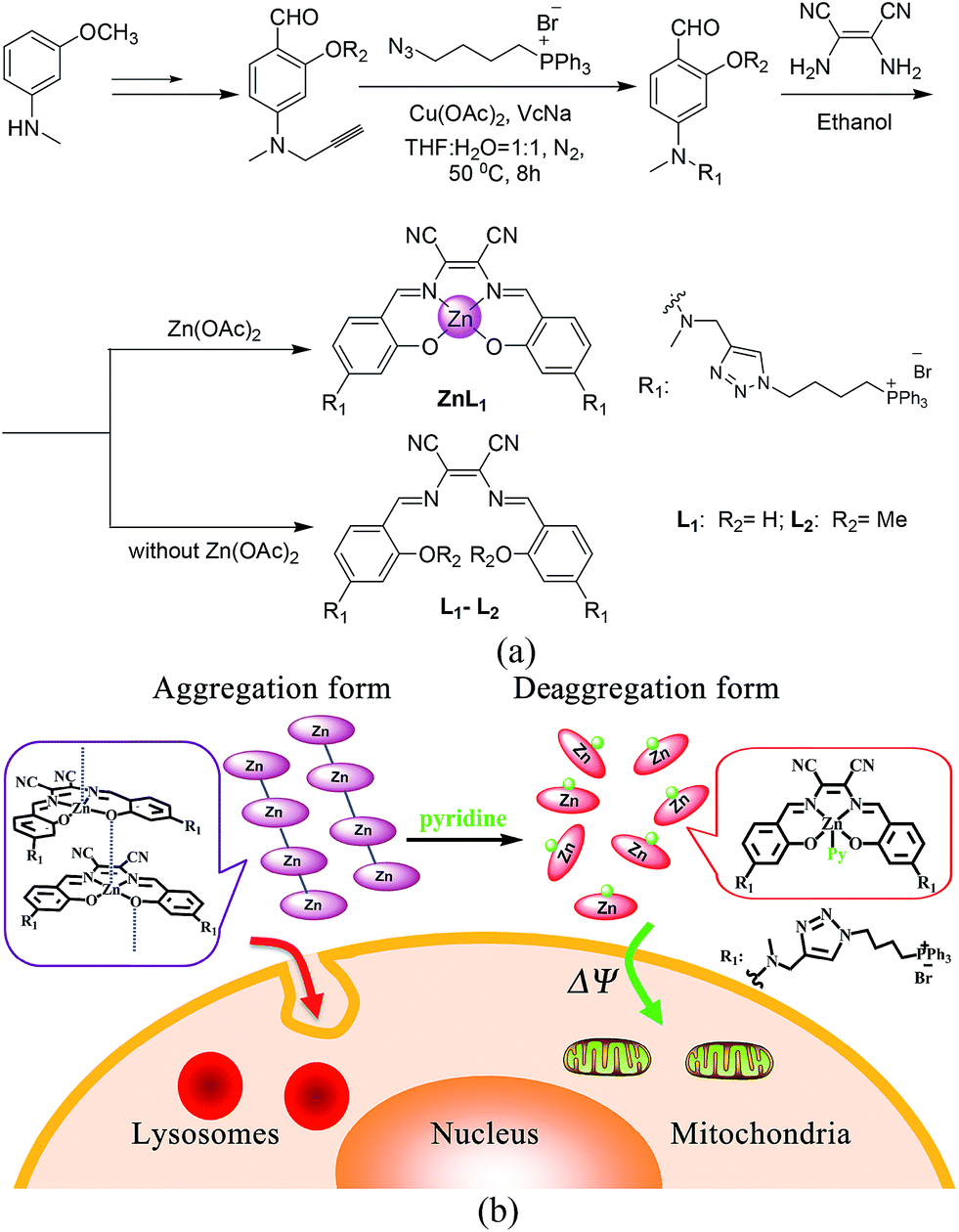 | ||
| Scheme 1 (a) Synthetic routes and chemical structures of ZnL1, L1 and L2. (b) Aggregation and deaggregation forms of ZnL1 in aqueous media lead to different cellular uptake pathways and subcellular localization. | ||
Results and discussion
ZnL1 and L1 were synthesized according to Scheme 1a. N-substituted salicylaldehyde was obtained from 3-methoxyl-N-methylaniline and propargyl bromide, followed by Vilsmeier–Haack formylation. TPP was introduced using the copper-catalyzed “click” reaction. ZnL1 was synthesized through a “one pot” reaction of salicylaldehyde, diimine and zinc acetate, while L1 was obtained in the absence of zinc acetate. To increase the lipophilicity of L1, we prepared L2 as a control using salicylaldehyde with a methyl-group-protected phenol moiety (ESI†). The detailed synthetic procedure and characterization using 1H NMR, 13C NMR, ESI-MS, UV-vis and IR are given in the ESI.†As shown in Fig. 1a, the absorption spectra of ZnL1, L1 and L2 in DMSO solution (2 × 10−5 M) show two major absorption bands between 350–450 and 500–600 nm. The free bases L1 and L2 display broad absorption bands from 360 to 470 nm, whereas ZnL1 shows a sharp band centered at 385 nm with a low-energy shoulder at 435 nm. The low-energy bands are likely due to an internal charge transfer (ICT) transition. ZnL1, L1 and L2 exhibit red emission (λmax = 623, 631 and 624 nm for ZnL1, L1 and L2 in DMSO, respectively) with fluorescence quantum yields of 0.26, 0.29 and 0.21 (Table S1†). The 1H NMR spectra of ZnL1, L1 and L2 in d6-DMSO display sharp signals with the expected multiplicities for their molecular structures (Fig. S15–S17†). Following Di Bella's procedure,21 we used diffusion-ordered spectroscopy (DOSY) NMR to estimate the molecular weight of ZnL1 in d6-DMSO (Table S4 and Fig. S18†). The obtained molecular weight is ca. 1260, close to the calculated molecular weight. These results clearly suggest that monomeric ZnL1 is the main species in the coordinating solvent DMSO. In addition, the lipophilicities of ZnL1, L1 and L2 were measured using the logarithm of their octanol/water partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ps 0.12, −0.77, 0.09) according to Leo's method.43 This suggests that Zn coordination increases the lipophilicity of L1, by an amount comparable to the methoxylation of the phenolic group of L1 (Table S2†).
Ps 0.12, −0.77, 0.09) according to Leo's method.43 This suggests that Zn coordination increases the lipophilicity of L1, by an amount comparable to the methoxylation of the phenolic group of L1 (Table S2†).
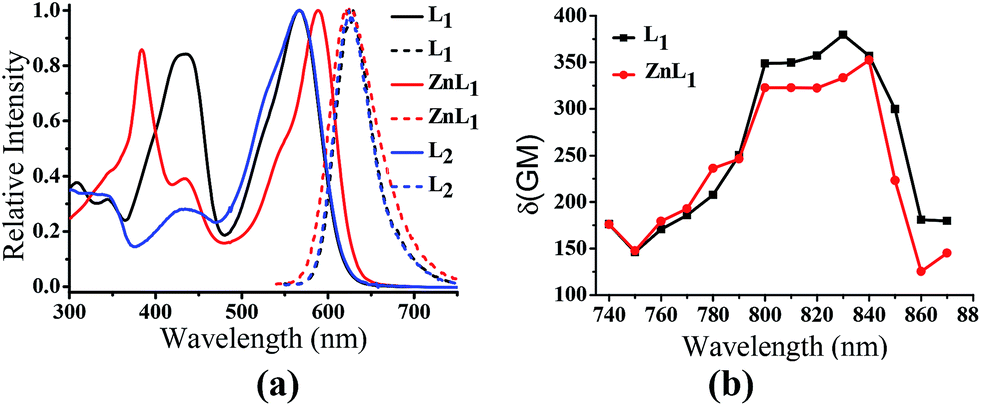 | ||
| Fig. 1 (a) Normalized UV-vis and fluorescence spectra of ZnL1, L1 and L2 in DMSO. λex = 380 nm. (b) Two photon induced absorption cross-section of ZnL1 and L1 in DMSO using Rhodamine B as a reference. | ||
Zn coordination affects cellular uptake and subcellular distribution
To demonstrate the effect of Zn coordination on biological behaviours, we investigated the intracellular distribution of ZnL1, L1 and L2 in HeLa cells using confocal laser scanning microscopy (CLSM). FYVE-EGFP, EHD1-EGFP, LysoTracker® Green DND-26 and MitoTracker Green served as markers for the early endosome, late endosome, lysosome and mitochondria organelles, respectively. As shown in Fig. 2a and S1–3,†ZnL1 showed good cell permeability and was mainly distributed in the lysosomal/endosomal compartments. It exhibited a co-localization level of approximately 0.56 with LysoTracker® Green DND-26. Interestingly, L1 and L2 particularly stained mitochondria with Pearson's correlation coefficients of 0.92 and 0.83 (Fig. 2b and c). The similar subcellular distributions of L1 and L2 clearly suggested that the lipophilicity of the salen ligands is not the main factor that affects their subcellular distribution. | ||
| Fig. 2 Co-localization analysis of (a) ZnL1 with lysosome tracker and (b and c) L1 or L2 with mitochondria tracker. (1) Fluorescence images of ZnL1, L1 and L2, ex: 543 nm; (2) fluorescence images of commercial trackers LysoTracker® Green DND-26 in (a) or MitoTracker Green in (b and c), ex: 488 nm; (3) merged images of (1) and (2); (4) differential interference contrast channel. Scale bar: 10 μm. | ||
To explore the effect of Zn coordination on the cellular uptake pathway, the temperature-dependent cellular uptake of ZnL1 or L1 in HeLa cells was carried out at 4 or 37 °C, and analysed using flow cytometry. As shown in Fig. 3a and b, the cellular internalization of ZnL1 at 4 °C showed a significantly lower intracellular level compared to that of the cells incubated in parallel at 37 °C, whereas L1 was less affected by temperature. This indicates that the cellular internalization of ZnL1 is an active process despite the low intracellular luminescence in cells incubated at 4 °C. In contrast, passive diffusion might be the dominant mechanism for the free base L1, although a background level of active transport also occurs.
 | ||
| Fig. 3 Cellular uptake of ZnL1 and L1, analysed using flow cytometry. Internalization of ZnL1 (a) or L1 (b) was investigated at 4 or 37 °C. HeLa cells were treated with cytochalasin D (5 μg mL−1), chlorpromazine (10 μg mL−1) or genistein (100 μM) for 30 min and then incubated with inhibitor and ZnL1 or L1 (2 μM) for 1 h. Cells treated with ZnL1 or L1 only were used as controls. Mean relative intracellular fluorescence intensities of the intracellular uptake of ZnL1 (c) and L1 (d) are shown as histograms (n = 3, *P < 0.001). | ||
To demonstrate the cellular internalization of ZnL1, we then used flow cytometry, applying the endocytosis inhibitors chlorpromazine (inhibitor of clathrin-mediated endocytosis),44–46 genistein (inhibitor of caveolae-mediated endocytosis),44,47,48 or cytochalasin D (inhibitor of macropinocytosis).49–53 As Fig. 3c and d show, the treatment with 100 μg mL−1 genistein resulted in a drastic decrease of 80% in the cellular uptake of ZnL1, suggesting that ZnL1 is mainly internalized via caveolae-mediated endocytosis, which plays a general role in the internalization of large sized particles into cells.52,54,55 In addition, treatment with cytochalasin D also led to a 40% decrease in cellular uptake, suggesting that a macropinocytosis-mediated pathway might also be partly involved (Fig. 3c). In contrast, for L1, treatment with genistein only resulted in a 45% decrease in the cellular uptake, and no obvious effect was observed when treated with cytochalasin D or chlorpromazine, which indicates that L1 partly undergoes caveolae-mediated endocytosis and may mainly cross the cell membrane by passive transport, and then internalize in the mitochondria (Fig. 3d). Furthermore, to exclude the possibility that zinc coordination diminishes the mitochondrial permeation capability, we examined the ability of ZnL1 and L1 to stain isolated mitochondria. Following Schagger's procedure,56 we isolated mitochondrial fractions and confirmed the activity using JC-1, shown in Fig. S4.† After the incubation of ZnL1 and L1 with active mitochondria for 0.5 h, the fluorescence of the resuspended mitochondria was detected (Fig. S5†), indicating the mitochondrial permeation capability of ZnL1 and L1. To further verify this, we also used giant unilamellar phospholipid vesicles (GUVs) as mitochondria models following Pielak's method (Fig. S6†).57 As shown in Fig. 4, both ZnL1 and L1 permeated the mitochondria mimic membranes and localized in the GUV matrix after 20 min. These results suggested that Zn coordination does not affect the mitochondria permeation capability, and the different subcellular distributions of ZnL1 and L1 might be due to their cellular uptake pathways.
 | ||
| Fig. 4 Mitochondria permeability investigation of ZnL1 and L1 with GUVs as mitochondria membrane mimics. Confocal images of (a) ZnL1 and (b) L1 confined in GUVs. (1) Fluorescence images of ZnL1 and L1; (2) merged images of (1) and (3); and (3) differential interference contrast channel. Scale bar: 10 μm. | ||
Photophysical properties and morphology in aqueous media
Since cellular uptake of ZnL1 is related to caveolae-mediated endocytosis and macropinocytosis, we envisioned that the behaviour of ZnL1 in aqueous media might influence the cellular uptake and subcellular distribution. To understand this behaviour, we carried out UV-vis absorption and emission spectroscopy of ZnL1 and L1 (Fig. 5b and c, S7 and S8†) in aqueous media. With increasing water content in the DMSO solution, ZnL1 and L1 displayed blue shifts of 15 nm and 26 nm in the low-energy absorption spectrum, and the emission was quenched gradually, with blue shifts of 6 and 11 nm, respectively. The lack of a clear isosbestic point in the absorption spectra and the quenched fluorescence for ZnL1 and L1 suggested the formation of aggregates in aqueous media.19,58–60 Interestingly, plotting the emission intensities of ZnL1 and L1versus the water contents (%) showed a logistic regression with P values of 2.71 and 1.04 (Fig. 5d), respectively. The larger P value for ZnL1 indicated a cooperative effect on the formation of ZnL1 aggregates in aqueous media. | ||
| Fig. 5 (a) The change of speciation of ZnL1 in water solution. (b) UV-vis and (c) fluorescence spectra (λex = 380 nm) of ZnL1 (20 μM) in mixed solutions of H2O/DMSO. The content of H2O was varied from 0% to 99%. (d) Fluorescence changes of ZnL1 and L1 with different water percentage. (e) DLS analysis of ZnL1 and L1 in water containing 0.1% DMSO. (f) TEM images of self-assembled (i) L1 and (ii) ZnL1 (scale bar: 200 nm). (g) SEM pictures of two assemblies of (i) L1 and (ii) ZnL1 (scale bar: 4 μm). | ||
The 1H NMR spectra of ZnL1 and L1 (concentration: 1 mM) in the mixtures of D2O and d6-DMSO solvents (Fig. S11†) show that the signals of the protons of the salen skeleton became broadened and their intensity decreased progressively with increasing D2O content, which is probably due to restricted motion of the hydrophobic salen moiety, whereas the signals of the triphenylphosphonium group did not significantly change. In addition, diffusion-ordered spectroscopy (DOSY) NMR for ZnL1 in d6-DMSO solution containing 30% D2O showed an average molecular mass of 1602 (Table S4 and Fig. S19†), indicating partial aggregation of ZnL1 under these conditions. However, for the broad and weak signals, we cannot obtain reliable DOSY data at higher concentrations of D2O, which are predicted to form larger aggregates. Nevertheless, optical spectroscopy data and NMR spectroscopic studies clearly indicate the “deaggregation to aggregation” transition of ZnL1 when the solvent was switched from DMSO to water.
To further examine the morphology of ZnL1 or L1 in aqueous media, dynamic light scattering (DLS) experiments were performed. As shown in Fig. 5e, L1 (1 μM) has a smaller hydrodynamic diameter (Dr, ca. 60 nm) and narrower distribution than ZnL1 (Dr, ca. 720 nm). No change of the ZnL1 or L1 aggregates were observed in cell culture media (Fig. S9†), indicating that the ingredients in the cell culture media have little effect on the aggregates. Then, the morphologies of ZnL1 and L1 aggregates were studied by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) by drop-casting the aqueous solutions of ZnL1 or L1 onto Cu–C (200) substrates. As shown in Fig. 5f and g, the TEM and SEM images clearly show the formation of a fibrous nanostructure (with dimensions of 43.5 ± 2.6 nm and more than 1 μm in length) for ZnL1, while L1 shows a distinct globular morphology with dimensions ranging from 20 nm to 70 nm, which are consistent with the DLS results. To exclude the effect of lipophilicity, we used L2 as a control, and L2 exhibited a similar particle size and distribution to those of L1 in both aqueous solution and cell culture medium (Fig. S9 and 10†). Given the similar structures of ZnL1, L1 and L2, we ascribed the different morphologies in aqueous media to the intermolecular Zn⋯O interaction in ZnL1 aggregates (Scheme 1b).
Although the aggregation of Znsalen complexes through the intermolecular Zn⋯O interaction in non-coordinating solvents is a usual feature, such studies in water have much less been reported. There are several examples of water soluble Znsalens that were found to be monomeric species in aqueous solutions,27,28,61 and thus the effect of water on the aggregation of ZnL1 in water needs to be further discussed. Generally, water is a Lewis base and a coordinating solvent, and can compete with the intermolecular Zn⋯O interaction, and hence may prevent aggregation. However, another facet of water as solvent, in which the hydrophobic effect of Znsalen promotes such aggregation, cannot be neglected. Obviously, the role of water is complicated and highly dependent on the different binding affinities of the H2O–Zn and the intermolecular phenolic O–Zn interaction, the structure and lipophilicity of Znsalens, the properties of the conjugates and the solvent effect. In this work, the formation of ZnL1 aggregates in water, confirmed by UV-vis absorption, fluorescence and NMR spectroscopic studies, together with morphological studies clearly indicates that the intermolecular Zn⋯O interaction is more prominent than the coordination of water. On the other hand, since lipophilic triphenylphosphonium cations were introduced, ZnL1 is to some extent hydrophobic (logP 0.12). The flexible spacer (C4 chain and triazole linker) promotes the aggregation of the Znsalen moiety and the triphenylphosphonium cations as head groups located in the interfaces, which is in agreement with the changes of the proton signals in the 1H-NMR spectra. Thus, the hydrophobic effect is another important factor that determines ZnL1 aggregation in water.
Stability of ZnL1 aggregates in aqueous media
To understand the contribution of the intermolecular Zn⋯O interaction to ZnL1 aggregation, we carried out competitive pyridine binding experiments. According to previous studies,15,17–19 the competitive binding of Lewis bases such as pyridine would dissociate the aggregates if the intermolecular Zn⋯O interaction was the main driving force for the self-assembly process in non-coordinating solvents. As shown in Fig. 6b, when pyridine was added to an aqueous solution of ZnL1, the bands at around 380 and 550 nm were red shifted to 387 and 590 nm, indicating the dissociation of the ZnL1 aggregates. As expected, the fluorescence intensity increases along with the increasing amount of pyridine (Fig. 6c). Fluorescence lifetimes were recorded before and after pyridine addition. In water, the luminescence decays are fitted as double-exponential decays with lifetimes of 2.0 (76%) and 5.2 (24%) ns. After pyridine titration, the fluorescence lifetime is 4.0 ns fitted as a single-exponential decay, slightly shorter than that in DMSO (5.2 ns). The complete deaggregation of ZnL1 requires up to 2 × 105 equiv. pyridine (Fig. S12†), which is comparable to the extremely stable bimetallic Znsalen complex reported by Kleij and coworkers.20 Monitoring the pyridine titration using 1H NMR spectra demonstrated the sharpened proton signals of ZnL1 with downfield shifts (Fig. S13†), also supporting the deaggregation of ZnL1 by pyridine. Moreover, the dissociation of ZnL1 aggregates is evident from DLS results and TEM imaging. Fig. 6d shows the decrease in hydrodynamic radius from 720 nm to 27 nm with increasing pyridine from 0 to 300 thousand equiv. TEM imaging confirmed this trend and showed that the size of aggregates was reduced from 1250 ± 117 to 31 ± 2.3 nm (Fig. 6e).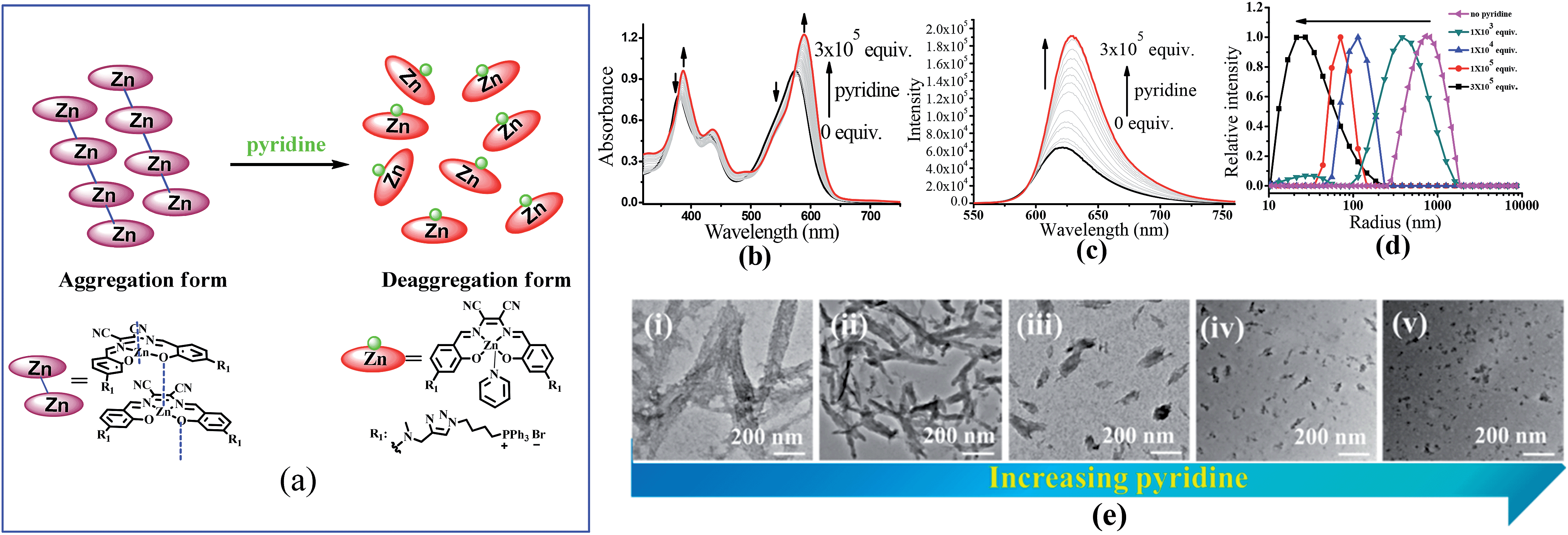 | ||
| Fig. 6 (a) The change of speciation of ZnL1 in the presence of pyridine. The dissociation of the aggregated state of 20 μM ZnL1 induced by competitive pyridine binding monitored using (b) UV-vis and (c) fluorescence spectra. (d) DLS analysis of ZnL1 (1 μM) in water with the addition of 0–300 thousand equiv. pyridine. (e) TEM images of self-assembled ZnL1 in the presence of (i–v) 0, 1 × 103, 1 × 104, 1 × 105, 3 × 105 equiv. pyridine. Scale bar is 200 nm. | ||
In addition to pyridine, we attempted to test the effect of anions on ZnL1 aggregation in aqueous media. Previous studies demonstrated that anions such as phosphate23,27,61 and Br−25 are able to coordinate Zn and dissociate Znsalen aggregates, however, with much smaller binding constants than pyridine.61,62 In this work, we chose PO43− (PBS buffer), Br− (nBu4NBr) and HBSS buffer (containing various inorganic anions), which are relevant to biological studies. As shown in Fig. 7a–c, with increasing the concentration of PBS, nBu4NBr and HBSS from 0 to 50 mM, the UV-vis spectra showed increasing bandwidths and blue-shifted with decreasing intensity at 500–600 nm, indicating increased aggregate formation. To understand this, we proposed that the high concentration of anions promoting the ZnL1 aggregation is due to a salt effect arising from PBS, nBu4NBr and HBSS. To test this, we measured the UV-vis spectra of ZnL1 in the presence of NaCl (0–49 mM). As shown in Fig. 7d, similar spectroscopic changes were observed. Thus, we concluded that the aggregation of ZnL1 might be affected by the ionic strength of these salts. This also could be used to explain why pyridine is very effective to disassociate ZnL1 for the high binding constant to Zn ion and negligible ionic strength of pyridine. Although the “aggregation-to-deaggregation” process for Znsalen/salophens has been extensively studied,15,18–21 few studies on the stability of such aggregates in aqueous media have been performed. This is important to verify whether the aggregates are stable enough to affect the biological behaviours. Following Kleij's method,18,20 we also estimated the stability constants of the ZnL1 aggregates in aqueous media. The relationship of the overall aggregation constant Kagg, stepwise aggregation constant Kn/n+1, and the aggregation number n can be described using the following equation:
| (Znsalen)n + nPy ⇌ nZnsalen(Py) K = KPyn/Kagg, Kagg = Kn/n+1n−1 | (1) |
 | ||
| Fig. 7 UV-vis spectra of 20 μM ZnL1 in the presence of (a) 0–50 mM PBS, (b) HBSS, (c) nBu4NBr, or (d) NaCl. | ||
Effect of the “aggregation/deaggregation” process on the biological behaviours
To confirm the hypothesis that ZnL1 aggregation affected its biological behaviours, we performed cell imaging experiments using ZnL1 in the presence or absence of pyridine. According to the above results, ZnL1 aggregates can be dissociated by the addition of pyridine, which would exhibit different cellular uptake pathways and subcellular distributions from the sole use of ZnL1. The intracellular luminescence and subcellular distribution of ZnL1 were investigated in HeLa cells using confocal laser scanning microscopy (CLSM). As shown in Fig. 8a, in the absence of pyridine, ZnL1 exhibited punctate red fluorescence in the lysosomal/endosomal compartments, which were mainly co-localized with LysoTracker® Green DND-26 (Pearson's correlation coefficient 0.56). In contrast, in the presence of pyridine (about 6000 equiv.), ZnL1 was mainly localized in the mitochondria and co-localized with commercial MitoTracker Green (Pearson's correlation coefficient 0.60) (Fig. 8b). These results clearly demonstrate that the “aggregation/deaggregation” transition influences the subcellular distribution. | ||
| Fig. 8 Co-localization analysis of (a) ZnL1 in the absence of pyridine with lysosomes and (b) ZnL1 in the presence of pyridine with mitochondria: (1) fluorescence images of ZnL1, ex = 543 nm; (2) fluorescence of commercial trackers LysoTracker® Green DND-26 in (a) or MitoTracker Green in (b), ex = 488 nm; (3) merged picture of (1) and (2); (4) differential interference contrast channel. Scale bar: 10 μm. | ||
To understand whether the deaggregation of ZnL1 in aqueous solution influences the internalization mechanism, we examined the temperature-dependent cellular uptake using flow cytometry. As shown in Fig. 9a, a significantly lower intracellular fluorescence intensity was observed for ZnL1 in the presence of pyridine at 4 °C than that at 37 °C, indicating that the ZnL1 species were internalized by cells via a temperature-dependent process. Then, we employed the endocytosis inhibitors chlorpromazine (inhibitor of clathrin-mediated endocytosis),44–46 genistein (inhibitor of caveolae-mediated endocytosis),44,47,48 and cytochalasin D (inhibitor of macropinocytosis)49–53 as described above, to investigate whether ZnL1 internalizes into the cell through endocytosis. As shown in Fig. 9b, in the presence of pyridine, the treatments with genistein, cytochalasin D or chlorpromazine only resulted in a moderate decrease of intracellular fluorescence (27%, 17% and 14%, respectively) and thus cellular uptake of ZnL1 in the presence of pyridine is almost not correlated to endocytosis, which is markedly different from the endocytosis pathway of ZnL1 in the aggregated state.
 | ||
| Fig. 9 Cellular uptake of ZnL1 analysed by flow cytometry and CLSM. (a) Effect of incubation temperature on the internalization of ZnL1 in the presence of pyridine was investigated at 4 or 37 °C. (b) Effect of endocytosis on the internalization of ZnL1 in the presence of pyridine: HeLa cells were treated with cytochalasin D (5 μg mL−1), chlorpromazine (10 μg mL−1) or genistein (100 μM) for 30 min and then incubated with inhibitor and ZnL1 (2 μM) for 1 h. Cells treated with ZnL1 only were used as controls. (c) and (d) Effect of the plasma membrane potential and organic cation transporter inhibitor on the internalization of ZnL1: HeLa cells treated with ZnL1 in the presence of pyridine buffered in (1) HBSS, (2) high K+-HBSS, (3) 5 μM nigericin-HBSS (cells were treated with 5 μM nigericin first for 30 min) or (4) 1 mM tetrabutylammonium bromide-HBSS (cells were treated with 1 mM tetrabutylammonium bromide first for 20 min). Mean relative intracellular fluorescence intensities of intracellular uptake are shown in histograms (n = 3, *P < 0.001). Scale bar: 10 μm. | ||
Given that the TPP conjugate possesses a positive charge, the internalization of ZnL1 with pyridine may be facilitated by organic cation transporters (OCT) or driven by the plasma membrane potential (−50 to −70 mV, negative inside).63–66 When using tetrabutylammonium bromide as the OCT inhibitor,67 HeLa cells showed no obvious intracellular fluorescence change (Fig. 9c and d). To investigate the effect of the plasma membrane potential, we examined the cellular uptake of ZnL1 in the presence of pyridine under the conditions of high potassium buffer (K+-HBSS, 170 mM K+, depolarization)65,67,68 or nigericin-HBSS (10 μM, hyperpolarization).65,69 As shown in Fig. 9c and d, the K+-HBSS-treated cells displayed a remarkable decrease in intracellular fluorescence (80%), while the nigericin-treated cells showed an intracellular fluorescence increase of nearly 2 fold. This clearly demonstrated that ZnL1 in the presence of pyridine is internalized mostly by membrane potential dependent passive diffusion. Since we have demonstrated that PBS, HBSS, and Br− cannot lead to ZnL1 deaggregation and couldn't disturb pyridine coordination, combined with its photophysical properties and morphology in aqueous media, we hypothesized that the distinctive internalization pathways may be due to the different morphologies between the “aggregation/deaggregation” transition of ZnL1, because the caveolae-mediated or macropinocytosis-mediated endocytosis is related to the uptake of large sized particles into cells.44,66,68
Conclusions
Taking our results together, we demonstrated that the intermolecular Zn⋯O interaction between Znsalen played an important role in determining its cellular uptake pathway and subcellular distribution. Through comparative studies between ZnL1 and the free bases L1 and L2, Zn coordination was found to lead to a distinctive cellular uptake pathway and subcellular distribution. More importantly, the photophysical and morphology studies on ZnL1 and the free bases in aqueous media suggest that the different aggregation states arising from the intermolecular Zn⋯O interaction play a critical role in influencing the biological behaviours. This hypothesis was confirmed by cell imaging experiments using ZnL1 in the presence or absence of pyridine, which clearly demonstrated the effect of tuning the “aggregation/deaggregation” transition of ZnL1 in aqueous media on the cellular uptake pathway and subcellular distribution. These results point to a new factor, “metal induced aggregation”, which effectively influences cellular uptake and subcellular distribution, and which should not be overlooked in designing luminescent metal complexes as biological probes.Acknowledgements
This project was supported by the National Scientific Foundation of China and National Key Basic Research Support Foundation of China (NKBRSFC) (2013CB933402, 2015CB856300).Notes and references
- K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS PubMed.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- E. Baggaley, J. A. Weinstein and J. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS PubMed.
- M. Schäferling, Angew. Chem., Int. Ed., 2012, 51, 3532–3554 CrossRef PubMed.
- D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Leung, Coord. Chem. Rev., 2012, 256, 3087–3113 CrossRef CAS PubMed.
- D. L. Ma, H. Z. He, K. H. Leung, D. S. H. Chan and C. H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed.
- K. K.-W. Lo, A. W.-T. Choi and W. H.-T. Law, Dalton Trans., 2012, 41, 6021–6047 RSC.
- V. Fernández-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12–21 CrossRef CAS PubMed.
- D.-L. Ma, D. S.-H. Chan and C.-H. Leung, Acc. Chem. Res., 2014, 47, 3614–3631 CrossRef CAS PubMed.
- Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef CAS.
- E. Meggers, Chem. Commun., 2009, 1001–1010 RSC.
- M. Frezza, S. Hindo, D. Chen, A. Davenport, S. Schmitt, D. Tomco and Q. P. Dou, Curr. Pharm. Des., 2010, 16, 1813–1825 CrossRef CAS.
- R. P. Haugland, The handbook: a guide to fluorescent probes and labeling technologies, Molecular probes, 2005 Search PubMed.
- C. T. L. Ma and M. J. MacLachlan, Angew. Chem., Int. Ed., 2005, 44, 4178–4182 CrossRef CAS PubMed.
- J. K. H. Hui, Z. Yu and M. J. MacLachlan, Angew. Chem., Int. Ed., 2007, 46, 7980–7983 CrossRef CAS PubMed.
- A. W. Kleij, Dalton Trans., 2009, 4635–4639 RSC.
- M. M. Belmonte, S. J. Wezenberg, R. M. Haak, D. Anselmo, E. C. Escudero-Adan, J. Benet-Buchholz and A. W. Kleij, Dalton Trans., 2010, 39, 4541–4550 RSC.
- G. Consiglio, S. Failla, P. Finocchiaro, I. P. Oliveri, R. Purrello and S. Di Bella, Inorg. Chem., 2010, 49, 5134–5142 CrossRef CAS PubMed.
- G. Salassa, M. J. J. Coenen, S. J. Wezenberg, B. L. M. Hendriksen, S. Speller, J. A. A. W. Elemans and A. W. Kleij, J. Am. Chem. Soc., 2012, 134, 7186–7192 CrossRef CAS PubMed.
- G. Consiglio, S. Failla, P. Finocchiaro, I. P. Oliveri and S. Di Bella, Inorg. Chem., 2012, 51, 8409–8418 CrossRef CAS PubMed.
- I. P. Oliveri, S. Failla, G. Malandrino and S. Di Bella, J. Phys. Chem. C, 2013, 117, 15335–15341 CAS.
- M. Cano, L. Rodríguez, J. C. Lima, F. Pina, A. Dalla Cort, C. Pasquini and L. Schiaffino, Inorg. Chem., 2009, 48, 6229–6235 CrossRef CAS PubMed.
- I. P. Oliveri and S. Di Bella, J. Phys. Chem. A, 2011, 115, 14325–14330 CrossRef CAS PubMed.
- I. P. Oliveri, G. Malandrino and S. Di Bella, Dalton Trans., 2014, 43, 10208–10214 RSC.
- E. C. Escudero-Adán, J. Benet-Buchholz and A. W. Kleij, Inorg. Chem., 2008, 47, 4256–4263 CrossRef PubMed.
- S. Khatua, S. H. Choi, J. Lee, K. Kim, Y. Do and D. G. Churchill, Inorg. Chem., 2009, 48, 2993–2999 CrossRef CAS PubMed.
- A. D. Cort, P. De Bernardin and L. Schiaffino, Chirality, 2009, 21, 104–109 CrossRef PubMed.
- S. J. Wezenberg, D. Anselmo, E. C. Escudero-Adán, J. Benet-Buchholz and A. W. Kleij, Eur. J. Inorg. Chem., 2010, 29, 4611–4616 CrossRef.
- Y.-B. Cai, J. Zhan, Y. Hai and J.-L. Zhang, Chem.–Eur. J., 2012, 18, 4242–4249 CrossRef CAS PubMed.
- J. Jing, J. Tang, D. Xie, Y.-B. Cai, J.-J. Chen and J.-L. Zhang, Sci. Sin.: Chim., 2014, 44, 191–203 CAS.
- Y. Hai, J.-J. Chen, P. Zhao, H. Lv, Y. Yu, P. Xu and J.-L. Zhang, Chem. Commun., 2011, 47, 2435–2437 RSC.
- J. Jing, J.-J. Chen, Y. Hai, J. Zhan, P. Xu and J.-L. Zhang, Chem. Sci., 2012, 3, 3315–3320 RSC.
- D. Xie, J. Jing, Y.-B. Cai, J. Tang, J.-J. Chen and J.-L. Zhang, Chem. Sci., 2014, 5, 2318–2327 RSC.
- J.-J. Chen, J. Jing, H. Chang, Y. Rong, Y. Hai, J. Tang, J.-L. Zhang and P. Xu, Autophagy, 2013, 9, 894–904 CrossRef CAS PubMed.
- J. Jing and J.-L. Zhang, Chem. Sci., 2013, 4, 2947–2952 RSC.
- R. A. J. Smith, C. M. Porteous, A. M. Gane and M. P. Murphy, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5407–5412 CrossRef CAS PubMed.
- A. T. Hoye, J. E. Davoren, P. Wipf, M. P. Fink and V. E. Kagan, Acc. Chem. Res., 2008, 41, 87–97 CrossRef CAS PubMed.
- B. C. Dickinson and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 9638–9639 CrossRef CAS PubMed.
- S. Marrache and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9445–9450 CrossRef CAS PubMed.
- S. K. Bae, C. H. Heo, D. J. Choi, D. Sen, E.-H. Joe, B. R. Cho and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 9915–9923 CrossRef CAS PubMed.
- R. K. Pathak, S. Marrache, D. A. Harn and S. Dhar, ACS Chem. Biol., 2014, 9, 1178–1187 CrossRef CAS PubMed.
- A. Leo, C. Hansch and P. Y. C. Jow, J. Med. Chem., 1976, 19, 611–615 CrossRef CAS.
- J. Rejman, V. Oberle, I. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- J. P. Richard, K. Melikov, H. Brooks, P. Prevot, B. Lebleu and L. V. Chernomordik, J. Biol. Chem., 2005, 280, 15300–15306 CrossRef CAS PubMed.
- E. Blanchard, S. Belouzard, L. Goueslain, T. Wakita, J. Dubuisson, C. Wychowski and Y. Rouillé, J. Virol., 2006, 80, 6964–6972 CrossRef CAS PubMed.
- I. R. Nabi and P. U. Le, J. Cell Biol., 2003, 161, 673–677 CrossRef CAS PubMed.
- J. Rejman, A. Bragonzi and M. Conese, Mol. Ther., 2005, 12, 468–474 CrossRef CAS PubMed.
- J. S. Wadia, R. V. Stan and S. F. Dowdy, Nat. Med., 2004, 10, 310–315 CrossRef CAS PubMed.
- I. M. Kaplan, J. S. Wadia and S. F. Dowdy, J. Controlled Release, 2005, 102, 247–253 CrossRef CAS PubMed.
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2010, 39, 1159–1170 RSC.
- S.-F. Peng, M. T. Tseng, Y.-C. Hoc, M.-C. Wei, Z.-X. Liao and H.-W. Sung, Biomaterials, 2011, 32, 239–248 CrossRef CAS PubMed.
- F. Madani, S. Lindberg, Ü. Langel, S. Futaki and A. Gräslund, J. Biophys., 2011, 2011, 1–10 CrossRef PubMed.
- R. G. W. Anderson, Annu. Rev. Biochem., 1998, 67, 199–225 CrossRef CAS PubMed.
- S. Trabulo, A. L. Cardoso, M. Mano and M. C. P. De Lima, Pharmaceuticals, 2010, 3, 961–993 CrossRef CAS PubMed.
- K. Minezaki, M. Suleiman and R. Chapman, J. Physiol., 1994, 476, 459–471 CrossRef CAS.
- I. G. Zigoneanu, Y. J. Yang, A. S. Krois, M. E. Haque and G. J. Pielak, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 512–519 CrossRef CAS PubMed.
- D. M. Cohen and E. Fisher, J. Chem. Soc., 1962, 3044–3052 RSC.
- J. Brynestad and G. P. Smith, J. Phys. Chem., 1968, 72, 296–300 CrossRef CAS.
- G. Consiglio, S. Failla, P. Finocchiaro, I. P. Oliveri and S. D. Bella, Dalton Trans., 2012, 41, 387–395 RSC.
- O. Jurček, M. Cametti, M. Pontini, E. Kolehmainen and K. Rissanen, Org. Biomol. Chem., 2013, 11, 4585–4590 Search PubMed.
- I. P. Oliveri, G. Maccarrone and S. Di Bella, J. Org. Chem., 2011, 76, 8879–8884 CrossRef CAS PubMed.
- S. Davis, M. Weiss, J. Wong, T. J. Lampidis and L. B. Chen, J. Biol. Chem., 1985, 260, 13844–13850 CAS.
- D. Piwnica-Worms, J. F. Kronauge and M. L. Chiu, Circulation, 1990, 82, 1826–1838 CrossRef CAS.
- M. L. Chiu, J. F. Kronauge and D. Piwnica-Worms, J. Nucl. Med., 1990, 31, 1646–1653 CAS.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
- C. A. Puckett and J. K. Barton, Biochemistry, 2008, 47, 11711–11716 CrossRef CAS PubMed.
- J. Wang, J. D. Byrne, M. E. Napier and J. M. DeSimone, Small, 2011, 7, 1919–1931 CrossRef CAS PubMed.
- E. E. Bellocchio, R. J. Reimer, R. T. Fremeau and R. H. Edwards, Science, 2000, 289, 957–960 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03824j |
| This journal is © The Royal Society of Chemistry 2015 |