
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of a C–C double bond from two aliphatic carbons. Multiple C–H activations in an iridium pincer complex†
Alexey V.
Polukeev
a,
Rocío
Marcos
b,
Mårten S. G.
Ahlquist
b and
Ola F.
Wendt
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, PO Box 124, 22100 Lund, Sweden. E-mail: ola.wendt@chem.lu.se
bDivision of Theoretical Chemistry & Biology, School of Biotechnology, KTH Royal Institute of Technology, SE-106 91 Stockholm, Sweden
First published on 26th January 2015
Abstract
The search for novel, atom-economic methods for the formation of C–C bonds is of crucial importance in synthetic chemistry. Especially attractive are reactions where C–C bonds are formed through C–H activation, but the coupling of unactivated, alkane-type Csp3–H bonds remains an unsolved challenge. Here, we report iridium-mediated intramolecular coupling reactions involving up to four unactivated Csp3–H bonds to give carbon–carbon double bonds under the extrusion of dihydrogen. The reaction described herein is completely reversible and the direction can be controlled by altering the reaction conditions. With a hydrogen acceptor present a C–C double bond is formed, while reacting under dihydrogen pressure leads to the reverse process, with some of the steps representing net Csp3–Csp3 bond cleavage. Mechanistic investigations revealed a conceptually-novel overall reactivity pattern where insertion or deinsertion of an Ir carbene moiety, formed via double C–H activation, into an Ir–C bond is responsible for the key C–C bond formation and cleavage steps.
Introduction
The selective activation of C–H bonds by transition-metal complexes is one of the main goals in organometallic chemistry, as it may allow for efficient, low-waste methods for the functionalization of various organic molecules, not the least of cheap, but relatively inert alkanes.1 In particular, cross-coupling reactions where a C–C bond is formed via activation of C–H bonds of one2 or both2d,3 coupling partners have been intensively studied in recent years as a potential alternative to the traditional palladium-catalyzed couplings, where pre-functionalization of both substrates is required.4 Numerous methods have been developed for the connection of Csp–H, Csp2–H and Csp3–H bonds with each other,2d,3 with coupling of two Csp3–H bonds being the most challenging.3d However, in the latter case protocols are rather substrate-specific and the scope is limited to substrates with activated Csp3–H bonds; strong oxidants like peroxides are used to drive the reaction.3d Typically, one coupling partner bears a heteroatom adjacent to the Csp3–H bond, which facilitates oxidation and stabilizes the resulting electrophilic species, while the other partner with an acidic C–H bond is responsible for generating the nucleophile. In this respect, the coupling of unactivated Csp3–H bonds may provide a more universal procedure with a high potential in all branches of synthetic chemistry.Benzene-based iridium pincer complexes have been shown to be effective catalysts for various dehydrogenation reactions, including those that involve non-activated Csp3–H bonds.5 Previously, we reported on the synthesis of the complex trans-(PCyP)IrHCl (PCyP = {cis-1,3-bis-[(di-tert-butylphosphino)-methyl]cyclohexyl}) (1) and the attempted synthesis of (POCyOP)IrHCl (POCyOP = {cis-1,3-bis-[(di-tert-butyl-phosphinoxy)]cyclohexyl}), which resulted in complete dehydrogenation and aromatization of the ligand and formation of the known benzene-based (POCOP)IrHCl complex.6 We have also explored the potential of these aliphatic pincer complexes as dehydrogenation catalysts.7
The introduction of a cyclohexane moiety instead of a benzene ring into the pincer complex affects its thermal stability. This thermal instability is most likely related to the behavior of the ligand, and given the aliphatic nature of all the ligand C–H bonds, we reasoned that coordinatively-unsaturated species could give rise to interesting reactivity. One way to probe this was to investigate the ligand transformations using the iridium hydrido-phenyl complex, which closely resembles the elusive hydrido-alkyl intermediates involved in dehydrogenation reactions. Here we report on C–H activation reactions with the aliphatic ligand involving both the ligand backbone and the tert-butyl groups on the phosphine. This two-site activation is shown to lead to the first example of an intramolecular ring-closing reaction where two carbon atoms with non-activated, alkane-like Csp3–H bonds are joined to form a carbon–carbon double bond under extrusion of dihydrogen. The reaction is mediated by a pincer carbene complex and thus proceeds via quadruple C–H activations at a single metal center.
Results and discussion
Dynamic oxidative addition of benzene in a phenyl-hydride complex
In the presence of tBuONa, (PCyP)IrHCl (1) reacts with benzene, leading to the formation of extremely labile phenyl-hydride 2 (Scheme 1). At room temperature, complex 2 demonstrates dynamic behavior and its NMR signals are broad. Thus, a solution of 2 in methylcyclohexane-d14 reveals a broadened singlet at 63.1 ppm in the 31P{1H} NMR spectrum and a very broad signal at −47.8 ppm in the 1H NMR spectrum. At temperatures below ca. −10 °C, a P–H coupling with the high-field hydride resonance appears in the 31P{1H} NMR spectrum, while in the 1H NMR spectrum the hydride is observed as a triplet at −48.22 ppm (−40 °C, 2JPH = 12.3 Hz), shifted somewhat upfield at lower temperature. The slightly distorted shapes of the hydride and phosphorus resonances indicate the presence of a small amount of another compound with very close chemical shift values, most likely an isomer of 2 (in mesitylene-d12, the separation between the signals is better, and therefore two distinct hydride resonances are observed at −40 °C, and the amount of the isomer is bigger). It should be noted that the hydride signal remains significantly broadened (Δν1/2 = 16 Hz in the 1H{31P} spectrum at −40 °C) down to −90 °C, which may indicate the existence of an additional dynamic process (likely a rotation of the tBu groups). Also, two doublets and three triplets each integrating as 1H are observed in the aromatic region at low temperatures, consistent with η1-coordination of the phenyl group. Complex 2 readily reacts with nitrogen (even with traces in low-quality argon) to give (PCyP)IrN2,6 and is sensitive to moisture. Hence, it can be considered as a source of a 14e (PCyP)Ir species. In this regard, 2 resembles the benzene-based complex [2,6-(tBu2PCH2)2C6H3]Ir(H)Ph, which was found to undergo fast dissociative arene exchange at room temperature, with the rate being independent of the concentration of free benzene.8 Treating the coalescence of the two branches of the hydride-coupled doublet in the 31P{1H} NMR spectrum as a result of a simple exchange between two states of equal population, the barrier for the reductive elimination of benzene from 2 can be estimated as ΔG‡ = 14.0 kcal mol−1 at −3 °C, which is very close to the value for the complex [2,6-(tBu2PCH2)2C6H3]Ir(H)Ph (13.9 kcal mol−1 at −4 °C).8 The activation energy barrier for the reductive elimination of benzene from 2 was calculated by DFT, confirming the experimental data (ΔG‡ = 16.0 kcal mol−1 at −3 °C).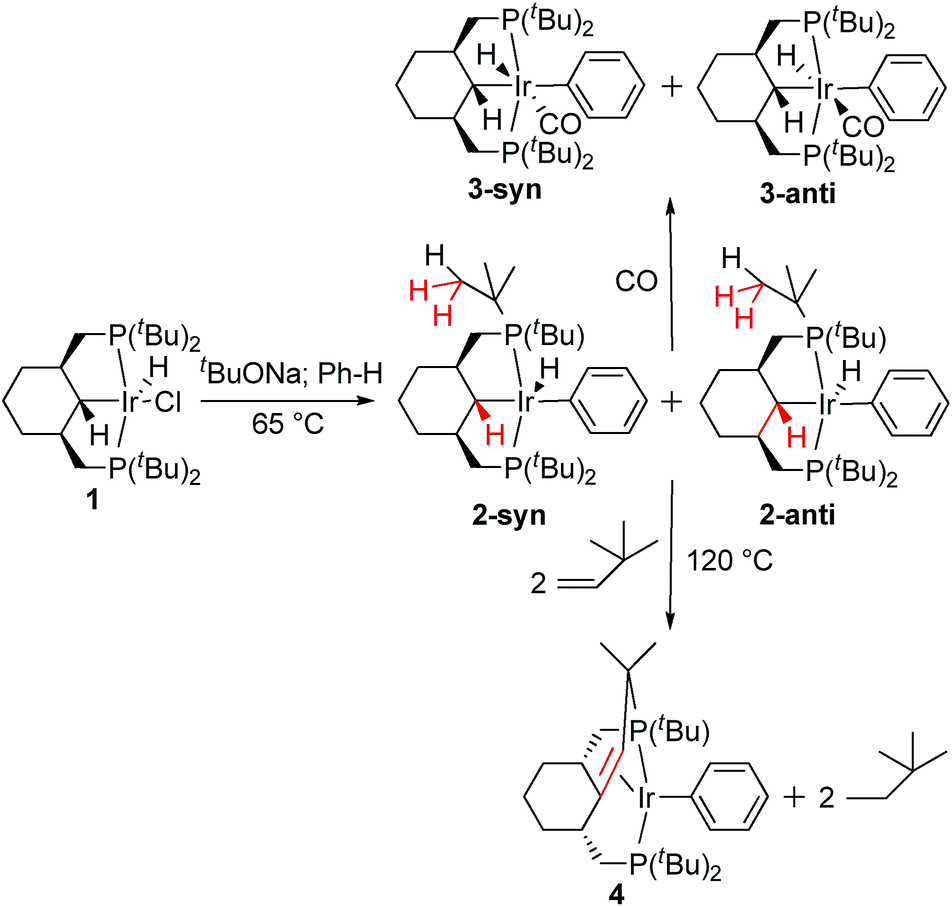 | ||
| Scheme 1 Reactions of complex 1 with benzene. | ||
The existence of two hydride signals for 2 at low temperatures can be explained by the lack of a horizontal plane of symmetry and the presence of two isomeric compounds – syn and anti with respect to the mutual orientation of C(α)–H and Ir–H. These isomers can be trapped by the addition of CO, giving 18e adducts 3-syn and 3-anti, characterized by signals at 45.1 and 49.1 in the 31P{1H} NMR spectra as well as signals at −9.27 (td, 3JPH = 17.0, 3JHH = 1.6 Hz) and −8.76 (td, 3JPH = 19.0 Hz, 3JHH = 2.3 Hz) ppm in the 1H NMR spectra, correspondingly. At room temperature, three broadened signals from the phenyl group are observed for 3-syn; these do not demonstrate exchange with deuterobenzene up to 80 °C and therefore the broadening seems to be a result of phenyl group rotation. Interestingly, while for the major isomer (syn–anti ratio = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) the ortho protons appear as a very broad signal almost at the coalescence point (ΔG‡ = 14.0 kcal mol−1 at 25 °C), for the minor isomer two doublets are observed, as expected for the slow limit of rotation of the phenyl group. As far as the rate of rotation reflects the degree of crowding around the metal, it seems reasonable that the preference for the syn isomer is a result of mainly steric factors. In the IR spectrum, two intense bands are observed at 1951 cm−1 (3-syn) and 1966 cm−1 (3-anti), corresponding to C–O stretching vibrations, as well as two more broad, low-intensity bands at 2124 cm−1 (3-syn) and 2199 cm−1 (3-anti), from Ir–H. The latter bands moved to lower frequencies (but could not be unambiguously assigned) when deuterated benzene was used to prepare complex 3, while the νCO bands moved to 1970 and 1983 cm−1, correspondingly.
:1) the ortho protons appear as a very broad signal almost at the coalescence point (ΔG‡ = 14.0 kcal mol−1 at 25 °C), for the minor isomer two doublets are observed, as expected for the slow limit of rotation of the phenyl group. As far as the rate of rotation reflects the degree of crowding around the metal, it seems reasonable that the preference for the syn isomer is a result of mainly steric factors. In the IR spectrum, two intense bands are observed at 1951 cm−1 (3-syn) and 1966 cm−1 (3-anti), corresponding to C–O stretching vibrations, as well as two more broad, low-intensity bands at 2124 cm−1 (3-syn) and 2199 cm−1 (3-anti), from Ir–H. The latter bands moved to lower frequencies (but could not be unambiguously assigned) when deuterated benzene was used to prepare complex 3, while the νCO bands moved to 1970 and 1983 cm−1, correspondingly.
To confirm the arrangement of ligands around Ir, a 13CO-labelled sample of 3 was prepared. In the 1H NMR spectrum, the hydride resonances appear as doublets of triplets of doublets; the same multiplicity is observed for the CO signals in the non-decoupled 13C NMR spectrum. Large two-bond 1H–13C couplings of 42.9 Hz and 42.5 Hz for 3-syn and 3-anti, correspondingly, indicate a mutual trans arrangement of the hydride and CO ligands.9 Therefore, the phenyl group occupies the position opposite to the pincer ligand. A three-bond 1H–13C coupling of 3.3 Hz is consistent with an anti mutual orientation of C(α)–H and Ir–CO in 3-syn, and the smaller coupling of 1.1 Hz implies a syn arrangement for 3-anti, according to the well-known dependence of couplings on the dihedral angle. The same trend is observed for 3JHH, which is 1.6 Hz for 3-syn (syn arrangement of C(α)–H and Ir–H) and 2.3 Hz for 3-anti (anti arrangement of C(α)–H and Ir–H). Finally, despite a lower content in the mixture, X-ray quality crystals of 3-anti were obtained, and the structure determination verified the conclusions based on spectroscopic data (Fig. 1).
 | ||
| Fig. 1 Molecular structure of complex 3-anti, with thermal ellipsoids at the 30% probability level. Hydrogen atoms, except for the one at C1, have been omitted for clarity. Selected bonds (Å) and angles (°): Ir1–C1 2.193(7), Ir1–C11 2.155(8), Ir1–C9 1.904(7), Ir1–P1 2.365(2), Ir1–P2 2.358(2), C9–O1 1.145(9), C1–Ir1–C11 176.5(3), P1–Ir–P2 158.06(7). | ||
C–C coupling reaction in 2
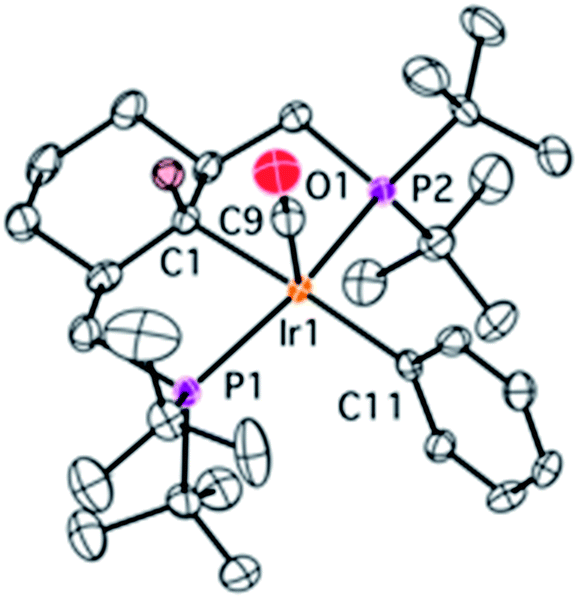
Upon heating a benzene solution of 2 to 120 °C, the NMR signals of 2 decrease in intensity and a new AX system appears in the 31P{1H} NMR spectrum. Gratifyingly, upon addition of tert-butylethylene as a hydrogen acceptor, complex 4 is formed in an almost quantitative yield. The 1H and, to some extent, 13C{1H} NMR spectra of compound 4 are very complex due to a number of overlapping signals. Doublets at 1.90 (3JPH = 13.0 Hz) and 0.91 (d, 3JPH = 9.7 Hz) ppm from methyl groups point to the activation of one of the tert-butyl groups. The![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– proton resonates at 1.95 ppm (d, 3JPH = 20.5 Hz). In the 13C{1H} NMR spectrum the olefinic signals are observed at 76.6 (2JP2C = 13.1 Hz, 2JP1C = 4.8 Hz) and 49.5 (2JP2C = 12.3 Hz, 2JP1C = 1.3 Hz) ppm as doublets of doublets. An estimation (hampered due to overlap) of 1JCH, which is around 156 Hz, is consistent with the expected sp2 hybridization of the olefinic carbons. The structure of complex 4 was confirmed using X-ray crystallography (Fig. 2). This unambiguously shows that the α-carbon and one of the methyl groups of the t-butyl have been coupled to form a new olefin functionality that the iridium(I) coordinates. The iridium atom has a distorted square-planar arrangement; average (from two molecules in the asymmetric unit) Ir–C bonds lengths of 2.16 and 2.20 Å as well as a CC bond length of 1.42 Å fall in the range observed for electron-rich Ir olefin complexes.10 During the formation of 4 from 1, three Csp3–H bonds are activated intramolecularly in addition to one external Csp2–H bond. Cyclometallation of tert-butyls11 or other groups12,13 bound to phosphorus is not unprecedented in the chemistry of iridium pincer complexes, but in the case of 4 it results in the formation of a new C–C double bond. To the best of our knowledge, this is the first example where the main product is the result of the formation of a C–C double bond from non-activated Csp3–H bonds.14
CH– proton resonates at 1.95 ppm (d, 3JPH = 20.5 Hz). In the 13C{1H} NMR spectrum the olefinic signals are observed at 76.6 (2JP2C = 13.1 Hz, 2JP1C = 4.8 Hz) and 49.5 (2JP2C = 12.3 Hz, 2JP1C = 1.3 Hz) ppm as doublets of doublets. An estimation (hampered due to overlap) of 1JCH, which is around 156 Hz, is consistent with the expected sp2 hybridization of the olefinic carbons. The structure of complex 4 was confirmed using X-ray crystallography (Fig. 2). This unambiguously shows that the α-carbon and one of the methyl groups of the t-butyl have been coupled to form a new olefin functionality that the iridium(I) coordinates. The iridium atom has a distorted square-planar arrangement; average (from two molecules in the asymmetric unit) Ir–C bonds lengths of 2.16 and 2.20 Å as well as a CC bond length of 1.42 Å fall in the range observed for electron-rich Ir olefin complexes.10 During the formation of 4 from 1, three Csp3–H bonds are activated intramolecularly in addition to one external Csp2–H bond. Cyclometallation of tert-butyls11 or other groups12,13 bound to phosphorus is not unprecedented in the chemistry of iridium pincer complexes, but in the case of 4 it results in the formation of a new C–C double bond. To the best of our knowledge, this is the first example where the main product is the result of the formation of a C–C double bond from non-activated Csp3–H bonds.14
 | ||
| Fig. 2 Molecular structure of complex 4 (one of two molecules from the asymmetric unit is shown) with thermal ellipsoids at the 30% probability level. Hydrogen atoms have been omitted for clarity. Selected bonds (Å) and angles (°): Ir2–C41 2.200(4), Ir2–C62 2.156(4), Ir2–C51 2.092(5), Ir2–P3 2.291(1), Ir2–P4 2.290(1), C41–C62 1.425(7), C41–Ir1–C51 164.0(2), P1–Ir–P2 160.05(5). | ||
A few examples of a somewhat related, stoichiometric, dehydrogenative cross-coupling of C–H bonds to give olefin moieties have been reported in the literature, but they are all based on significantly more reactive benzylic C–H bonds of substituted arylphosphines.15
Hydrogenation of the olefin complex 4: C–C bond cleavage
Interestingly, the process is reversible and the CC bond in 4 can be cleaved under certain conditions. Thus, exposure of a solution of 4 in C6D6 at room temperature to 1 atm of hydrogen results in the simultaneous formation of a mixture of complexes 5 and 6 in a ca. 93:7 ratio (Scheme 2). This ratio is temperature and hydrogen pressure dependent, with the amount of 6 being raised under conditions favoring the solubility of H2 in benzene, and the equilibrium is established quite fast. For example, under 1.5 atm of H2, the proportion 5:6 is 74:26 at 4 °C, 86:14 at 25 °C and 98:2 at 80 °C. The 31P{1H} NMR spectrum of 5 consists of an AX system (75.7, 7.2 ppm, 2JP1P2 = 344.0 Hz), while in the 1H NMR spectrum three separate hydride signals appear as rather complex multiplets, due to couplings with two non-equivalent phosphorus nuclei, two hydrides and the olefin moiety. From those, a 2JHH = 10.7 Hz should be mentioned as a rare example of a coupling between two mutually trans non-equivalent hydride ligands.16 The hydrides in complex 5 do not exchange positions up to 80 °C. In accordance with the increased oxidation state of the Ir atom, the NMR signals of the olefin moiety in 5 are shifted downfield compared to 4; for example the –CH proton resonates at 2.51 ppm (dd, 3JP2H = 21.7 Hz, 3JP1H = 3.3 Hz) and the quaternary carbon signal is observed at 81.3 ppm (m).
 | ||
| Scheme 2 Reactions of complex 4 with hydrogen. | ||
Complex 6 is characterized by an AB system (69.8, 63.3 ppm, 2JP1P2 = 312.9 Hz) in the 31P{1H} NMR spectrum as well as a triplet resonance at −10.07 ppm (2JPH(avg) = 9.5 Hz) in the 1H NMR, integrating as 4H. 2D NMR spectra strongly argue that 6 is a product of insertion of the olefin moiety into one of the Ir–H bonds, which added one molecule of hydrogen. For instance, in contrast to 5, no –CH– type carbons reveal a cross-peak with the hydride resonance in 1H–13C HMBC, while three correlations with –CH2– type carbons are observed, with two of them belonging to –CH2P– groups and one to the former –CH group.
When a mixture of 5 and 6 is heated at 140 °C for 24 h under an atmosphere of hydrogen, a quantitative conversion to tetrahydride complex 7 is observed, where the initial structure of the pincer ligand is recovered. Hence, the observed formation of a C–C double bond via Csp3–H activation is fully reversible, and proceeds in the forward direction when a hydrogen acceptor like tert-butylethylene is used, and in the reverse direction when H2 pressure is applied. Remarkably, the transformation from 5 to 7 corresponds to a net rupture of a C–C double bond, while the transformation from 6 to 7 is a net cleavage of a nearly unstrained, unactivated Csp3–Csp3 bond, with few precedents previously reported.17 Together with the tandem catalytic systems for alkane metathesis,18 this process is a rare example of an organometallic compound capable of a net cleavage of unactivated Csp3–Csp3 bonds.
Computational studies
To understand each step of the whole transformation we performed DFT calculations on the key reactions for forming and breaking C–C bonds, based on the experimentally observed species. Several mechanistic scenarios were considered for the formation of complex 7; the one with the lowest barrier and that is fully consistent with the experimental conditions is depicted in Fig. 3. First, from complex 5, a double bond insertion into the Ir–H bond leads to intermediate 8, which can be transformed to the experimentally observed Ir(V) complex 6via the oxidative addition of dihydrogen. The calculated barrier of ca. 20 kcal mol−1 is in qualitative agreement with the rate of establishment of the equilibrium (a few minutes) at ambient temperature. Intermediate 8 undergoes an α-alkyl elimination and carbene 9 is formed. The activation barrier is calculated at 31.4 kcal mol−1via transition state TS8–9. Complex 9 proceeds through an α-hydride insertion to form intermediate 10, which, under hydrogen atmosphere conditions, undergoes an H2 addition that finally forms the Ir(V) complex 11. The transition state TS9–10 is calculated at 25.8 kcal mol−1 on the free energy surface. This step is followed by C–H reductive elimination by Ir–Csp3 and Ir–H, which leads to formation of a new Csp3–H bond in the pincer ligand. The free energy barrier via transition state TS11–12 is calculated at 11.8 kcal mol−1.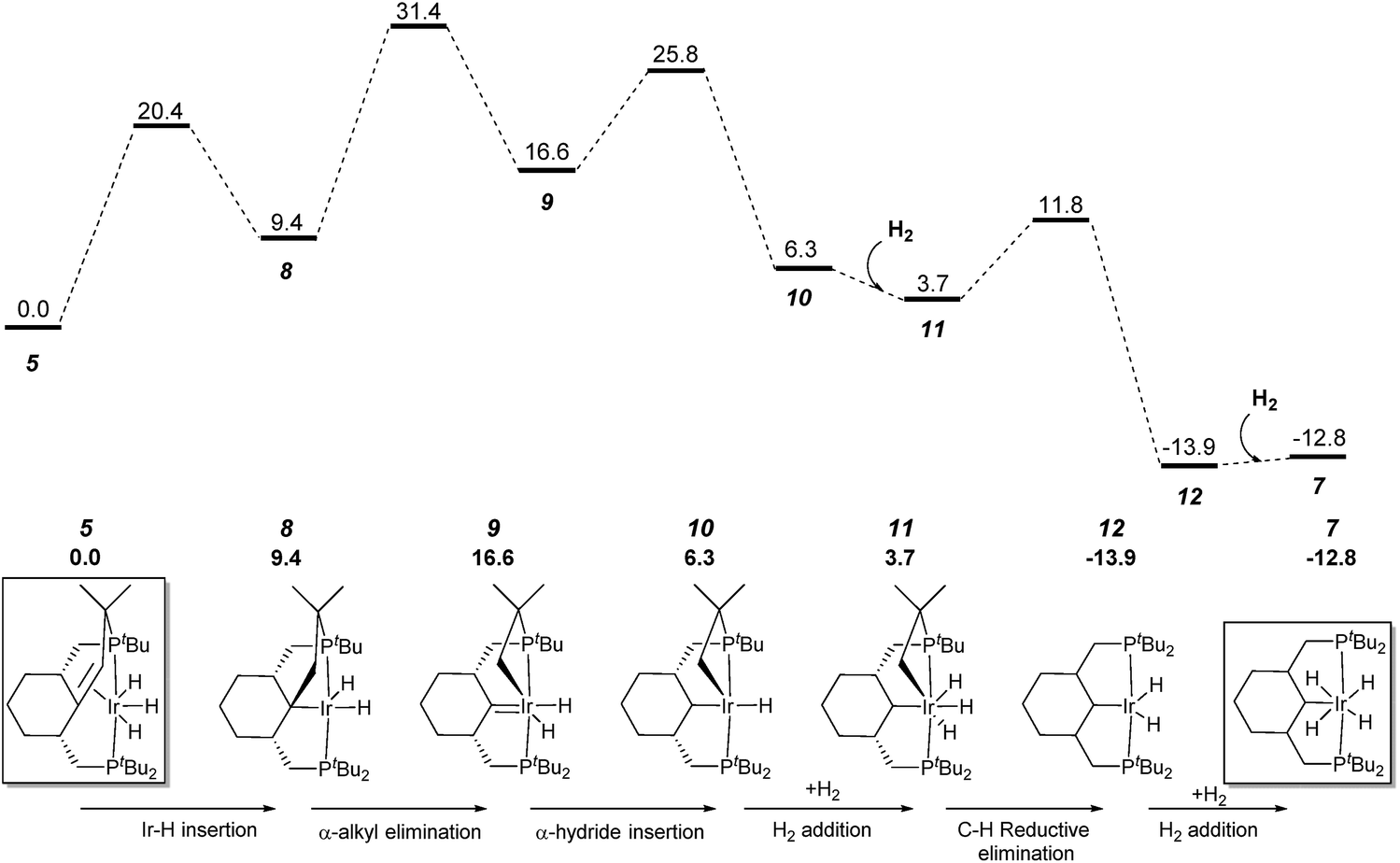 | ||
| Fig. 3 Profile of the calculated relative G (kcal mol−1) for the formation of complex 7 from complex 5via carbene complex 9. | ||
Ultimately, the process finishes with an oxidative addition of one dihydrogen molecule, leading to the Ir(V)H4 complex 7. The overall activation free energy corresponds to the C–C bond cleavage step (31.4 kcal mol−1).
We also considered alternative mechanisms from 8, involving inter alia C–H reductive elimination to give an alkane intermediate, which could be followed by Csp3–Csp3 oxidative addition (Fig. 4, red and blue pathways). However, the activation energies associated with this process were found to be prohibitively high. C–C bond oxidative addition/elimination at the metal have high barriers at least in part due to repulsion between the bulky alkyl ligands. The suggested mechanism is supported by the experimental observation of an α-hydrogen migration to Ir during the formation of a carbene complex (see below). Also, a number of R1 migrations from an R1MCR2R3 fragment to M–CR1R2R3 are known,19 with a few precedents reported for vinylidene pincer complexes.20 It should be noted that for the somewhat-related coupling reaction involving benzylic C–H bonds, a similar reaction sequence was also proposed.15b,c Interestingly, the previous examples where unactivated Csp3–Csp3 bonds were cleaved proceeded via another mechanism, namely β-carbon elimination.17 In addition, several examples of 1,2-shifts of methyl and benzyl groups to give arenium complexes (organometallic analogs of Wheland intermediates), which likely proceed through electrophilic attack of R+ on Cipso, are known; these may be followed by subsequent β-elimination.21,22
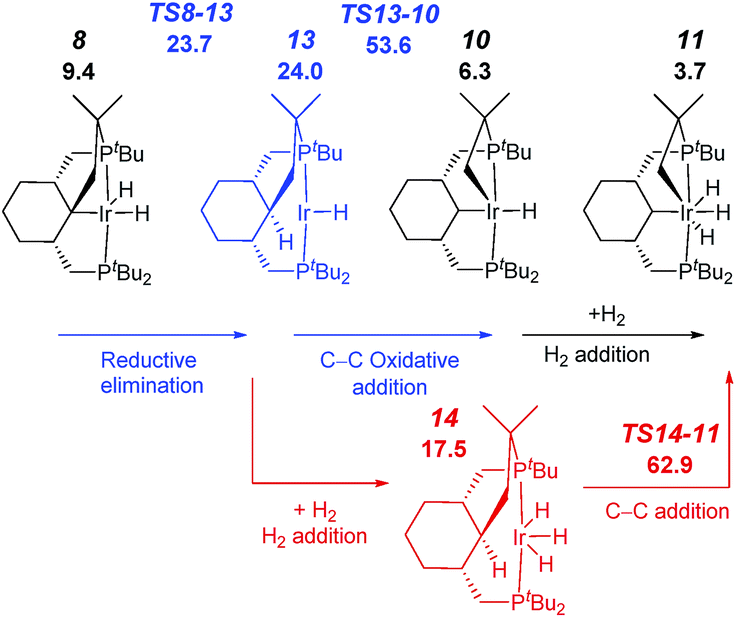 | ||
| Fig. 4 Profile of the calculated relative G (kcal mol−1) for the formation of complex 7 from complex 5via reductive elimination from Ir(III) (blue) or Ir(V) (red). | ||
As mentioned before, the formation of the C–C double bond is reversible, proceeding in one direction or other depending on the reaction conditions. In agreement with the experiment, under hydrogen atmosphere, formation of complex 7 is favored, while in the presence of a hydrogen acceptor, such as tert-butylethylene, the reverse reaction occurs leading to the olefin complex 5 (Fig. 5). In the reaction with tert-butylethylene, the overall activation energy barrier corresponds to the C–C bond formation step (TS9–8) and is calculated at 28.0 kcal mol−1.
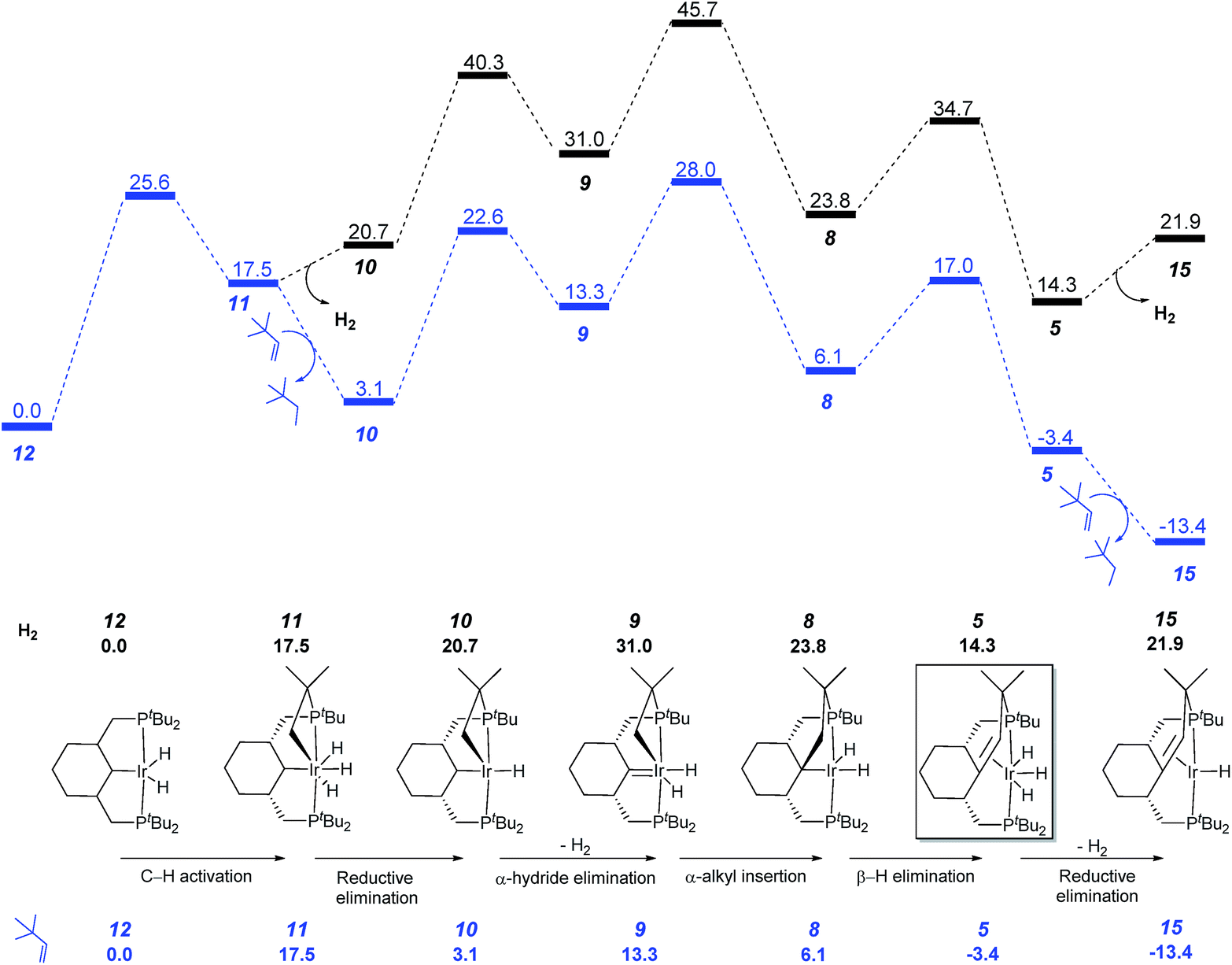 | ||
| Fig. 5 Profile of the calculated relative G (kcal mol−1) for the formation of complex 5 with and without hydrogen acceptor. | ||
Dehydrogenation of the cyclohexyl ring
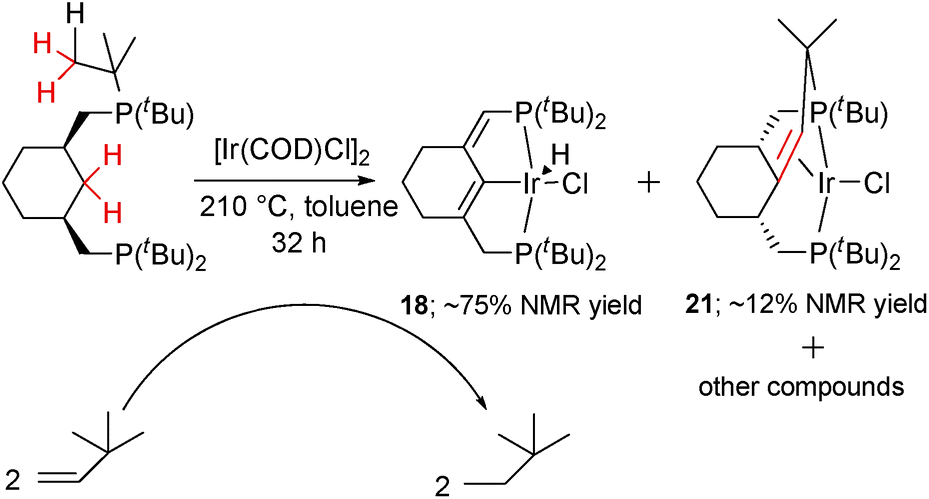
For comparison and to gain more insight into the process, we attempted the thermolysis of the parent, less electron-rich hydrido-chloride complex 1. Heating (PCyP)IrHCl (1) in toluene at higher temperatures reveals that it undergoes aromatization, and several intermediates were detected by NMR spectroscopy during this process (Scheme 3). All steps before aromatization are reversible, and carrying out the reaction in a sealed flask results in the simultaneous presence of compounds 1, 16, 17, 18, and 19 in the reaction mixture; to drive the process to completion it is necessary to purge the flask with Ar several times or to add a hydrogen acceptor like tert-butylethylene. In a control experiment, a mixture of 1, 16, 17, and 18 was indeed converted to 1 under a H2 atmosphere at 155 °C. The ratio of compounds 1, 16, 17, 18, and 19 depends on the conditions, with the percentages of the more dehydrogenated products increasing with increasing temperature. Despite a number of attempts, including solution and solid-state thermolysis of 1 under various conditions, we were unable to obtain complex 16 in a high yield and isolate it from other products, and so far 16 and 17 have been characterized in situ.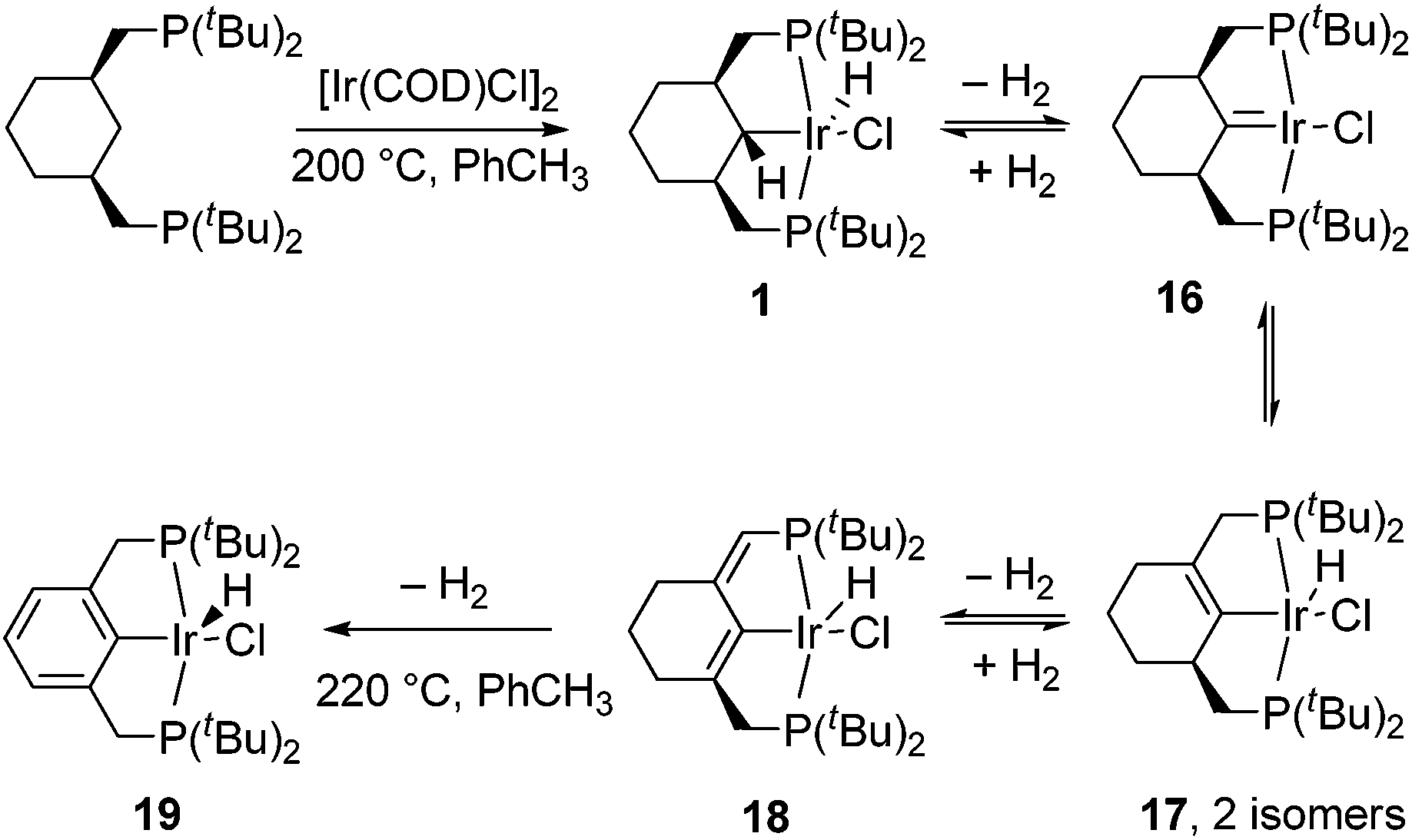 | ||
| Scheme 3 Aromatization of the cyclohexane-based pincer ligand. | ||
The aromatization process begins with the formation of carbene complex 16, which exhibits a singlet in the 31P{1H} NMR spectrum at 66.8 ppm and a remarkable high-field signal at −4.18 ppm in the 1H NMR spectrum from the β-C–H groups, probably due to the magnetic anisotropy effect of the CIr bond. The 13C{1H} NMR spectrum reveals a resonance at 246.9 ppm, corresponding to the α-carbon. This value dramatically differs from the 66.6 ppm reported by Shaw,23 who proposed the contribution of an ylide-type structure for the related complex 20 (Scheme 4) in order to explain the deviation of the observed chemical shift from that expected for carbene complexes at around 200 ppm. The ylide character of Shaw's complex has been mentioned in several papers and has been the subject of some discussion.24 Based on our data, we suggest that there is no reason to invoke an ylide structure for 16, and probably not for 20 either. Complex 20 was characterized in a mixture where its concentration was fairly low, and the overall number of 13C signals observed by Shaw was three instead of the five expected from the symmetry of complex 20. Therefore, given the similarity of complexes 20 and 16, it could very well be that the signal at 66.6 ppm comes from the –CH2– groups adjacent to the carbene moiety since the analogous –CH–groups in complex 16 resonate at 75.0 ppm, while the true low-intensity, low-field signal of the carbene carbon atom of 20 was not observed. In fact, according to the calculations, the bonding pattern in complex 16 is close to those in Fisher-type carbenes and thus 16 is better described as an Ir(I) compound (see ESI†).
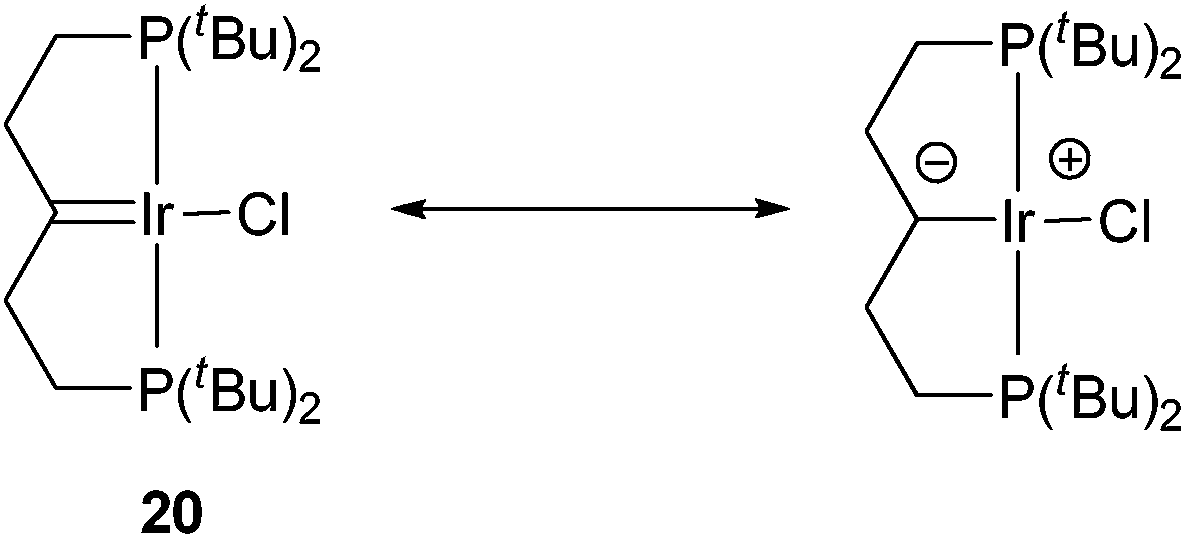 | ||
| Scheme 4 Shaw's carbene complex and the proposed contribution of the ylide structure. | ||
The formation of complex 16 requires high temperature and at these conditions, 16 readily turns into the isomeric compounds 17, with the equilibrium shifted to the side of the latter complexes. The 31P{1H} NMR spectrum reveals two isomers of 17, each with an ABX system (67.7, 64.9 ppm, 2JP1P2 = 336.8 Hz and 66.6, 63.0 ppm, 2JP1P2 = 335.0 Hz; high-field hydride signals are not decoupled), which indicates the presence of two non-equivalent phosphorus nuclei and a lack of the usual symmetry of the molecule. The corresponding hydride signals are observed at −41.25 (t, 2JP1H = 2JP2H = 13.2 Hz) and −44.63 (t, 2JP1H = 2JP2H = 12.3 Hz) ppm as apparent triplets, due to virtually equal coupling constants with both P atoms.25 The 13C{1H} NMR spectrum confirms the presence of one tetra-substituted double bond per molecule.
Further dehydrogenation results in the formation of diene complex 18, which again is characterized by an ABX system in the 31P{1H} NMR spectrum (67.7, 60.3 ppm, dd, 2JP1P2 = 334.3), as well as a doublet (2JPH = 8.0 Hz) at 5.61 ppm from the olefinic proton and an apparent triplet at −43.30 (2JP1H = 2JP2H = 12.7 Hz) ppm from the hydride. The CH– group appears at 117.4 in the 13C{1H} spectrum and a large 13C–31P coupling of 44.4 Hz, together with 2D spectra, confirms the position of the double bond near the phosphorus atom. Under the conditions specified in Scheme 5, the relative kinetic stability of 18 allows an accumulation (up to 75%) in the reaction mixture, from which 18 can be isolated by chromatography.
 | ||
| Scheme 5 Quadruple C–H activation of the cyclohexane-based pincer ligand. | ||
C–C coupling reaction in complex 1
During NMR spectroscopic monitoring of the formation of 18, the appearance of a new AX system (66.3, 23.7 ppm, 2JP1P2 = 370.0 Hz) was detected in the 31P{1H} spectrum. The yield of the new compound 21 was low, but we were able to isolate it by crystallization. The NMR features of 21 are similar to those of 4, with signals for the olefinic proton and carbons being significantly high-field shifted, and the X-ray structure of 21 (Fig. 6) confirms that a similar olefin complex has formed. The C–C distance (1.441(5) Å) is comparable to that in 4 and the Ir–C bonds (2.142(4) and 2.112(4) Å) are somewhat shorter. As shown in Scheme 5, four non-activated Csp3–H bonds are cleaved during the synthesis of 21 (starting from the ligand precursor), making this process a rare example of a quadruple C–H activation at a single metal center. It also shows that the C–C coupling is a general reaction for this class of compounds.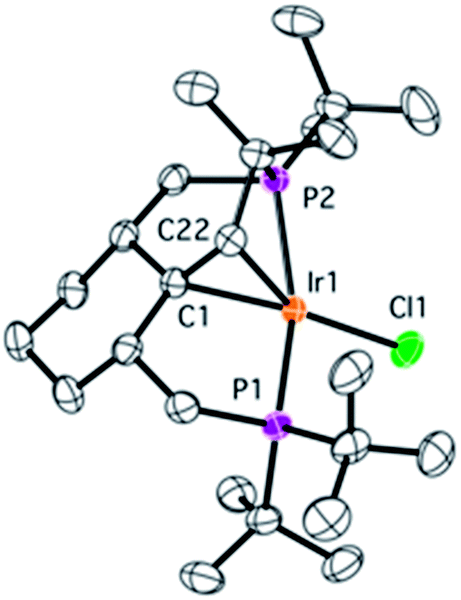 | ||
| Fig. 6 Molecular structure of complex 21 with thermal ellipsoids at 30% probability level. Hydrogen atoms have been omitted for clarity. Selected bonds (Å) and angles (°): Ir1–C1 2.142(4), Ir1–C22 2.112(4), Ir1–Cl1 2.380(1), Ir1–P1 2.2857(9), Ir1–P2 2.3153(9), C1–C22 1.441(5), Cl1–Ir1–C1 163.4(1), P1–Ir–P2 160.24(3). | ||
While for complexes 4 and 21 high ligand binding energy did not allow the determination of a catalytic cycle, we believe that this reactivity pattern can form a basis for future catalytic cross-couplings of non-activated Csp3–H and possibly other C–H bonds under relatively mild conditions. In addition, whereas one of the reasons for the unreactive nature of Csp3–Csp3 bonds is their low exposure to metal atoms due to the screening by C–H or other bonds, we here show that at least in some cases such screening can be used in a beneficial way, during which simple metallation of one of the C–H bonds leads to a rearrangement where a purely aliphatic fragment is cut into two others.
Conclusions
In conclusion, we have presented the C–H activation reactivity of cyclohexane-based iridium pincer complexes. The major difference between these systems and their arene-based counterparts is the non-innocent character of the pincer ligand, where both α- and β-hydrogens can be eliminated. This opened up an unprecedented reactivity, where non-activated Csp3–H bonds extrude two molecules of dihydrogen in the formation of a C–C double bond. This process is reversible and upon pressurizing with H2 the resulting C–C bond can be hydrogenated to recover the ligand structure; remarkably, this happens via a net Csp3–Csp3 bond cleavage. Mechanistic studies indicate that a key step to open up such reactivity is the formation of the carbene complex, which, via migratory insertion or deinsertion into an Ir–C bond, is responsible for C–C bond formation and cleavage. This knowledge will hopefully enable a catalytic variety of this transformation allowing the intermolecular formation of double bonds from the coupling of two non-activated aliphatic carbons.Acknowledgements
Financial support from the Swedish Research Council, the Knut and Alice Wallenberg Foundation and the Crafoord Foundation is gratefully acknowledged. Computational resources have been provided by the National Supercomputer Centre in Linköping, Sweden.Notes and references
- (a) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154 CrossRef CAS; (b) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed; (c) R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437 RSC; (d) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed; (e) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed.
- (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (e) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed.
- (a) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704 RSC; (b) X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253 RSC; (c) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC; (d) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed.
- Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed.
- A. Arunachalampillai, D. Olsson and O. F. Wendt, Dalton Trans., 2009, 8626 RSC.
- A. V. Polukeev, R. Gritcenko, K. J. Jonasson and O. F. Wendt, Polyhedron, 2014, 84, 63 CrossRef CAS PubMed.
- M. Kanzelberger, B. Singh, M. Czerw, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2000, 122, 11017 CrossRef CAS.
- (a) M. Montag, I. Efremenko, R. Cohen, L. J. W. Shimon, G. Leitus, Y. Diskin-Posner, Y. Ben-David, H. Salem, J. M. L. Martin and D. Milstein, Chem. –Eur. J., 2010, 16, 328 CrossRef CAS PubMed; (b) A. V. Polukeev, P. V. Petrovskii, A. S. Peregudov, M. G. Ezernitskaya and A. A. Koridze, Organometallics, 2013, 32, 1000 CrossRef CAS.
- (a) A. Friedrich, R. Ghosh, R. Kolb, E. Herdtweck and S. Schneider, Organometallics, 2009, 28, 708 CrossRef CAS; (b) C. Bianchini, E. Farnetti, M. Graziani, G. Nardin, A. Vacca and F. Zanobini, J. Am. Chem. Soc., 1990, 112, 9190 CrossRef CAS.
- (a) H. A. Y. Mohammad, J. C. Grimm, K. Eichele, H.-G. Mack, B. Speiser, F. Novak, M. G. Quintanilla, W. C. Kaska and H. A. Mayer, Organometallics, 2002, 21, 5775 CrossRef CAS; (b) A. V. Polukeev, S. A. Kuklin, P. V. Petrovskii, F. M. Dolgushin, M. G. Ezernitskaya and A. A. Koridze, Russ. Chem. Bull., 2010, 59, 745 CrossRef CAS.
- (a) M. Yamashita, Y. Moroe, T. Yano and K. Nozaki, Inorg. Chim. Acta, 2011, 369, 15 CrossRef CAS PubMed; (b) M. A. Esteruelas, M. Olivan and A. Velez, Inorg. Chem., 2013, 52, 5339 CrossRef CAS PubMed.
- For migration of Ir on a benzene ring, assisted by metallation of a chelating –CH2–N(CH3)2 group, see: A. A. H. van der Zeijden, G. van Koten, R. Luijk, R. A. Nordemann and A. L. Spek, Organometallics, 1988, 7, 1549 CrossRef CAS.
- It should be noted that the formation of olefins from saturated carbons takes place during alkane metathesis, but they are not major products because of thermodynamical constraints. For details, see ref. 18.
- (a) J. Campos, M. F. Espada, J. Lopez-Serrano and E. Carmona, Inorg. Chem., 2013, 52, 6694 CrossRef CAS PubMed; (b) J. Campos, J. Lopez-Serrano, E. Alvarez and E. Carmona, J. Am. Chem. Soc., 2012, 134, 7165 CrossRef CAS PubMed; (c) W. Baratta, M. Ballico, A. Del Zotto, E. Zangrando and P. Rigo, Chem. –Eur. J., 2007, 13, 6701 CrossRef CAS PubMed; (d) W. Baratta, E. Herdtweck, P. Martinuzzi and P. Rigo, Organometallics, 2001, 20, 305 CrossRef CAS; (e) M. A. Bennett and P. A. Longstaff, J. Am. Chem. Soc., 1969, 91, 6266 CrossRef CAS.
- S. Gründemann, H.-H. Limbach, G. Buntkowsky, S. Sabo-Etienne and B. Chaudret, J. Phys. Chem. A, 1999, 103, 4752 CrossRef.
- (a) K. Ruhland, Eur. J. Org. Chem., 2012, 2683 CrossRef CAS; (b) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed.
- (a) A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257 CrossRef CAS PubMed; (b) M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2012, 45, 947 CrossRef CAS PubMed and references therein.
- (a) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed; (b) Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 10024 CrossRef CAS PubMed.
- (a) J. D. Hackenberg, S. Kundu, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2014, 136, 8891 CrossRef CAS PubMed; (b) H. M. Lee, J. Yao and G. Jia, Organometallics, 1997, 16, 3927 CrossRef CAS; (c) G. Jia, H. M. Lee, H. P. Xia and I. D. Williams, Organometallics, 1996, 15, 5453 CrossRef CAS.
- For a 1,2 shift to an aryl group in Pt pincer complexes, see: (a) J. Terheijden, G. van Koten, I. C. Vinke and A. L. Spek, J. Am. Chem. Soc., 1985, 107, 2891 CrossRef CAS; (b) M. Albrecht, R. A. Gossage, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 1999, 121, 11898 CrossRef CAS; (c) M. Albrecht, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 2001, 123, 7233 CrossRef CAS PubMed.
- For a 1,2-shift to aryl groups in Rh and Ir pincer complexes, followed by olefin formation, see: (a) A. Vigalok and D. Milstein, J. Am. Chem. Soc., 1997, 119, 7873 CrossRef CAS; (b) A. Vigalok, B. Rybtchinski, L. J. W. Shimon, Y. Ben-David and D. Milstein, Organometallics, 1999, 18, 895 CrossRef CAS.
- (a) H. D. Empsall, E. M. Hyde, R. Markham, W. S. McDonald, M. C. Norton, B. L. Shaw and B. Weeks, J. Chem. Soc., Chem. Commun., 1977, 589 RSC; (b) C. Crocker, H. D. Empsall, R. J. Errington, E. M. Hyde, W. S. McDonald, R. Markham, M. C. Norton, B. L. Shaw and B. Weeks, J. Chem. Soc., Dalton Trans., 1982, 1217 RSC.
- (a) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (b) W. Leis, H. A. Mayer and W. C. Kaska, Coord. Chem. Rev., 2008, 252, 1787 CrossRef CAS PubMed; (c) H. Werner, Angew. Chem., Int. Ed., 2010, 49, 4714 CrossRef CAS PubMed; (d) R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949 CrossRef CAS.
- Spin simulations suggest that these signals are not virtual triplets, which one may expect to see in the X part of the ABX system; each of them can be described as doublet of doublets, which collapsed into a triplet due to equal coupling constants.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, Cartesian coordinates, additional graphs and computational details. CCDC 1024127–1024129. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03839h |
| This journal is © The Royal Society of Chemistry 2015 |