
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic/inorganic double-layered shells for multiple cytoprotection of individual living cells†
Daewha
Hong
a,
Hojae
Lee
a,
Eun Hyea
Ko
a,
Juno
Lee
a,
Hyeoncheol
Cho
a,
Matthew
Park
a,
Sung Ho
Yang
b and
Insung S.
Choi
*a
aCenter for Cell-Encapsulation Research, Department of Chemistry, KAIST, Daejeon 305-701, Korea. E-mail: ischoi@kaist.ac.kr
bDepartment of Chemistry Education, Korea National University of Education, Chungbuk 363-791, Korea
First published on 1st October 2014
Abstract
The cytoprotection of individual living cells under in vitro and daily-life conditions is a prerequisite for various cell-based applications including cell therapy, cell-based sensors, regenerative medicine, and even the food industry. In this work, we use a cytocompatible two-step process to encapsulate Saccharomyces cerevisiae in a highly uniform nanometric (<100 nm) shell composed of organic poly(norepinephrine) and inorganic silica layers. The resulting cell-in-shell structure acquires multiple resistance against lytic enzyme, desiccation, and UV-C irradiation. In addition to the UV-C filtering effect of the double-layered shell, the biochemical responses of the encapsulated yeast are suggested to contribute to the observed UV-C tolerance. This work offers a chemical tool for cytoprotecting individual living cells under multiple stresses and also for studying biochemical behavior at the cellular level.
Introduction
The protection and preservation of individual living cells under in vitro conditions have been intractable challenges in various cell-based applications including cell therapy, cell-based sensors, tissue engineering, and even renewable energy sources.1 In cell therapy, the cytoprotection of therapeutic cells, under daily-life conditions and in the body, is required for a prolonged shelf-life and for administering on the spot therapy, respectively. Field-deployable cell-based sensors also benefit from the effective protection of living cells against a multitude of external harmful aggressors. In this regard, the simple structural mimicry of bacterial endospores,2 the rigid and tough coats of which protect inner cells from extreme stresses, has previously been attempted to endow the cells with enhanced tolerance against a certain stress depending upon the materials used for cell encapsulation.3 For example, enhanced thermotolerance was achieved by the heat-dissipating properties of inorganic silica or silica–titania hybrid coats,3a–c and resistance against lytic enzymes was achieved by the small pore size of organic poly(dopamine) shells.3d Besides living cells, viruses have also been encapsulated within ionic calcium phosphate shells for the development of thermotolerant vaccines.4 However, the reported enhanced tolerance has mainly been limited to a single type of stress, which precludes the wider application of encapsulated cells to the areas where multiple stresses are unavoidable.In the case of natural bacterial endospores, both structural transformation and biochemical alteration occur during the sporulation process, which provide multiple resistance against lethal stresses, including UV radiation, desiccation, heat, malnutrition, and toxic chemicals, over an extended period of time.5 Their multi-layered shell structure, composed of cortex, spore coat, and exosporium, plays an important role in the multifunctional features of the endospores, and the biochemical alteration contributes in a complementary way to the enhanced tolerance of the endospores.6 In this work, we formed an organic/inorganic, double-layered shell on individual Saccharomyces cerevisiae (baker's yeast) for multiple resistance against stresses.
Results and discussion
The double-layered shell was designed to be composed of organic poly(norepinephrine) and inorganic silica (Fig. 1a). We chose norepinephrine, a dopamine derivative with an additional hydroxyl group, for the organic layer, because poly(norepinephrine) (PN) makes a uniform, conformal contact with a substrate.7 In addition, the cytocompatibility of PN is superior to that of poly(dopamine), which has been previously used in cell encapsulation and has been shown to be effective in the prevention of enzymatic attack.3d,8 The PN layer was formed on individual yeast cells by gently shaking a yeast suspension in a Tris–HCl buffer solution (pH 8.5) containing PN (2 mg mL−1) for 6 h at room temperature, and the cell viability was investigated by a fluorescein diacetate (FDA) assay, assessing the esterase activities in the cell.9 The relative cell viability after the PN-layer formation was measured to be 94%, while it was 66% after 3 h of polydopamine-layer formation (see ESI, Fig. S1†). The PN layer was hardly seen in transmission electron microscopy (TEM) images, but its thickness was estimated to be about 30 nm based on PN films on gold that were formed under the same reaction conditions.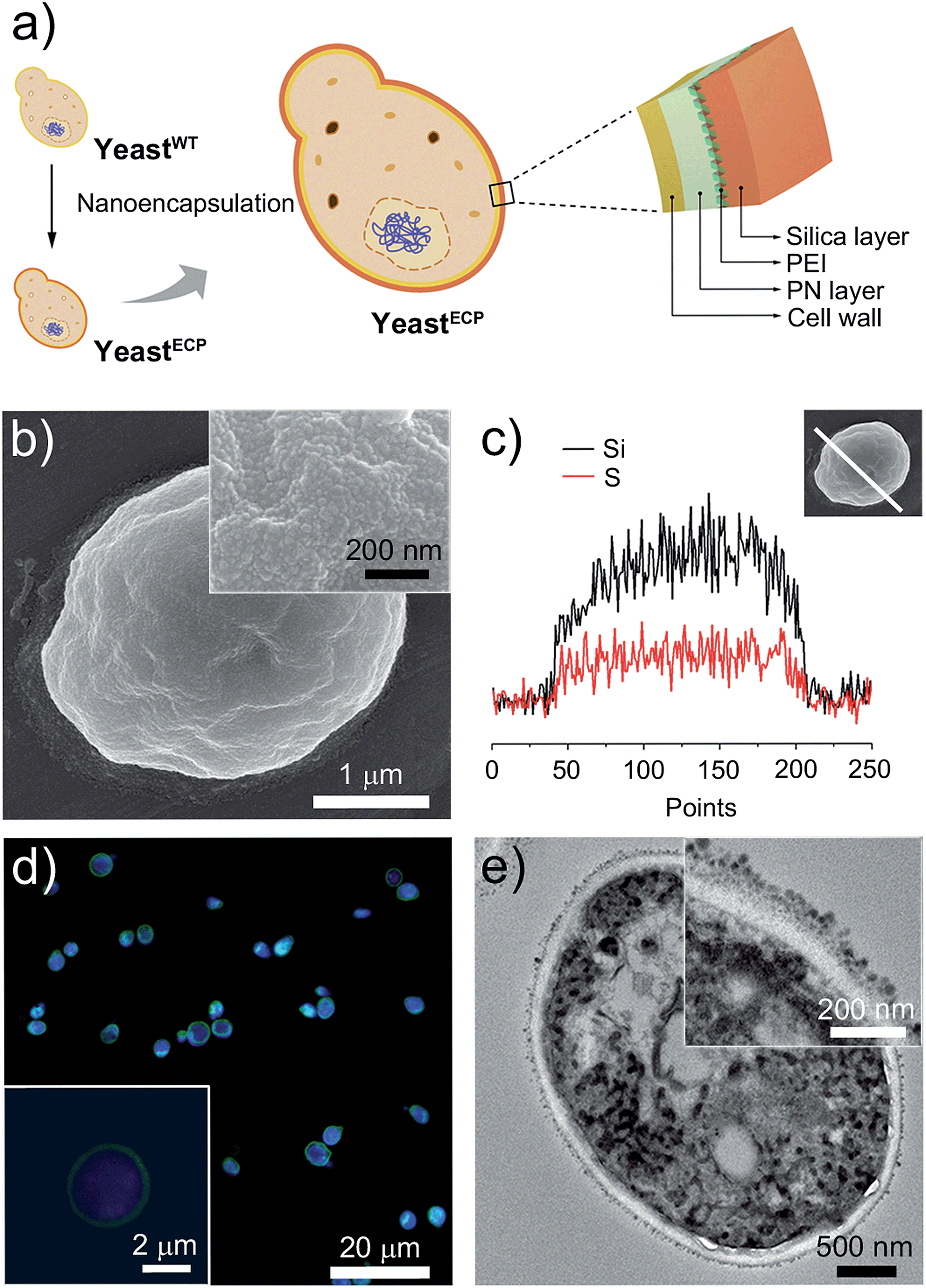 | ||
| Fig. 1 (a) Schematic representation of the artificial shell, composed of organic poly(norepinephrine) (PN) and inorganic silica layers (yeastWT: wild-type yeast; yeastECP: encapsulated yeast). (b and c) SEM micrographs and EDX line-scan analysis of yeastECP confirming the presence of an SH-bearing silica shell. The double-layered shell was mechanically tough enough to maintain the original shape of the yeast. (d) CLSM micrographs of yeastECP after staining the cells with 4′,6-diamidino-2-phenylindole (DAPI) (blue) and functionalizing the shells with N-(5-fluoresceinyl)maleimide (green). (e) TEM micrographs of the microtome-sliced yeastECP. | ||
The hydroquinone moiety in the PN layer was used for grafting poly(ethyleneimine) (PEI), where the amine group in PEI reacted with the hydroquinone moiety via nucleophilic 1,4-conjugate addition. The PN-coated yeast (yeast@PN) was incubated in a Tris–HCl buffer solution (pH 8.5) of PEI (2 mg mL−1) for 2 h at room temperature. An inorganic silica layer was then formed on top of the PN layer by bioinspired silicification with PEI as a catalytic template.10 The PEI-grafted yeast cells were incubated for 30 min in a 100 mM silicic acid derivative solution that had been made by hydrolysing tetramethyl orthosilicate (TMOS, 1 M) and (3-mercaptopropyl)trimethoxysilane (MPTMS, 1 M), respectively, in an aqueous HCl solution (1 mM) at room temperature for 20 min and adding the resulting solutions to a sodium phosphate buffer (50 mM, pH 5.8) with 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75:900 (v/v/v) ratio.10 After double-layer formation, the relative cell viability was measured to be up to 92.6%, clearly indicating that the encapsulation processes were highly cytocompatible.
:75:900 (v/v/v) ratio.10 After double-layer formation, the relative cell viability was measured to be up to 92.6%, clearly indicating that the encapsulation processes were highly cytocompatible.
The scanning electron microscopy (SEM) images show uniformly coated shells composed of fine silica particulates (Fig. 1b), the presence of which was further confirmed by energy-dispersive X-ray (EDX) spectroscopy line-scan analysis for Si and S elements (Fig. 1c). Although the PN and silica layers were not distinguished clearly, the TEM images of microtome-sliced samples showed about a 60 nm-thick shell outside the cell wall (Fig. 1e; also see ESI, Fig. S2†). Based on the TEM images, the size of the silica nanoparticulates is mostly in the range 15–25 nm. To investigate the encapsulation efficiency, the thiol (SH) group in the silica layer was coupled with N-(5-fluoresceinyl)maleimide (green). The cell-in-shell structure was clearly observed for all yeast cells by confocal laser-scanning microscopy (CLSM), indicating a high encapsulation efficiency (Fig. 1d).
Barrett–Joyner–Halenda (BJH) analysis showed that the pore size of the PN/silica hybrid shell was mostly less than 10 nm (see ESI, Fig. S3†), which would preclude the permeation of large macromolecules, enzymes, and macrophages. As expected, lyticase, a cell wall-lysing enzyme complex, was effectively screened out by the PN/silica shell (Fig. 2a). For the cell-lysis test, wild-type yeast cells (yeastWT) and encapsulated yeast cells (yeastECP) were incubated in a Tris–EDTA buffer solution containing lyticase at 37 °C, and the optical density was measured at 600 nm by UV-visible spectroscopy for the evaluation of cell density. After a 1 h treatment of lyticase, the majority of yeastWT were lysed (ca. 83%), while about 75% of yeastECP survived. A 4 h treatment lysed about 90% of yeastWT, but half of yeastECP were still alive. Resistance against desiccation was more striking (Fig. 2b). The yeastECP or yeastWT were dried after filtering water out with a hydrophilic membrane to confirm resistance under water-free conditions. After 2 h drying at 30 °C, most of yeastWT (∼90%) were dead, but the viability of yeastECP was 86%, indicating that the enhancement in the survival ratio was 12.5.
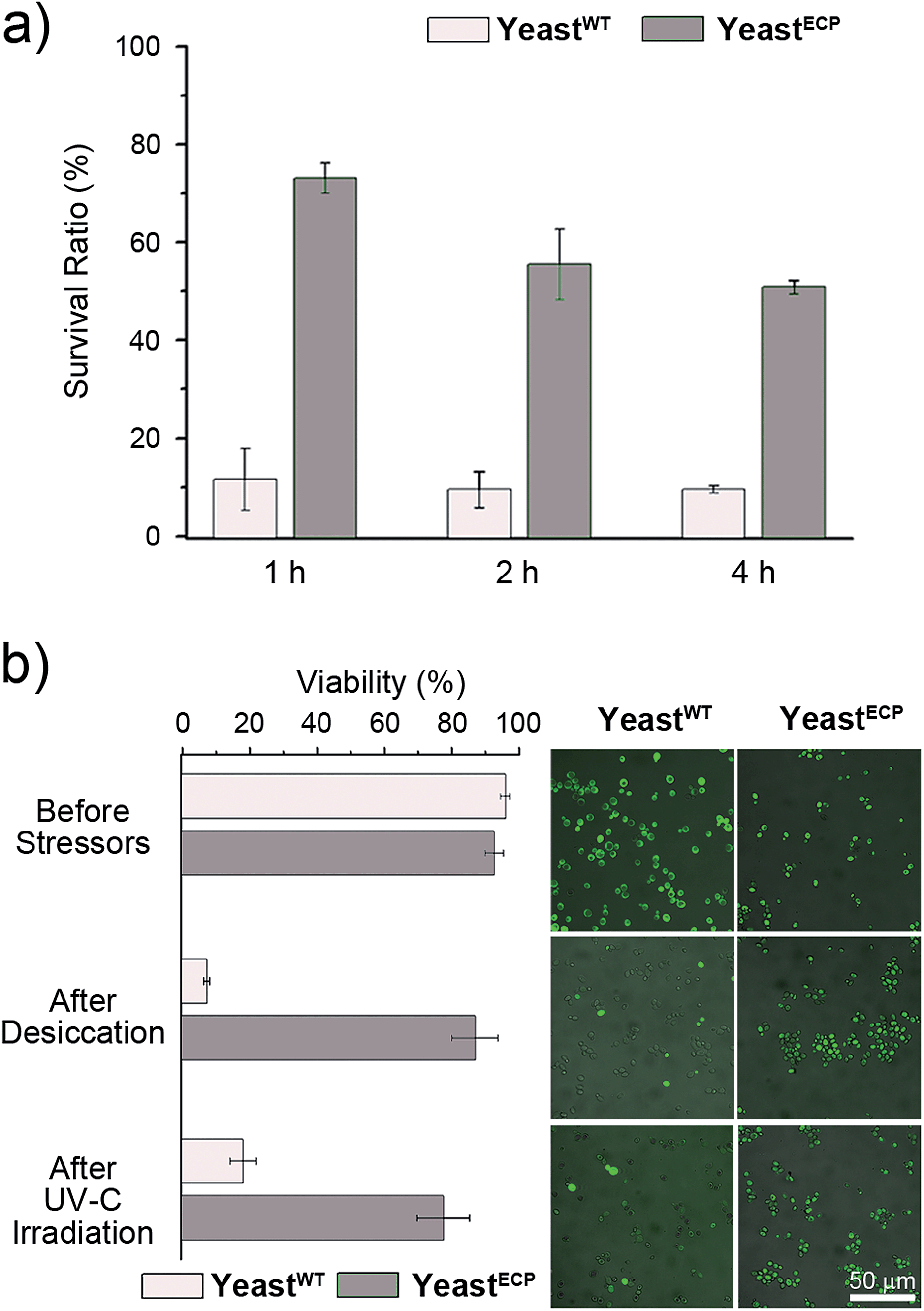 | ||
| Fig. 2 (a) Survival ratios of yeastWT and yeastECP after a treatment of lyticase. The survival ratio = OD600 at a predetermined time/OD600 before the treatment of lyticase × 100. (b) The enhanced tolerance of yeastECP against 2 h desiccation (30 °C) and 500 s UV-C irradiation (254 nm, 4 W). The cells in green after FDA treatment were considered alive. | ||
UV-C (wavelength: 100–280 nm) is the most germicidal in the UV range, and most microbes, including yeast, are killed upon a short exposure to UV-C. As expected, more than 80% of yeastWT were dead after 500 s irradiation of 254 nm light (Fig. 2b). In stark contrast, ∼75% of yeastECP still survived after the same irradiation. We believe that the UV-C filtering property of the PN/silica hybrid shells contributed to the observed enhancement, because the UV-visible spectrophotometric measurements show that the absorbance of the PN/silica film on quartz at 254 nm was 1.63 times higher than that of the PN film (see ESI, Fig. S4†). Accordingly, ∼80% of the yeast cells coated only with PN were dead after UV-C irradiation (see ESI, Fig. S5†).
In addition to the UV-C filtering property of the hybrid shell, we also investigated the biochemical responses of yeastECP at the protein level by mass spectrometry-based proteomic analysis with yeastWT as a comparison. Briefly, yeastWT and yeastECP before and after UV-C irradiation were prepared (i.e., four samples). Each sample was lysed, and the total obtained proteins from each sample were subjected to proteomic analysis. For a comparative study, Fisher's exact test was performed between the two target samples, and the proteins, the p-values of which were less than 0.05, were accepted to be expressed differently: 33 of the analyzed proteins (789) were expressed differently for the yeastWTvs. the UV-C irradiated yeastWT; 34 of the analyzed proteins (482) for the yeastECPvs. the UV-C irradiated yeastECP (Fig. 3a). The protein expression levels were totally different between yeastWT and yeastECP after UV-C irradiation, and the up- and down-regulated proteins did not overlap with each other between yeastWT and yeastECP. The results imply that the yeastECP was not exposed directly to UV-C, but rather faced manageable stresses in different forms (e.g., light with longer wavelengths or heat). In contrast, the yeastWT were dead upon direct UV-C exposure. It was notable that the proteins involved in protein folding (ECM10 and SSA1) were found to be up-regulated for yeastECP after UV-C irradiation; they belong to the HSP70 family that assists in the proper folding of proteins and prevents the aggregation of denatured proteins.11 Given the fact that these chaperones were not up-regulated after UV-C irradiation in the case of yeastWT, we think that molecular chaperones could be strong candidates for providing the enhanced tolerance of yeastECP against UV-C irradiation.
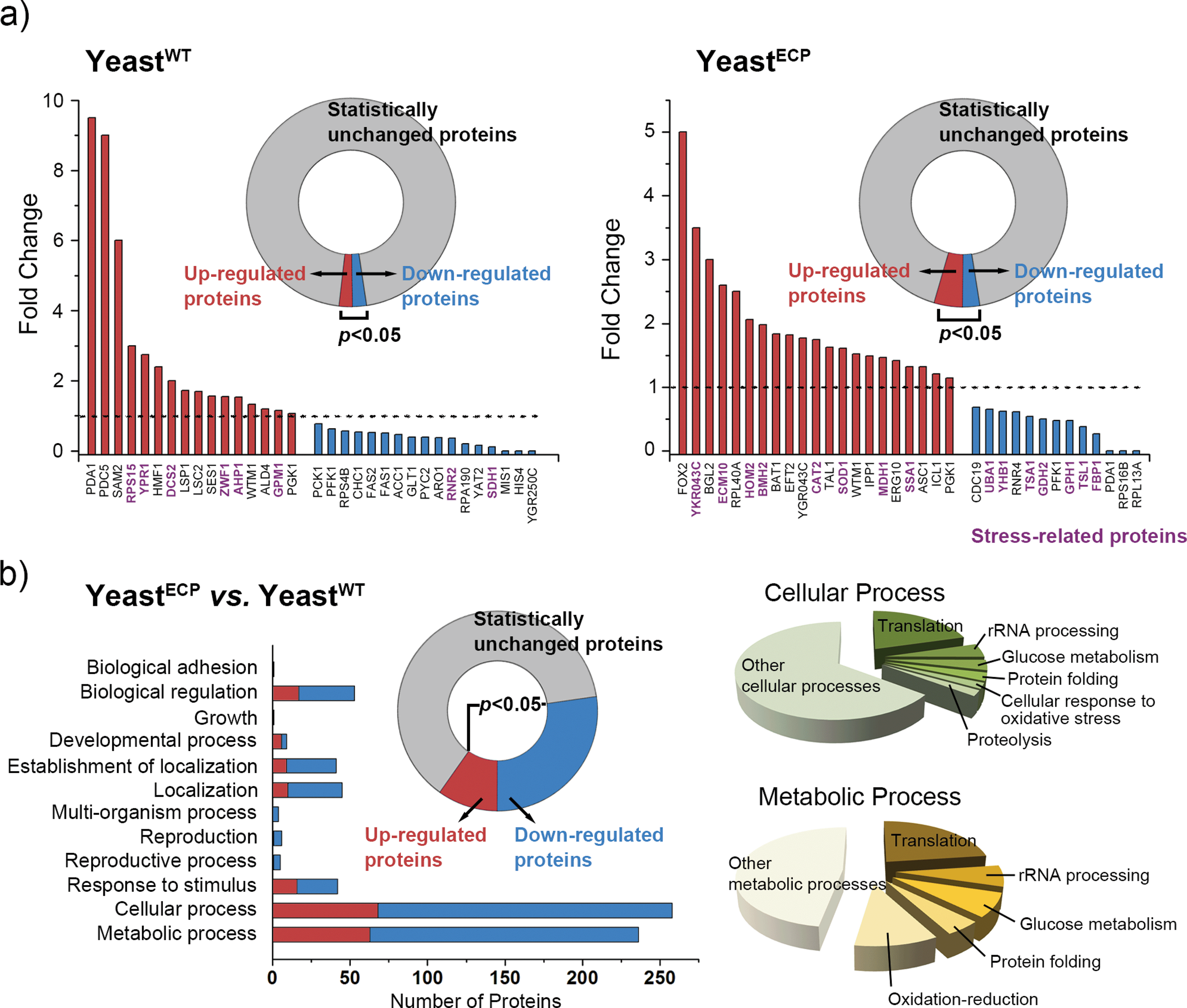 | ||
| Fig. 3 Mass spectrometry-based proteomic analysis of yeastWT and yeastECP. (a) Proteome-wide analysis of yeastWT and yeastECP after UV-C irradiation. The expression change was evaluated based on the fold change (Fc) value. The Fc value = spectral count of yeastECP/spectral count of yeastWT; up-regulated with 1 < Fc and down-regulated with 1 > Fc. The known stress-related proteins are denoted in purple. (b) Proteome-wide analysis of yeastECPvs. yeastWT before UV-C irradiation. Left bar graph: the number of changed proteins in each category of biological process. Right pie charts: sub-categorization of the cellular process or metabolic process. | ||
A comparative analysis between yeastWT and yeastECP before UV-C irradiation showed that 272 of the analyzed proteins (734) were expressed differently after encapsulation processes, and the majority of them were found to be involved in a cellular process or a metabolic process (Fig. 3b). The proteins that are involved in translation, rRNA processing, glucose metabolism, protein folding, and oxidation-reduction were found to be the main ones that changed (for the full data set for the cellular or metabolic processes, see ESI, Fig. S6†). Noticeably, the proteins that participate in protein synthesis (translation and rRNA processing) were all down-regulated in the case of yeastECP (compared with yeastWT), indicating that the majority of protein synthesis was arrested at least transiently during and after encapsulation processes (see ESI, Fig. S7†). While suppressing the protein synthesis, the yeastECP up-regulated many stress-related proteins in glucose metabolism (glyceraldehyde-3-phosphate dehydrogenase (TDH1 and TDH3), enolase (ENO1 and ENO2), and phosphoglycerate kinase (PGK1)),12 in protein folding (HSP104 and the HSP90 family (HSP82 and HSC82)),13 and in oxidation–reduction (thiol-specific antioxidants (PRX) and NADPH-generating enzymes during the pentose phosphate shuttle (ZWF1)) (see ESI, Fig. S7†).14 These data imply that the chemical processes for encapsulation act as a certain sub-lethal stress to yeast, and yeastECP might acquire biochemical flexibility for dealing with stresses more effectively. For example, ATP accumulation via a glycolytic pathway could be utilized effectively and synergistically for the activity of molecular chaperones.
Conclusions
In summary, this work showed that the multiple resistance of encapsulated cells could be achieved by cytocompatibly forming organic/inorganic, double-layered shells that mimic the hierarchical, multi-layered shells of bacterial endospores. The synergistic property-combination of poly(norepinephrine) and silica layers led to enhanced tolerance against enzymatic attack, desiccation, and UV-C irradiation. Of more importance is the new finding that the encapsulated cells were biochemically flexible in facing stresses. The mass spectrometry-based proteomic analysis implied that the encapsulation process did not harm the yeast cells but made them deal favorably with the stresses, presumably by inducing general stress-based responses and metabolic alterations. We believe that our work suggests a chemical strategy for manipulating and controlling cellular activities at the single-cell level, and also provides a platform for the practical development of single-cell based sensors, cell therapy, and tissue engineering by synergistically combining the properties of organic PN and inorganic silica.Experimental procedures
Materials
Dopamine hydrochloride (Sigma), DL-norepinephrine hydrochloride (≥97%, Aldrich), poly(ethyleneimine) (PEI, branched, Mn: ∼10000, Aldrich), tetramethyl orthosilicate (TMOS, ≥99%, Aldrich), (3-mercaptopropyl)trimethoxysilane (MPTMS, 95%, Aldrich), hydrochloride (HCl, 35%, Junsei), formic acid (Fluka), acetone (≥99.8%, Merck), acetonitrile (Burdick & Jackson), ammonium bicarbonate (NH4HCO3, Sigma), dithiothreitol (DTT, Sigma), iodoacetamide (Sigma), glycerol (≥99%, Sigma), dimethyl sulfoxide (DMSO, ≥99.9%, Sigma), lyticase (≥2000 units per mg protein, Sigma), fluorescein diacetate (FDA, Sigma), trypsin (Promega), 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories), N-(5-fluoresceinyl)maleimide (≥90%, Sigma), yeast-extract–peptone–dextrose broth (YPD broth, Duchefa Biochemistry), yeast-extract–peptone–dextrose agar (YPD agar, Duchefa Biochemistry), sodium chloride (NaCl, ≥99.5%, Jin Chemical Pharmaceutical), sodium phosphate dibasic (≥99%, Sigma-Aldrich), sodium phosphate monobasic (≥99%, Sigma-Aldrich), and tris(hydroxymethyl)aminomethane (Tris–HCl, pH 8.5, iNtRON Biotechnology), ethylenediaminetetraacetic acid (EDTA, Sigma), Tris–EDTA buffer solution (TE, Fluka), phenylmethylsulfonyl fluoride (PMSF, ≥99%, Sigma), protease inhibitor cocktail (Thermo Fisher Scientific), 3K-membrane filter (Thermo Fisher Scientific), glass beads (acid-washed, Sigma), NuPAGE® LDS sample buffer (4×, Life Technology), NuPAGE® sample reducing agent (10×, Life Technology), NuPAGE® Novex 4–12% bis-Tris protein gels (Life Technology), NuPAGE® MOPS SDS running buffer (20×, Life Technology), C18 Ziptips (Millipore), C18 reversed phase resin (Michrom Bioresources) were used as received. The NaCl solution was prepared by dissolving NaCl in distilled water (final concentration: 0.15 M).
Encapsulation of individual Saccharomyces cerevisiae (baker's yeast)
A single colony of yeast cells was picked from a YPD broth agar plate, suspended in a YPD broth, and cultured in a shaking incubator at 30 °C for 30 h. Aliquots of cells were washed with the NaCl solution and a Tris–HCl buffer solution to remove the YPD broth from the cells. The yeast cells were immersed in a Tris–HCl buffer solution of norepinephrine hydrochloride (2 mg mL−1) and shaken gently for 6 h. The resulting yeast cells coated with poly(norepinephrine) (yeast@PN) were washed with a Tris–HCl buffer solution. PEI was then grafted by immersing yeast@PN in a Tris–HCl buffer solution of PEI (2 mg mL−1) for 2 h. The PEI-grafted cells were washed with the Tris–HCl buffer solution, the NaCl solution, and the sodium phosphate buffer solution (50 mM, pH 5.8). For silicification, the PEI-grafted yeast@PN was placed in a 100 mM silicic acid derivative solution, which had been made by hydrolyzing TMOS (1 M) and MPTMS (1 M), respectively, in an aqueous HCl solution (1 mM) at room temperature for 20 min and adding the resulting solutions to a phosphate buffer (50 mM, pH 5.8) with 25:75:900 (v/v/v) ratio. After 30 min, the cells were washed with the NaCl solution.
Viability test
Cell viability was investigated by fluorescein diacetate (FDA) assay, where FDA was hydrolyzed to the green-fluorescent fluorescein by esterases in the metabolically active cells. The FDA stock solution (10 mg mL−1) was prepared in acetone due to the insolubility of FDA in water, and 2 μL of the stock solution was mixed with 1 mL of the sodium phosphate buffer solution (10 mM, pH 6.5) containing the yeast suspension. The mixture was incubated for 30 min at 30 °C, and then the cells were washed with the NaCl solution and characterized by confocal laser-scanning microscopy.Cell-lysis and cytoprotection experiments
For the cell-lysis test, a stock solution was prepared by dissolving lyticase (10 kU) in a mixture of glycerol (500 μL) and TE buffer solution (500 μL, pH 7.5). Then 10 μL of the stock solution was added to the yeast suspension (TE buffer solution, pH 7.5), and the suspension was placed in a shaking incubator at 37 °C. A small amount of the mixture was picked at the predetermined time, and the optical density was measured at 600 nm by UV-visible spectroscopy. For the UV-C irradiation experiment, yeastWT or yeastECP was suspended in the NaCl solution and transferred to a quartz tube. The quartz tube was placed in a sealed box, and UV light (254 nm, 4 W) was irradiated for 500 s. After irradiation, the cells were collected, and their viability was evaluated by the FDA assay. For the desiccation experiment, yeastWT or yeastECP was suspended in the NaCl solution, and the suspension was filtered through a hydrophilic membrane to remove the NaCl solution. The remaining cells were dried at 30 °C for 2 h, and the cells were then collected by washing with the NaCl solution, followed by the FDA assay to evaluate viability.Proteomic analysis
Cells were harvested at 4 °C by centrifugation (20 min, 14000 rpm). The harvested cell pellet was resuspended in a chilled lysis buffer (50 mM Tris–HCl (pH 8.0), 1% DMSO, 1 mM EDTA, 1 mM PMSF, 1× protease inhibitor cocktail) and vortexed 10 times with 80 mg of chilled glass beads for 1 min (the cells were kept on ice for 1 min between the vortexing steps). Glass beads and cell debris were removed at 4 °C by centrifugation (1 h, 14000 rpm). Desalted proteins from the supernatant and cell lysates were collected and dried with a speed vacuum dryer. The 27 μg of protein samples was mixed with a NuPAGE® LDS sample buffer and a NuPAGE® reducing agent. The mixture was heated at 100 °C for 5 min and loaded on a NuPAGE® Novex 4–12% bis-Tris gel. The gel was run at 150 V in a NuPAGE® MOPS SDS running buffer. The SDS-PAGE gel was removed from the cassette, rinsed 3 times with distilled water, incubated in a coomassie stain on a rocking table for 1 h, and rinsed with distilled water. The gel pieces containing the separated proteins were destained with an acetonitrile–water (1:1) solution of NH4HCO3 (50 mM) and vortexed until the coomassie stain was removed completely. The gel pieces were then dehydrated in acetonitrile and vacuum-dried for 20 min. For the digestion, the gel pieces were subjected to reduction conditions at 56 °C for 45 min by using an aqueous solution of DTT (10 mM) and NH4HCO3 (50 mM), followed by alkylation with an aqueous solution of iodoacetamide (55 mM) and NH4HCO3 (50 mM) for 30 min in the dark. Finally, each gel piece was treated with trypsin in the NH4HCO3-buffered aqueous solution (50 mM, pH 7.8) at 37 °C overnight. After digestion, the tryptic peptides were extracted in a 5% acetonitrile–water (1:1) solution of formic acid at room temperature for 20 min. The supernatants were collected and dried with a CentriVap® DNA centrifugal concentrator. The samples were purified and concentrated in an aqueous formic acid (0.1%) solution using C18 ZipTips before mass spectrometric (MS) analysis. The tryptic peptides were loaded onto a fused silica microcapillary column (12 cm × 75 μm) packed with C18 reversed phase resin (diameter: 5 μm; pore: 200 Å). Liquid chromatography (LC) separation was conducted under a linear-gradient (3–40% acetonitrile in a 0.1% aqueous formic acid solution with a flow rate of 250 nL min−1 for 60 min). The column was directly connected to a LTQ linear ion-trap mass spectrometer (Finnigan) equipped with a nano-electrospray ion source. The electrospray voltage was set to be 1.95 kV, and the threshold for switching from MS to MS/MS was 500. The normalized collision energy for MS/MS was 35% of the main radio frequency amplitude, and the duration of activation was 30 ms. All spectra were acquired in the data-dependent scan mode. Each full MS scan was followed by five consecutive MS/MS scans corresponding to the most intense to the fifth most intense peak of the full MS scan. The repeat count of peaks for dynamic exclusion was 1, and its repeat duration was 30 s. The dynamic exclusion duration was set to be 180 s, and the width of exclusion mass was ±1.5 Da. The list size of dynamic exclusion was 50. All MS/MS samples were analyzed by using Sequest (Thermo Fisher Scientific; version SRF v.5). Sequest was set up to search the SGD_Yeast_MaxQuant_Contaminants_FRFR.fasta.hdr database by assuming the digestion enzyme strict trypsin. Sequest was searched with a fragment ion mass tolerance of 1.00 Da and a parent ion tolerance of 2.0 Da. The carbamidomethyl group of cysteine was specified in Sequest as a fixed modification. The oxidation of methionine was specified in Sequest as a variable modification. Scaffold (version Scaffold_4.3.0, Proteome Software) was used to validate the MS/MS-based peptide and protein identifications. The protein identifications were accepted, if they could be established at greater than 99.0% probability to achieve a false discovery rate (FDR) less than 1.0% and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm. The proteins that contained similar peptides and could not be differentiated based on the MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins were annotated with GO terms from gene_association.sgd (downloaded on January 28, 2014).15
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (MSIP) (2012R1A3A2026403). We thank the Biomedical Mass Analysis Team (Diatech Korea Co., Ltd.) for the nano-LC-MS/MS analysis.Notes and references
- (a) M. T. Stephan, J. J. Moon, S. H. Um, A. Bershteyn and D. J. Irvine, Nat. Med., 2010, 16, 1035–1041 CrossRef CAS PubMed; (b) P. R. Leduc, M. S. Wong, P. M. Ferreira, R. E. Groff, K. Haslinger, M. P. Koonce, W. Y. Lee, J. C. Love, J. A. McCammon, N. A. Monteiro-Riviere, V. M. Rotello, G. W. Rubloff, R. Westervelt and M. Yoda, Nat. Nanotechnol., 2007, 2, 3–7 CrossRef CAS PubMed; (c) A. I. Zamaleeva, I. R. Sharipova, R. V. Shamagsumova, A. N. Ivanov, G. A. Evtugyn, D. G. Ishmuchametova and R. F. Fakhrullin, Anal. Methods, 2011, 3, 509–513 RSC; (d) R. F. Fakhrullin, J. García-Alonso and V. N. Paunov, Soft Matter, 2010, 6, 391–397 RSC; (e) W. Xiong, Z. Yang, H. Zhai, G. Wang, X. Xu, W. Ma and R. Tang, Chem. Commun., 2013, 49, 7525–7527 RSC; (f) B. Wang, P. Liu, Z. Liu, H. Pan, X. Xu and R. Tang, Biotechnol. Bioeng., 2013, 111, 386–395 CrossRef PubMed.
- (a) S. H. Yang, D. Hong, J. Lee, E. H. Ko and I. S. Choi, Small, 2013, 9, 178–186 CrossRef CAS PubMed; (b) D. Hong, M. Park, S. H. Yang, J. Lee, Y.-G. Kim and I. S. Choi, Trends Biotechnol., 2013, 31, 442–447 CrossRef CAS PubMed; (c) J. H. Park, S. H. Yang, J. Lee, E. H. Ko, D. Hong and I. S. Choi, Adv. Mater., 2014, 26, 2001–2010 CrossRef CAS PubMed; (d) R. F. Fakhrullin and Y. M. Lvov, ACS Nano, 2012, 6, 4557–4564 CrossRef CAS PubMed; (e) R. F. Fakhrullin, A. I. Zamaleeva, R. T. Minullina, S. A. Konnova and V. N. Paunov, Chem. Soc. Rev., 2012, 41, 4189–4206 RSC.
- (a) E. H. Ko, Y. Yoon, J. H. Park, S. H. Yang, D. Hong, K.-B. Lee, H. K. Shon, T. G. Lee and I. S. Choi, Angew. Chem., Int. Ed., 2013, 52, 12279–12282 CrossRef CAS PubMed; (b) X. Xu, B. Wang and R. Tang, ChemSusChem, 2011, 4, 1439–1446 CrossRef CAS; (c) G. Wang, L. Wang, P. Liu, Y. Yan, X. Xu and R. Tang, ChemBioChem, 2010, 11, 2368–2373 CrossRef CAS PubMed; (d) S. H. Yang, S. M. Kang, K.-B. Lee, T. D. Chung, H. Lee and I. S. Choi, J. Am. Chem. Soc., 2011, 133, 2795–2797 CrossRef CAS PubMed; (e) S. H. Yang, E. H. Ko, Y. H. Jung and I. S. Choi, Angew. Chem., Int. Ed., 2011, 50, 6115–6118 CrossRef CAS PubMed; (f) S. H. Yang, K.-B. Lee, B. Kong, J.-H. Kim, H.-S. Kim and I. S. Choi, Angew. Chem., Int. Ed., 2009, 48, 9160–9163 CrossRef CAS PubMed; (g) R. Kempaiah, S. Salgado, W. L. Chung and V. Maheshwari, Chem. Commun., 2011, 47, 11480–11482 RSC; (h) R. Magrassi, P. Ramoino, P. Bianchini and A. Diaspro, Microsc. Res. Tech., 2010, 73, 931–936 CAS.
- (a) G. Wang, R.-Y. Cao, R. Chen, L. Mo, J.-F. Han, X. Wang, X. Xu, T. Jiang, Y.-Q. Deng, K. Lyu, S.-Y. Zhu, E.-D. Qin, R. Tang and C.-F. Qin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7619–7624 CrossRef CAS PubMed; (b) G. Wang, X. Li, L. Mo, Z. Song, W. Chen, Y. Deng, H. Zhao, E. Qin, C. Qin and R. Tang, Angew. Chem., Int. Ed., 2012, 51, 10576–10579 CrossRef CAS PubMed; (c) X. Wang, Y. Deng, S. Li, G. Wang, E. Qin, X. Xu, R. Tang and C. Qin, Adv. Healthcare Mater., 2012, 1, 443–449 CrossRef CAS PubMed.
- W. L. Nicholson, N. Munakata, G. Horneck, H. J. Melosh and P. Setlow, Microbiol. Mol. Biol. Rev., 2000, 64, 548–572 CrossRef CAS.
- P. T. McKenney, A. Driks and P. Eichenberger, Nat. Rev. Microbiol., 2013, 11, 33–44 CrossRef CAS PubMed.
- S. Hong, J. Kim, Y. S. Na, J. Park, S. Kim, K. Singha, G.-I. Im, W. J. Kim and H. Lee, Angew. Chem., Int. Ed., 2013, 52, 9187–9191 CrossRef CAS PubMed.
- B. Wang, G. Wang, B. Zhao, J. Chen, X. Zhang and R. Tang, Chem. Sci., 2014, 5, 3463–3468 RSC.
- Y. V. Nancharaiah, M. Rajadurai and V. P. Venugopalan, Environ. Sci. Technol., 2007, 41, 2617–2621 CrossRef CAS.
- J. Lee, J. Choi, J. H. Park, M.-H. Kim, D. Hong, H. Cho, S. H. Yang and I. S. Choi, Angew. Chem., Int. Ed., 2014, 53, 8056–8059 CrossRef CAS PubMed.
- S. Lindquist and E. A. Craig, Annu. Rev. Genet., 1988, 22, 631–677 CrossRef CAS PubMed.
- W. H. Mager and P. M. Ferreira, Biochem. J., 1993, 290, 1–13 CAS.
- J. R. Glover and S. Lindquist, Cell, 1998, 94, 73–82 CrossRef CAS.
- D. J. Jamieson, Yeast, 1998, 14, 1511–1527 CrossRef CAS.
- A. I. Nesvizhskii, A. Keller, E. Kolker and R. Aebersold, Anal. Chem., 2003, 75, 4646–4658 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, and other additional details. See DOI: 10.1039/c4sc02789b |
| This journal is © The Royal Society of Chemistry 2015 |