
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn organometallic structure-activity relationship study reveals the essential role of a Re(CO)3 moiety in the activity against gram-positive pathogens including MRSA†
Malay
Patra
a,
Michaela
Wenzel
c,
Pascal
Prochnow
c,
Vanessa
Pierroz
b,
Gilles
Gasser
b,
Julia E.
Bandow
*c and
Nils
Metzler-Nolte
*a
aLehrstuhl für Anorganische Chemie I – Bioanorganische Chemie, Fakultät für Chemie und Biochemie, Ruhr-Universität Bochum, Universitätsstrasse 150, D-44801 Bochum, Germany. E-mail: Nils.Metzler-Nolte@rub.de; Fax: +49-234-32-14378
bDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
cRuhr-Universität Bochum, Biologie der Mikroorganismen, Arbeitsgruppe Mikrobielle Antibiotikaforschung, Universitätsstrasse 150, D-44801 Bochum, Germany. E-mail: julia.bandow@rub.de; Fax: +49-234-32-14620; Tel: +49-234-32-23102
First published on 13th October 2014
Abstract
The worrying appearance of microbial resistance to antibiotics is a worldwide problem which needs to be tackled urgently. Microbial resistance to the common classes of antibiotics involving purely organic compounds unfortunately develops very rapidly and in most cases, resistance was detected soon after or even before release of the antibiotic to the market. Therefore, novel concepts for antibiotics must be investigated, and metal-containing compounds hold particular promise in that area. Taking a trimetallic complex (1a) which contains a ferrocenyl (Fc), a CpMn(CO)3 (cymantrene) and a [(dpa)Re(CO)3] residue as the lead structure, a systematic structure–activity relationship (SAR) study against various gram-positive pathogenic bacteria including methicillin-resistant Staphylococcus aureus (MRSA) strains was performed. The [(dpa)Re(CO)3] moiety was discovered to be the essential unit for the observed antibacterial activity of 1a. The ferrocenyl and CpMn(CO)3 units can be replaced one by one or both together by organic moieties such as a phenyl ring without loss of antibacterial activity. The most potent mono-metallic complex (9c′) has an antibacterial activity comparable to the well-established organic drugs amoxicillin and norfloxacin and importantly, only moderate cytotoxicity against mammalian cells. Microbiological studies on membrane potential, membrane permeabilization, and cell wall integrity revealed that 9c′ targets the bacterial membrane and disturbs cell wall integrity, but shows more efficient membrane permeabilization than the lead structure 1a.
Introduction
The steady increase of microbial resistance to commercial drugs is a serious health issue worldwide. One pathogen of high clinical importance is methicillin-resistant Staphylococcus aureus (MRSA), which often is not only resistant to methicillin but also to other clinically used antibiotics such as gentamycin, erythromycin, and neomycin.1,2 The treatment of bacterial infections with such multi-resistant germs is therefore highly difficult. Even in developed countries like the US, over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 life-threatening MRSA infections were reported in 2005.3 According to a report by the European Centre of Disease Control and Prevention, MRSA has reached an average prevalence of 50% in Europe.4 Microbial resistance to conventional organic antibiotics often develops and spreads amazingly fast.1 In the case of several antibiotics like linezolid or some β-lactams, resistance was observed already before the compound was commercialized for human use.5–7 In parallel with the first occurrence of noticeable antimicrobial resistance in the 1990s, the annual approval rate of antibiotic therapeutics decreased steadily. Currently, on average, only one antibiotic per year reaches the market.1,8 The most recent data on compounds in clinical trials in stages I–III have been compiled in an excellent review article.9 Although the concurrence of growing antimicrobial resistance and the decline of newly approved antibiotics may be accidental, in effect the number of new antibiotics that have been discovered or commercialized in the last two decades is insufficient to control infections with multi-resistant pathogens. These facts clearly emphasize the need for new classes of antimicrobial compounds. The best candidates for resistance-breaking antibacterials are compounds which either possess completely new modes of action, target more than one bacterial structure, or attack essential structures that are less prone to target mutation such as the bacterial cell envelope.10
000 life-threatening MRSA infections were reported in 2005.3 According to a report by the European Centre of Disease Control and Prevention, MRSA has reached an average prevalence of 50% in Europe.4 Microbial resistance to conventional organic antibiotics often develops and spreads amazingly fast.1 In the case of several antibiotics like linezolid or some β-lactams, resistance was observed already before the compound was commercialized for human use.5–7 In parallel with the first occurrence of noticeable antimicrobial resistance in the 1990s, the annual approval rate of antibiotic therapeutics decreased steadily. Currently, on average, only one antibiotic per year reaches the market.1,8 The most recent data on compounds in clinical trials in stages I–III have been compiled in an excellent review article.9 Although the concurrence of growing antimicrobial resistance and the decline of newly approved antibiotics may be accidental, in effect the number of new antibiotics that have been discovered or commercialized in the last two decades is insufficient to control infections with multi-resistant pathogens. These facts clearly emphasize the need for new classes of antimicrobial compounds. The best candidates for resistance-breaking antibacterials are compounds which either possess completely new modes of action, target more than one bacterial structure, or attack essential structures that are less prone to target mutation such as the bacterial cell envelope.10
It was earlier shown that derivatization with organometallic fragments is able to overcome the resistance problem of conventional organic drug candidates.11 The rationale behind this idea is that the metal-based compounds might offer metal-specific modes of action not available for organic drugs.12,13 This strategy was shown to be very successful for the antimalarial drug candidate chloroquine (CQ). Ferroquine (FQ, Fig. 1) is a ferrocene derivative of CQ. FQ is not only capable of overcoming the resistance problem of CQ but also showed enhanced antimalarial activity compared to CQ.14 Mechanistic investigation suggested that the activity of FQ against the CQ-resistant P. falciparum is related to increased lipophilicity, change in basicity, and introduction of redox-properties into the molecule by the ferrocene moiety.15,16
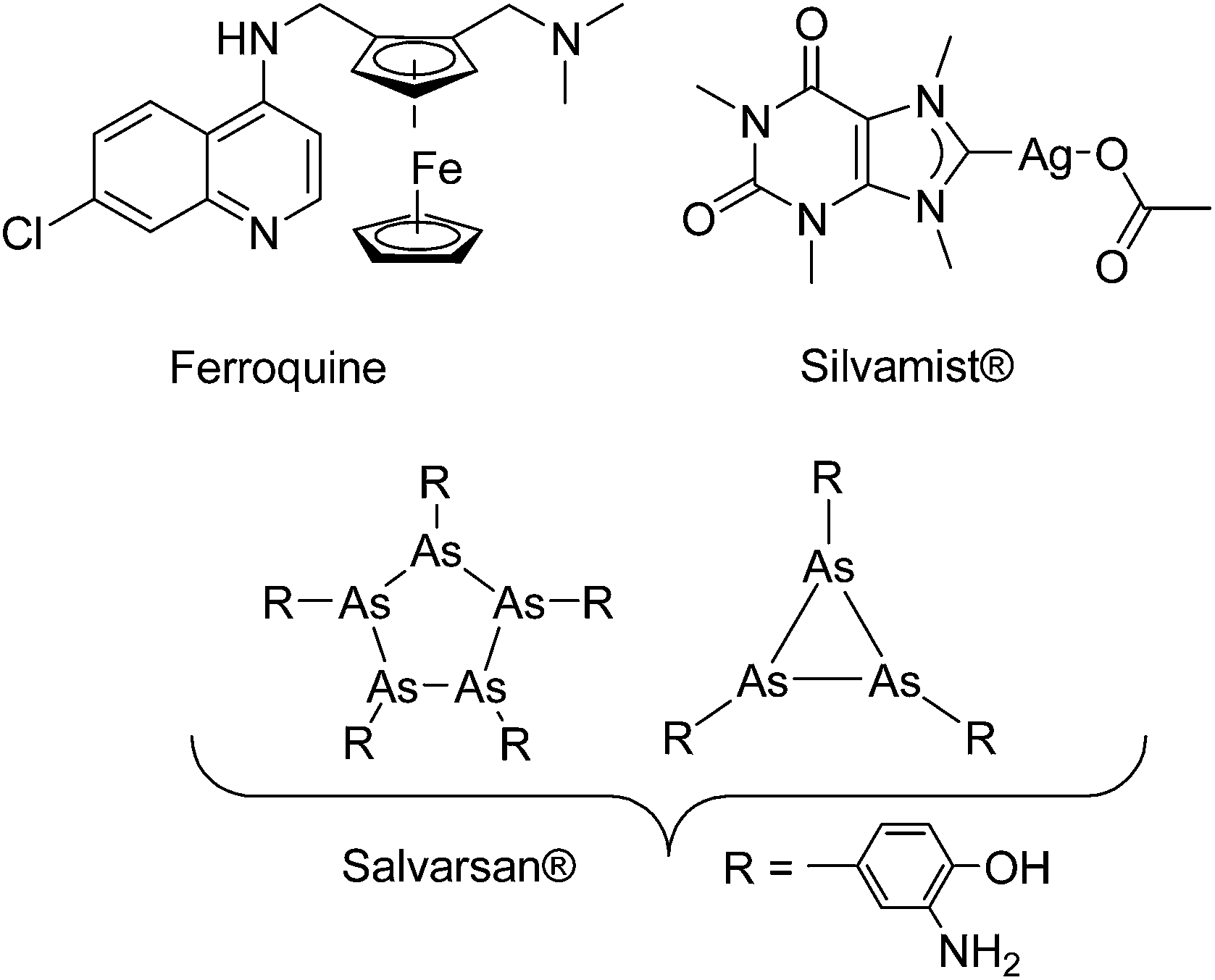 | ||
| Fig. 1 Structures of a few organometallic antimalarial and antibacterial drug candidates (Ferroquine and Silvamist©) or drugs (Salvarsan).17 | ||
Despite promising results in anticancer and antimalarial research,11,13,14,18 recent research into organometallic compounds with antibacterial activity is quite limited,19 although their potential for treatment of infectious diseases was already proven in the early 1900s by the discovery of salvarsan (Fig. 1), an organoarsenical antimicrobial agent.20,21 Importantly, at that time, salvarsan was the only treatment option available for the deadly bacterial infection Syphilis. As an exception, silver complexes with N-heterocyclic carbene (NHC) ligands are emerging as a class of promising compounds.22,23 For example Silvamist (Fig. 1), a caffeine derived Ag–NHC complex, exhibits high activity against tobramycin-resistant pathogenic bacteria in vitro as well as in vivo.22,24
Since a few years, our groups are involved in the development of organometallic-containing antibacterial agents.19,25–32 Very recently, we discovered the potent antibacterial activity of the hetero-tri-organometallic compound 1a (see Table 1 for structure).33 Being a structurally new class of antibacterial agents, 1a inhibits the growth of gram-positive bacteria, including MRSA, at a minimal inhibitory concentration (MIC) of 2 μg mL−1 (1.4 μM, Table 1). The activity of 1a against MRSA is 90 times higher than that of the commercial antibacterial agent amoxicillin (MIC 1/4 48 mg mL−1, 131 mM), and comparable to that of norfloxacin (MIC 1/4 0.5 mg mL−1, 1.6 mM). A ruthenocene derivative, 1b (see Table 1 for its structure), was found to be slightly less active than 1a. A mechanism-of-action study revealed that both compounds interact strongly with the bacterial membrane and interfere with membrane-associated processes such as cell wall biosynthesis and respiration.33 Additionally, 1a induces oxidative stress in bacteria, which is not the case for 1b. The solubility of both compounds was around 25 μg mL−1 in aqueous solution, thereby limiting pharmacological evaluation of the compounds. However, due to its impressive antibacterial activity, we decided to rationalize the activity of 1a. In addition, we aimed at obtaining easy-to-synthesize derivatives with higher solubility while retaining or improving antibacterial potency and selectivity over mammalian cells. Also, we considered it crucial to understand the importance of each organometallic moiety present in 1a, namely ferrocene, CpMn(CO)3, and [(dpa)Re(CO)3], and to define their individual contribution to antibacterial activity.
We now report on the synthesis and biological activities of a series of non-, mono-, and bi-metallic compounds derived from the structure of 1a. This study allows us to identify the individual contribution of each organometallic component to the antibacterial activity of this class of molecules.
Results and discussion
Synthesis and spectroscopic characterization
Compounds 9a–c were synthesized following a similar synthetic route to that previously reported for 1a (Scheme 1).34 As a representative example, the synthesis of 9c started with the CuI-catalyzed [3 + 2] cycloaddition reaction35 of azidomethyl benzene to the side chain of the alkyne-containing peptide nucleic acid (PNA) building block 2.36 Compound 3 was isolated in 82% yield. Formation of 3 was confirmed by the presence of the characteristic singlet from the triazole ring at 5.30 ppm (major isomer) and 5.39 ppm (minor isomer) in its 1H NMR spectrum. The presence of two rotamers around the central amide bond, usually denoted major (maj) and minor (min), is a signature for this type of compounds.37 Treatment of 3 with TFA, followed by amide coupling with p-iodo-aniline gave 5b in 52% yield. The Fmoc-group of 5b was then cleaved with 20% piperidine in DMF. HATU-mediated peptide coupling of the resulting residue with carboxylic acid 6a yielded compound 7c. A signal at 199 ppm for the keto carbonyl group present in the 13C NMR spectrum of 7c confirmed the presence of the expected compound. The last synthetic step involved the insertion of the alkyne-substituted (dpa)Re(CO)3 complex (8)34 into the iodo-substituted aromatic ring of 7c by Pd-catalyzed Sonogashira coupling to give compound 9c. The 13C NMR spectrum of 9c showed a signal at 195 ppm (rotamers present) corresponding to the Re(CO)3 moiety present. Furthermore, a peak at m/z = 1071.9 for the [M-PF6]+ species confirmed the formation of 9c. After silica column chromatography, 9c was further purified by semi-preparative RP-HPLC to remove trace amounts of impurity. The counter anion of 9c was therefore changed to CF3COO− and the compound was designated as 9c′. All new compounds were characterized by NMR, ESI-mass spectrometry, and IR spectroscopy. It is worth mentioning that all intermediates as well as the final compounds showed the presence of rotamers in solution leading to a sometimes tedious assignment of 1H and 13C NMR signals. Compounds 5a, 6b, 7d were synthesized following literature procedures.34,38 The purity of all compounds used for biological activity test was checked by reverse phase analytical HPLC and confirmed to be ≥95%.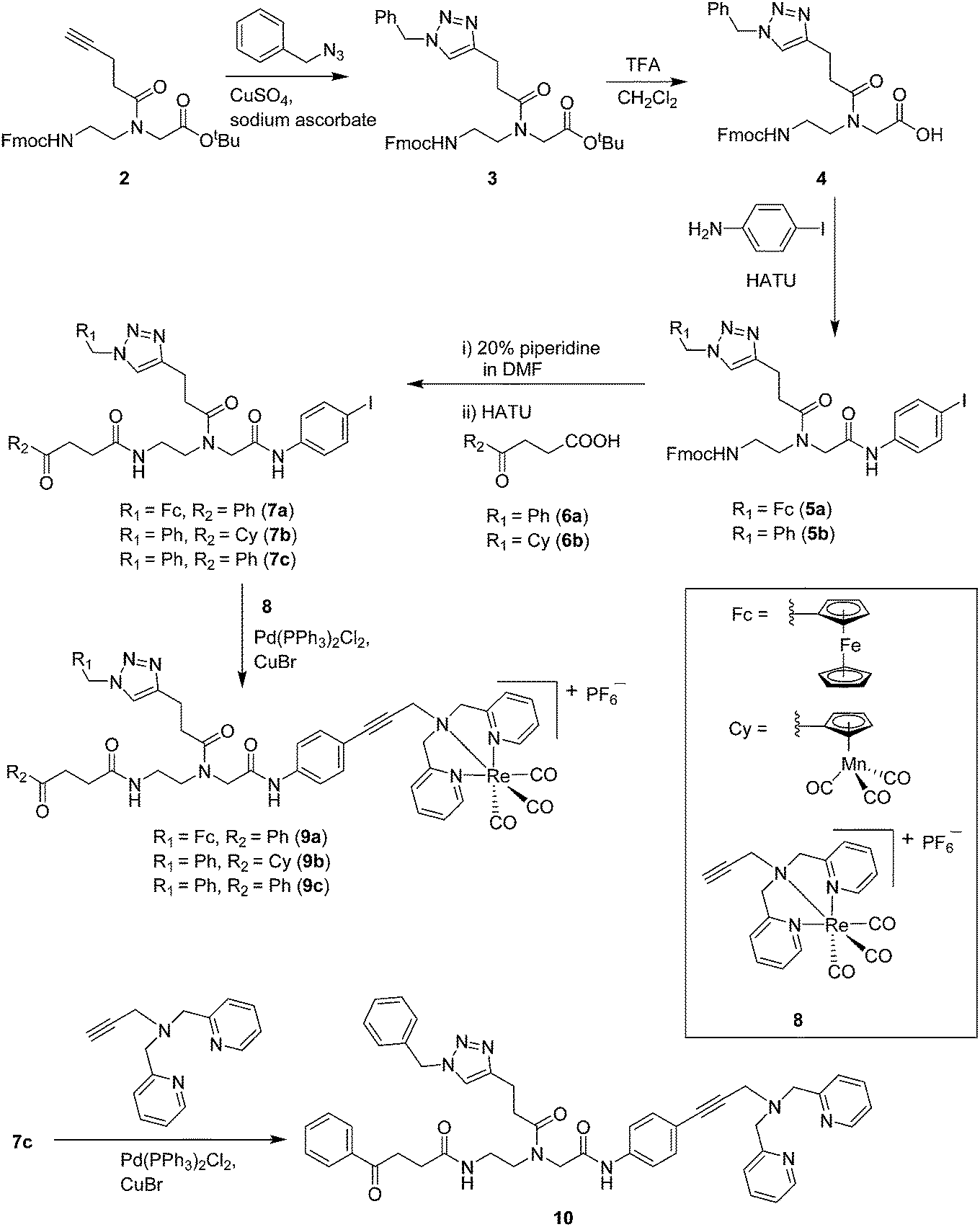 | ||
| Scheme 1 Synthesis of mono- and bi-metallic, as well as metal-free compounds used in this study. | ||
Antimicrobial activity
The antimicrobial activities of all newly synthesized non-, mono-, and bi-metallic derivatives were tested against gram-positive Bacillus subtilis and Staphylococcus aureus strains including an MRSA strain. The results are listed in Table 1. As reported recently, the tri-metallic 1a inhibited growth of gram-positive bacteria at MICs of 2 μg mL−1.33 The analogous tri-metallic compound 1b, in which the ferrocene moiety is replaced by a ruthenocene unit, was found to be slightly less active (MICs = 4–32 μg mL−1).33 Thus, oxidative stress by ferrocene seems to enhance antibacterial potency but is not the only component determining antibiotic activity.33 This is consistent with the similar modes of action of both tri-metallic compounds targeting the bacterial membrane.33 Among the three bi-metallic derivatives, 7d lacks the [(dpa)Re(CO)3] moiety but contains the ferrocenyl and CpMn(CO)3 moieties. Compounds 9a and 9b have the [(dpa)Re(CO)3] moiety in common. However, 9a contains a ferrocenyl moiety while 9b contains the CpMn(CO)3. When tested for antibacterial activity, 7d was found to be inactive up to 512 μg mL−1. However, both derivatives 8a and 8b exhibited potent antibacterial activity with MIC values in the range of 2–4 μg mL−1. The mono-metallic compounds lacking the [(dpa)Re(CO)3] moiety (5a, 7a, 7b) were also inactive. In contrast, the mono-metallic derivative 9c′, which contains the [(dpa)Re(CO)3] as the only organometallic residue while both ferrocenyl and CpMn(CO)3 were replaced by phenyl groups, was found to retain full antibacterial activity. It inhibits the growth of MRSA at a concentration of 2 μg mL−1 (Table 1). The activity is comparable to that of the parent compound 1a and the bi-metallic derivatives 9a and 9b. These results suggest that neither the ferrocene nor the CpMn(CO)3 moiety is essential, but the presence of the [(dpa)Re(CO)3] moiety is indispensable for antibacterial activity. On the other hand, the alkyne-substituted-[(dpa)Re(CO)3] complex 8 was found to be inactive up to 512 μg mL−1,33 demonstrating that the organometallic moiety alone is not sufficient for antibiotic activity but needs the pseudo-peptide backbone. The purely organic intermediate compounds 5b and 7c were also inactive. We then synthesized compound 10, the purely organic ligand of the active mono-metallic derivative 9c′. Compound 10 was found to have no antibacterial activity. This observation emphasizes that the organometallic {Re(CO)3} fragment is crucial for 9c′ to show antibacterial activity. Based on these results, we suggest that neither ferrocene nor CpMn(CO)3 but the {Re(CO)3} residue together with the pseudo-peptide backbone is essential for antibacterial activity. It is worth mentioning that none of the active compounds 1a, 1b, 9a, 9b, and 9c′ showed activity against gram-negative Pseudomonas aeruginosa, Escherichia coli, or Acinetobacter baumannii. This might be due to the presence of the outer membrane of gram-negative bacteria, which hinders access of many compounds to the cytoplasmic membrane where 1a exerts its activity.| Compound | Structure | MIC | |||||
|---|---|---|---|---|---|---|---|
| B. subtilis | S. aureus DSM 20231 | S. aureus ATCC43300 (MRSA) | |||||
| μg mL−1 | μM | μg mL−1 | μM | μg mL−1 | μM | ||
| 1a (ref. 33) |
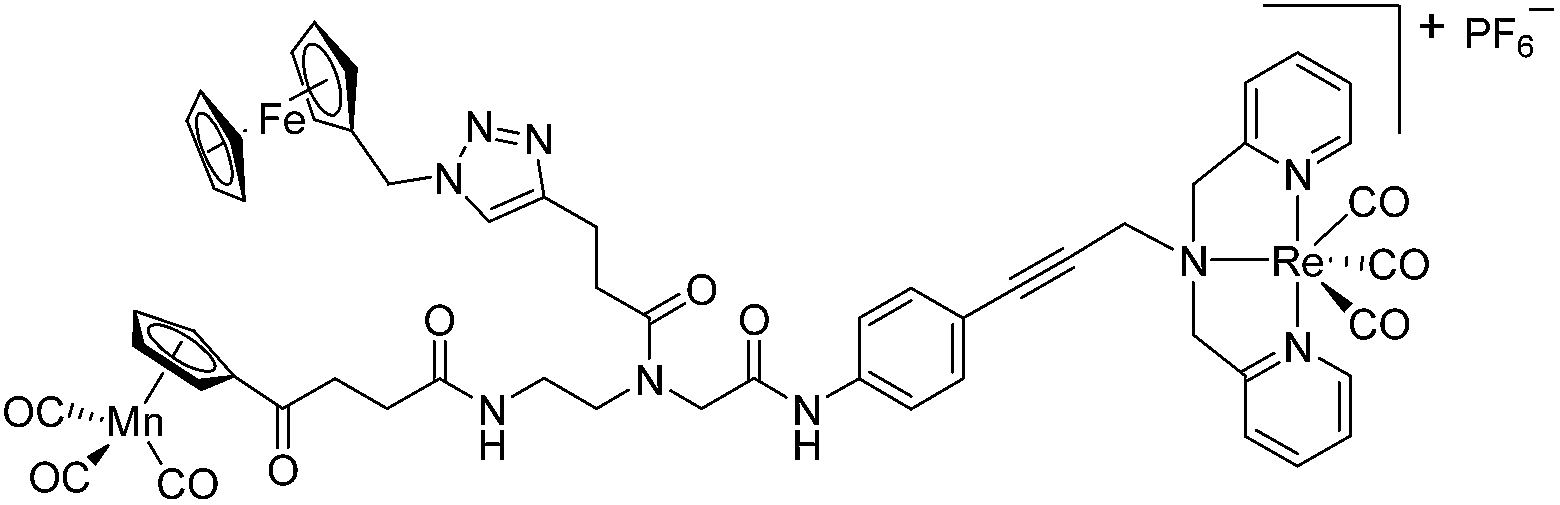
|
2 | 1.4 | 2 | 1.4 | 2 | 1.4 |
| 1b (ref. 33) |
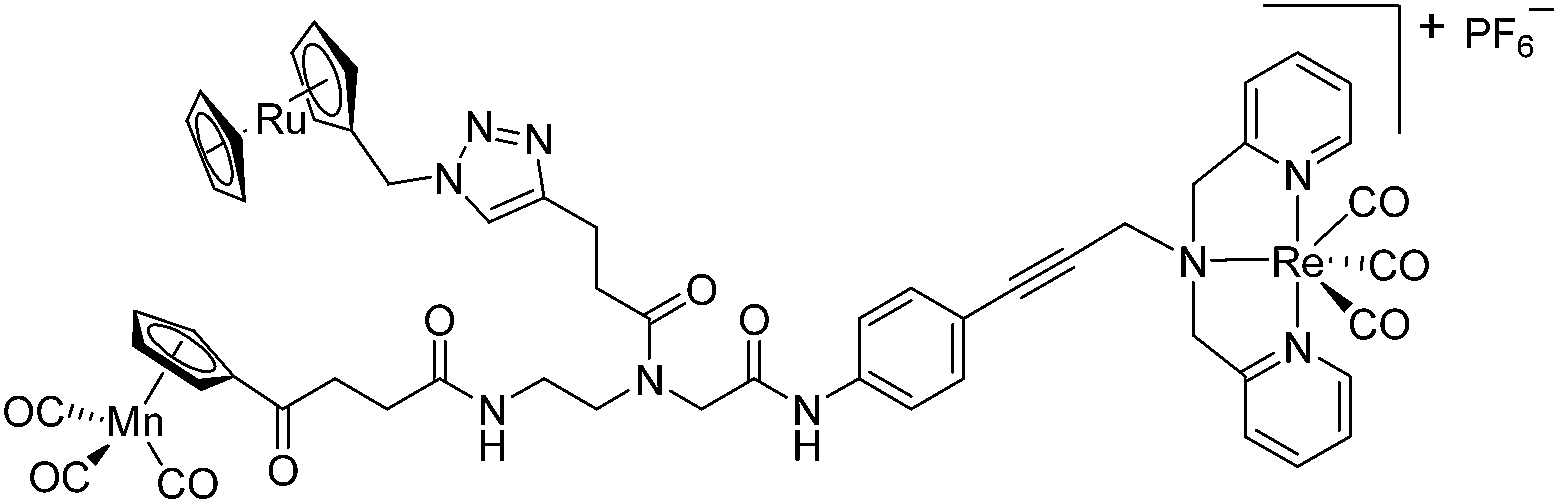
|
32 | 21 | 4 | 2.7 | 6 | 4 |
| 7d |
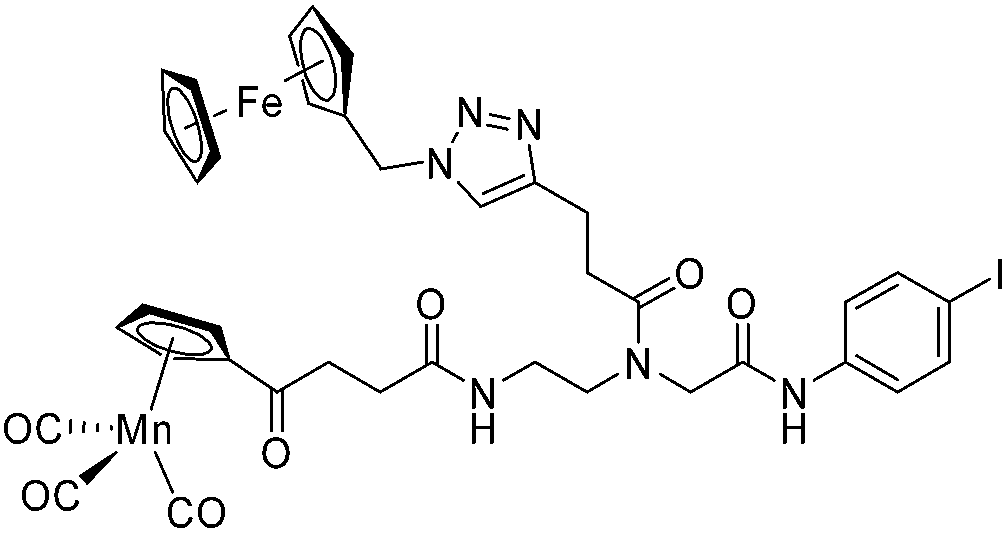
|
ia | ia | ia | |||
| 9a |
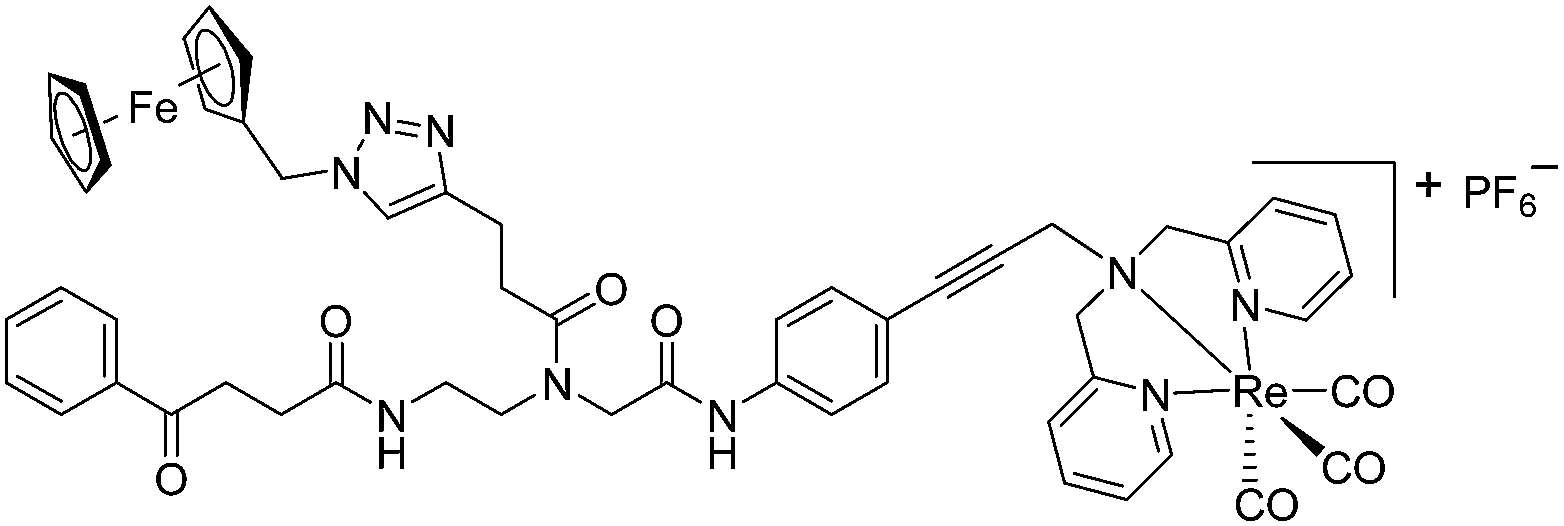
|
4 | 3 | 2 | 1.5 | 2 | 1.5 |
| 9b |
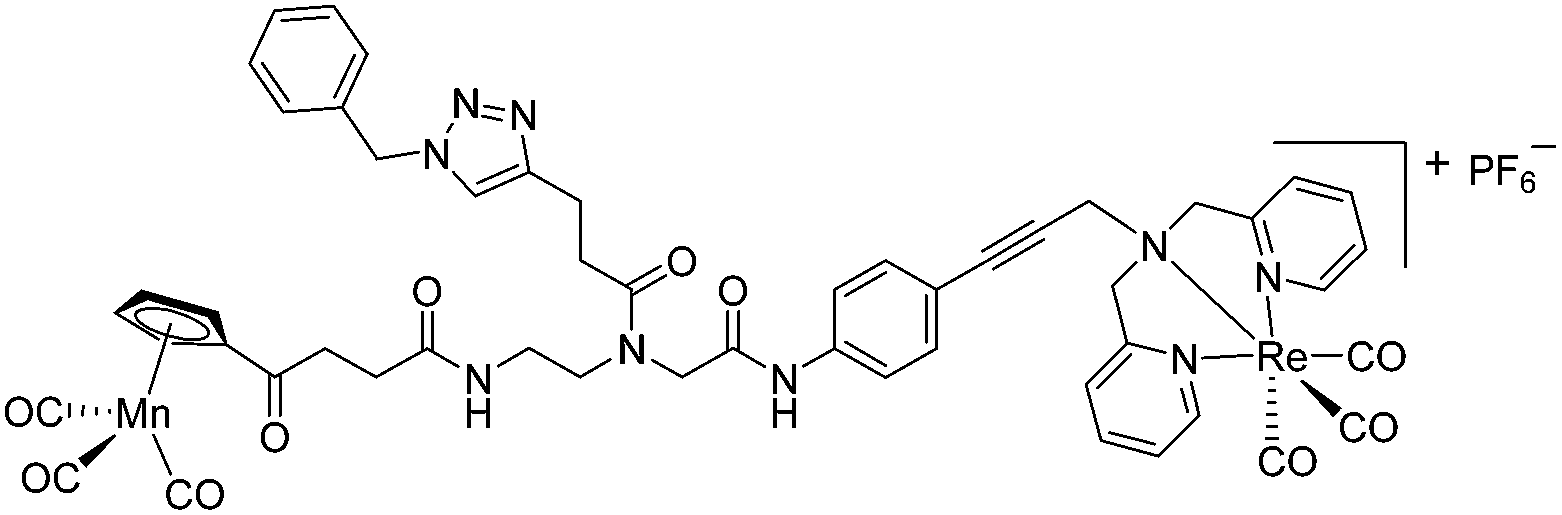
|
4 | 2.9 | 4 | 2.9 | 2 | 1.5 |
| 5a |
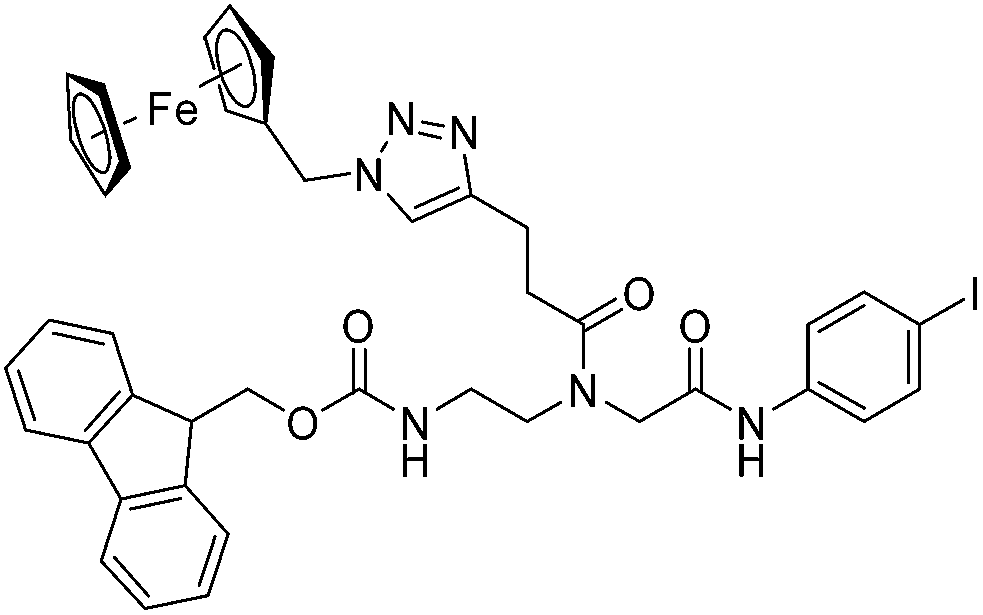
|
ia | ia | ia | |||
| 7a |
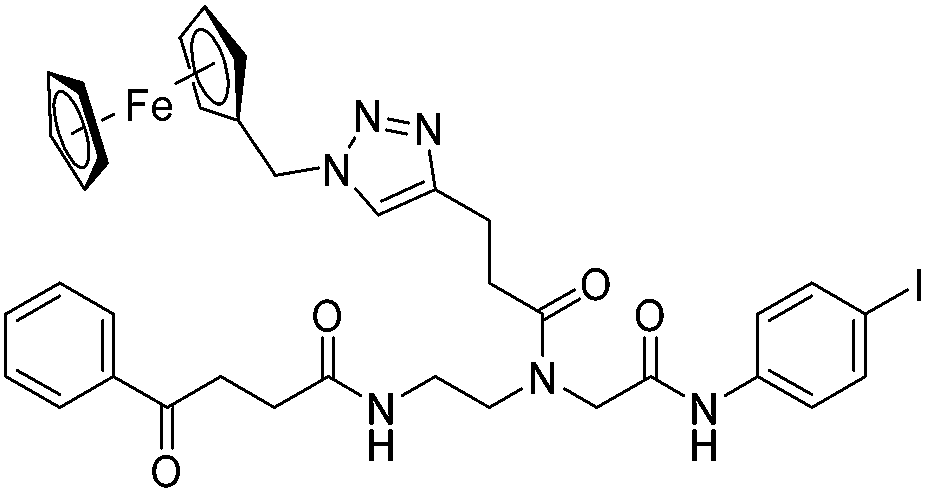
|
ia | ia | ia | |||
| 7b |
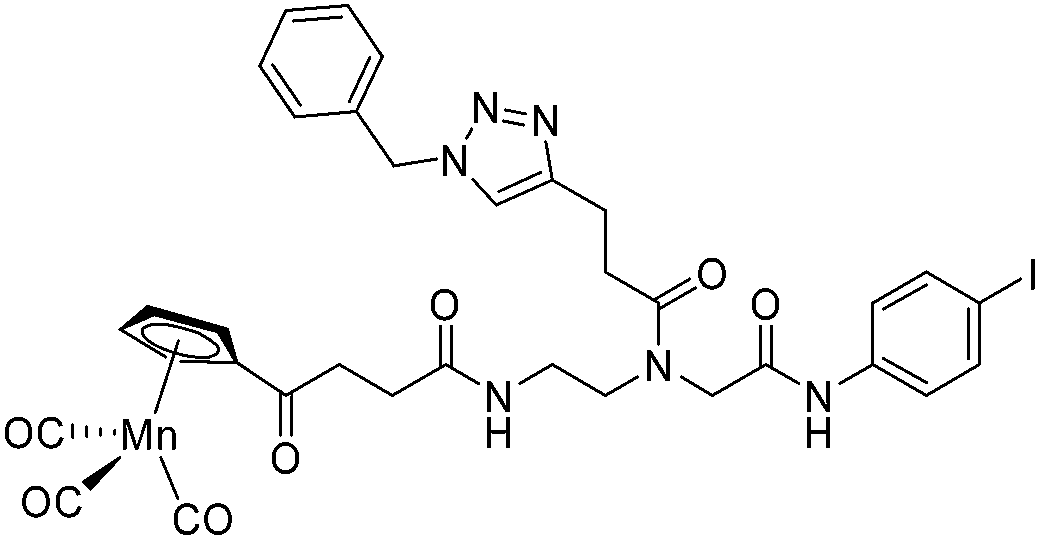
|
ia | ia | ia | |||
| 9c′ |
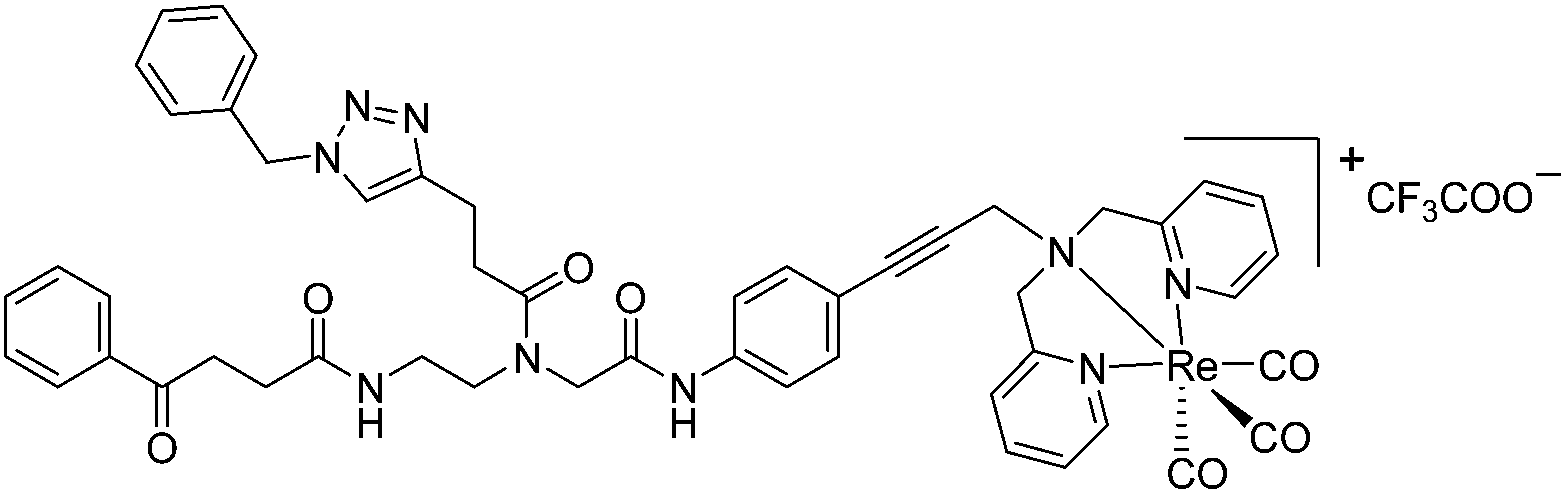
|
4 | 3.3 | 2 | 1.6 | 2 | 1.6 |
| 8 (ref. 33) |
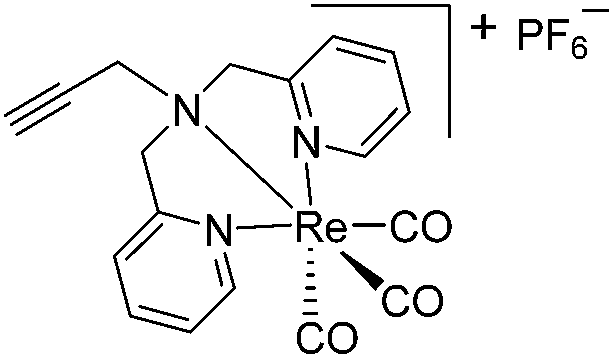
|
ia | ia | ia | |||
| 5b |
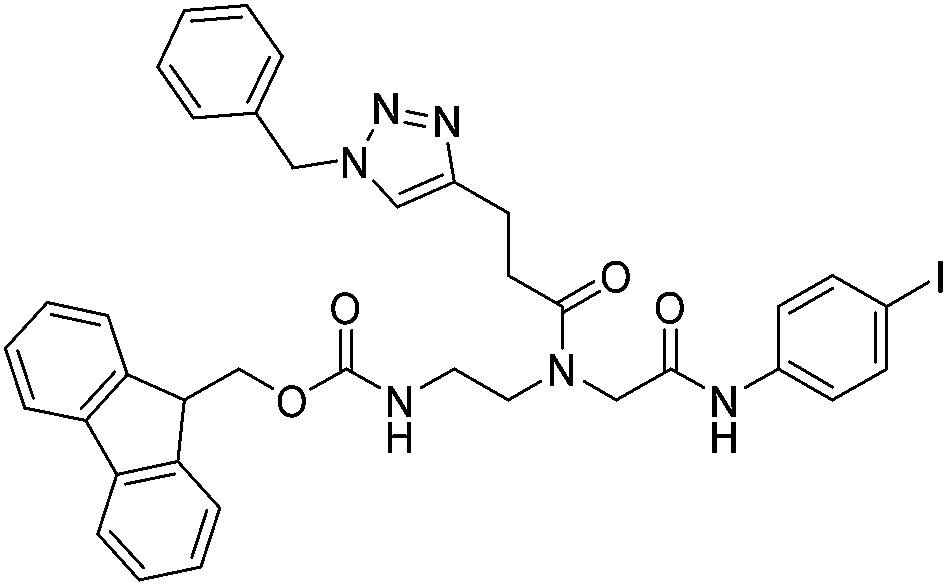
|
ia | ia | ia | |||
| 7c |
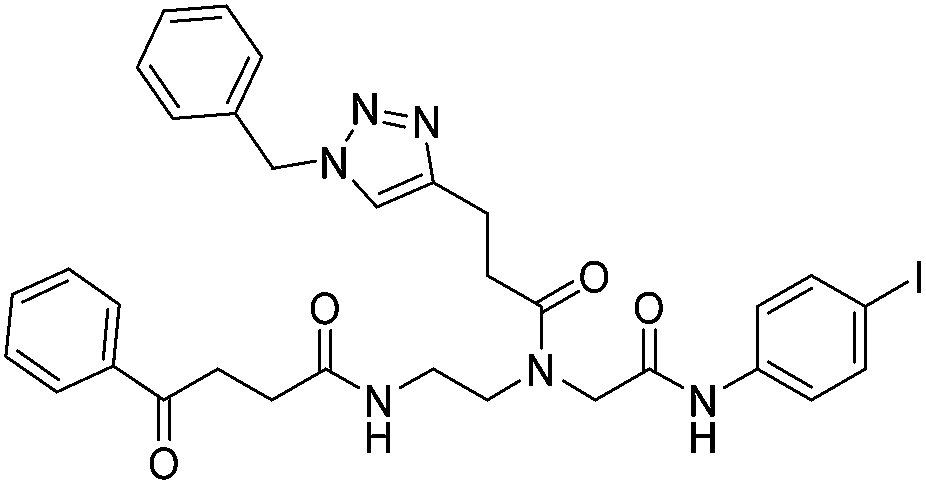
|
ia | ia | ia | |||
| 10 |
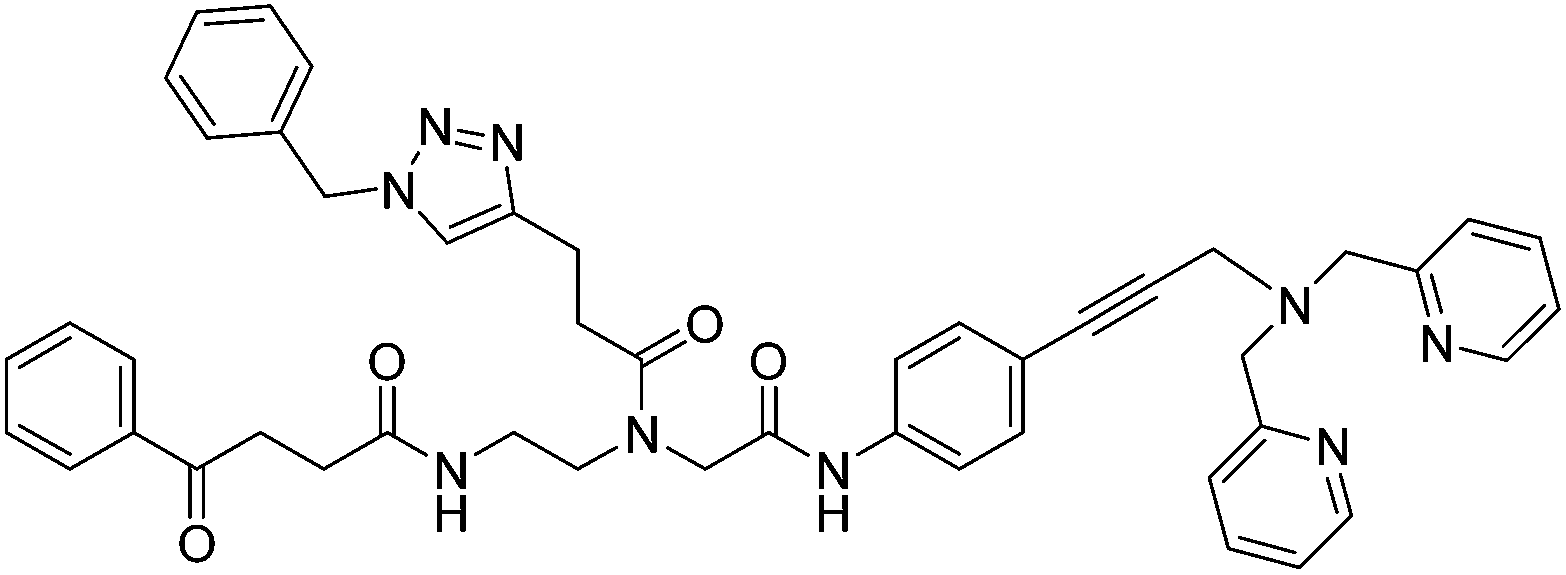
|
ia | ia | ia | |||
| Amoxicillin33 | 3 | 8.2 | 2 | 5.5 | 48 | 131 | |
| Norfloxacin33 | 1 | 3.1 | 0.5 | 1.6 | 0.5 | 1.6 | |
Determination of solubility
Maximum solubility of the active derivatives was determined in Belitzky minimal medium (BMM), a chemically defined bacterial culture medium, and in Dulbecco's modified Eagle's medium (DMEM), a standard growth medium for mammalian cells (Table 2). Starting from 10 mg mL−1 stock solutions of the compounds in DMSO, dilutions ranging from 1 μg mL−1 to 1 mg mL−1 were prepared in either BMM or DMEM. After 10 min incubation at room temperature, samples were centrifuged and checked for precipitated compounds visually and by light microscopy (for micro-precipitates). The highest compound concentration not leading to visible precipitates was defined as maximum solubility. Compared to the tri-metallic 1a, the di- and mono-metallic derivatives displayed two-fold higher solubility in DMEM. In BMM, solubility of the di-metallic compounds 9a and 9b was increased 4-fold. Solubility of the mono-metallic compound 9c′ was even 12-fold higher compared to that of 1a.| Compounds | In BMM (μg mL−1) | In DMEM (μg mL−1) |
|---|---|---|
| 1a | 25 | 25 |
| 9a | 100 | 50 |
| 9b | 100 | 50 |
| 9c′ | 300 | 50 |
Determination of toxicity against mammalian cell lines
A good antibacterial compound should be selective towards bacteria over mammalian cells. To obtain preliminary insight into the toxicity, the active compounds (9a–b and 9c′) were tested against two different mammalian cancerous (HeLa and HepG2) and a non-cancerous (MRC-5) cell lines. A metal-based anticancer drug, cisplatin, was used as positive control (Table 3). As reported for 1a,33 cytotoxicity was found to be highly dependent on the cell lines. For the tri-metallic 1a as well as the bi-metallic 9a and 9b, IC50 values of 11–14 μg mL−1 (8–10 μM), i.e. comparable to cisplatin were determined against HeLa cells. In contrast, the same compounds showed moderate toxicity against MRC-5 and were non-toxic to HepG2 cells up to 100 μM. However, 1a, 9a and 9b are highly active against gram-positive bacteria at a concentration of 2–4 μg mL−1 (1.4–3 μM). Interestingly, the mono-metallic derivative 9c′ exhibits only moderate toxicity towards all mammalian cell lines studied in this work with IC50 values of 31–63 μg mL−1 (26–52 μM), which is 15–30 times higher than the MIC against MRSA (1.6 μM, 2 μg mL−1). All in all, these results imply that the mono-metallic compound 9c′ is not only highly selective for gram-positive over gram-negative bacteria but also by at least one order of magnitude less toxic to mammalian than to bacterial cells.| Compounds | IC50 values | |||||
|---|---|---|---|---|---|---|
| HeLa | HepG2 | MRC-5 | ||||
| μg mL−1 | μM | μg mL−1 | μM | μg mL−1 | μM | |
| 1a | 12.7 ± 1 | 7.8 ± 1.9 | >145 | >100 | >145 | >100 |
| 9a | 13.5 ± 2.6 | 10.2 ± 2.0 | >133 | >100 | 30.2 ± 4.1 | 22.8 ± 3.1 |
| 9b | 10.7 ± 1.3 | 8.0 ± 1.0 | >134 | >100 | Not determined | |
| 9c′ | 31.2 ± 0.9 | 26.3 ± 0.8 | 62.5 ± 11.5 | 52.7 ± 9.7 | 43.7 ± 5.9 | 36.9 ± 5.0 |
| Cisplatin | 3.5 ± 0.9 | 11.5 ± 2.9 | 1.6 ± 0.01 | 5.5 ± 0.5 | 2.4 ± 0.4 | 7.9 ± 1.2 |
Influence of 9a, 9b and 9c′ on the bacterial cell envelope
We showed that the bacterial membrane is the target structure of compound 1a.33 Binding of 1a to the membrane leads to loss of the membrane potential resulting in substantial energy limitation. Depolarization is thereby thought to be caused by interference with membrane-bound processes like respiration. 1a has also been shown to affect membrane-bound steps of cell wall biosynthesis, which is observed after treatment with a number of membrane-integrating compounds, e.g. some lantibiotics.12 In order to establish whether the active derivatives 9a, 9b, and 9c′ still have the same antibacterial mechanism, we tested their influence on membrane potential, membrane permeabilization, and cell wall integrity. The antimicrobial peptide nisin, which binds to cell wall precursor lipid II and forms membrane pores, was used as a positive control.39Depolarization was indirectly monitored in B. subtilis using a strain carrying a GFP fusion to the cell division protein MinD. MinD depends on an intact membrane potential for correct localization at the cell poles and the cell division plane (Fig. 2, control in panel A).40 Upon depolarization, it delocalizes from these spots. As shown before for 1a and confirmed herein for the pore-forming compound nisin, all derivatives led to MinD delocalization pointing to membrane depolarization (Fig. 2, panels A). Subsequently, the BacLight assay was employed to investigate if depolarization is accompanied by disruption of the membrane structure. This staining method combines a green-fluorescent dye that crosses intact membranes and a red-fluorescent dye, which may only enter bacterial cells through membrane pores or larger holes. In fluorescence overlays, permeabilized cells appear orange. While the pore-forming nisin led to red fluorescencing cells, the compounds 9a, 9b, and 9c′ led to yellow to orange fluorescence (Fig. 2, panel C). This provides evidence for slight membrane permeabilization by 9a, 9b, and 9c′ rather than for organized pore formation. Permeabilization could be due to a detergent-like carpet mechanism of action as was proposed for some antimicrobial peptides.41 However, 1a was not shown to permeabilize the bacterial membrane in the BacLight assay33 suggesting slight differences in membrane interaction between the tri-metallic compound and the di- or mono-metallic derivatives.
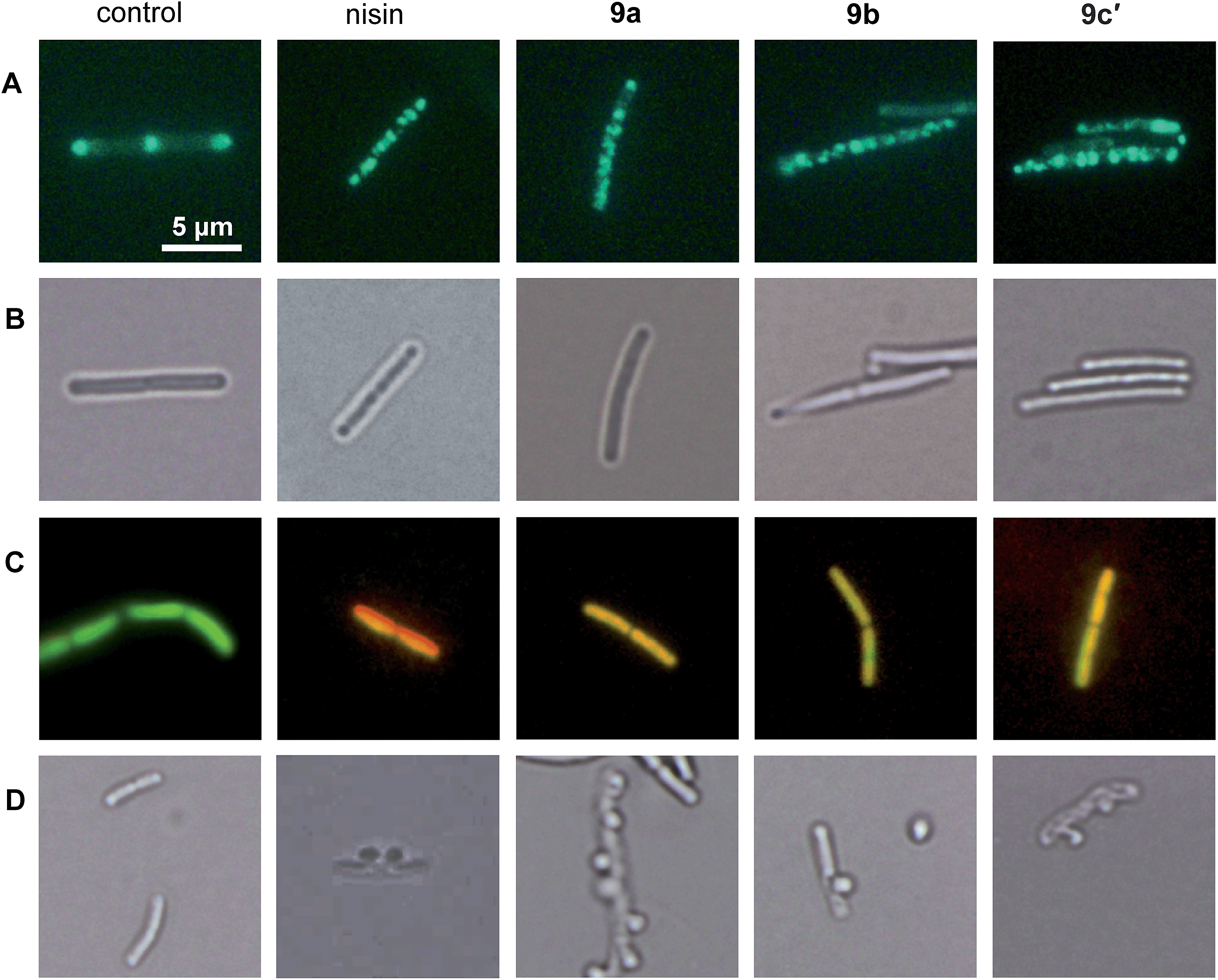 | ||
| Fig. 2 Influence of 9a, 9b, and 9c′ on B. subtilis membrane depolarization, membrane permeabilization, and cell wall integrity. (A) GFP-MinD localization after antibiotic treatment. Disruption of the septal and polar localization of MinD is indicative of membrane depolarization. (B) Light microscopy images of the cells shown above. (C) Antibiotic-treated cells stained with BacLight. The green dye is able to enter intact bacterial cells. The red dye can only cross the bacterial membrane through membrane pores. In a fluorescence overlay red or orange cells are indicative of pore formation. (D) Antibiotic-stressed cells fixed with acetic acid and methanol. Inhibition of the cell wall biosynthesis leads to holes in the cell wall resulting in membrane excrescences after fixation with acetic acid/methanol. | ||
1a has been shown to affect cell wall integrity, probably by disturbing membrane-bound cell wall biosynthesis steps.33 The effects of the derivatives 9a, 9b, and 9c′ on cell wall integrity were monitored by microscopic examination of the cell shape after fixation with acetic acid and methanol. This treatment reliably leads to membrane extrusions through cell wall holes, which occur upon inhibition of membrane-bound cell wall biosynthesis steps.42 Just like the positive control nisin, which inhibits cell wall biosynthesis by binding to precursor lipid II, all active compounds led to membrane extrusions (Fig. 2, panel D). This demonstrates the same, probably indirect effect on cell wall biosynthesis by all derivatives. Taken together, 9a, 9b, and 9c′ still target the bacterial membrane and influence cell wall integrity but show more efficient membrane permeabilization than the tri-metallic 1a.
Conclusion
The urgent need for novel classes of antimicrobial compounds is evidenced by rapidly growing bacterial resistance against the conventional chemotherapeutic options. The discovery of novel concepts is therefore highly desirable in order to identify new structural classes of antibacterial compounds without a pre-existing resistance history. We introduced (multi-)organometallic-containing complexes as new class of antimicrobial agents with a novel mode of action. Unlike our earlier belief that the trimetallic nature was essential for activity of compound 1a, we herein discovered the [(dpa)Re(CO)3] moiety as the crucial component for potent activity against gram-positive bacteria, including MRSA strains. Preliminary insight into the mode of action of the bimetallic 9a, 9b, and the monometallic derivative 9c′ suggests that these compounds target the bacterial membrane and have influence on the cell wall integrity, similarly to the trimetallic lead structure 1a. However, 9a, 9b and 9c′ showed even more efficient membrane permeabilization ability compared to 1a. Moreover, all active derivatives displayed higher solubility in culture media than the trimetallic 1a, with 9c′ having the highest solubility. Finally, a determination of toxicity against different mammalian cell lines shows that the monometallic derivative 9c′ is at least 15–30 times more toxic to gram-positive bacteria than to the mammalian cell lines tested. This differential activity is certainly encouraging enough for further development. So far, Re(I) tricarbonyl compounds are well-known for their luminescent properties and anticancer therapeutic potential.13,43–50 To the best of our knowledge, this is the first study reporting that a {ReI(CO)3} core with an appropriate organic ligand is a promising new lead structure for the development of antibacterial compounds.Experimental section
Materials
All chemicals were of reagent grade quality or better, obtained from commercial suppliers, and used without further purification. Solvents were used as received or dried over molecular sieves. All preparations were carried out using standard Schlenk techniques. Reactions involving CpMn(CO)3-containing compounds were carried out in the dark. N,N-bis(pyridine-2-ylmethyl)prop-2-yn-1-amine, compound 2, 5a, 6b, 7d, and 8 were synthesized following the literature procedure.34,36,38,51 The analytical data matched those previously reported. For all newly synthesized compounds, except 9c, detailed synthesis procedures and full analytical data are presented in the ESI.†Instrumentation and methods
1H and 13C NMR spectra were recorded in deuterated solvents on Bruker DRX 200, 250, 400, or 600 spectrometers at 30 °C. Chemical shifts δ are reported in ppm (parts per million), coupling constants J are given as absolute values in Hz. The residual solvent peaks have been used as an internal reference. Abbreviations for the peak multiplicities are as follows: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). Infrared spectra were recorded on an ATR unit using a Bruker Tensor 27 FTIR spectrophotometer at 4 cm−1 resolution. Signal intensity is abbreviated br (broad), s (strong), m (medium), and w (weak). ESI mass spectra were recorded on a Bruker Esquire 6000. Analytical HPLC was performed using a Nucleosil 100-5 C18 column (250 × 3 mm) on a Hitachi chromaster HPLC system at 25 °C. The flow rate was 0.5 mL min−1 and UV-absorption was measured at 250 nm. The runs were performed with a linear gradient of A acetonitrile (Sigma-Aldrich HPLC-grade) and B (distilled water containing 0.1% v/v TFA): t = 0–3 min, 5% A; t = 17–22 min, 100% A; t = 22–25 min, 5% A. Preparative HPLC was performed using a Nucleosil 100-7 C18 column (250 × 21 mm) on a Varian Prostar HPLC system at 25 °C. The flow rate was 15 mL min−1 and UV-absorption was measured at 250 nm. The runs were performed with a linear gradient of A acetonitrile (Sigma-Aldrich HPLC-grade) and B (distilled water containing 0.1% v/v TFA): t = 0–3 min, 10% A; t = 3–6 min, 50% A; t = 6–23 min, 100% A; t = 23–27 min, 100% A, t = 30 min, 100% A.Compound 9c
A solution of 7c (371 mg, 0.53 mmol) and 8 (350 mg, 0.53 mmol) in 12 mL DMF/NEt3 mixture (2/1, v/v) was degassed by two ‘freeze–pump–thaw’ cycles. Then CuBr (9.86 mg, 0.07 mmol) and cis-dichlorobis (triphenylphosphine) palladium(II) (7.7 mg, 0.01 mmol) were added under nitrogen atmosphere and the mixture was degassed again by one ‘freeze–pump–thaw’ cycle. The wine color solution thus obtained was allowed to stir 20 h at room temperature. The solvent was removed and the residue was diluted with 100 mL of CH2Cl2, washed with distilled water (4 × 60 mL) and brine (2 × 60 mL). The CH2Cl2 layer was dried over Na2SO4, filtered and concentrated. The solid was then loaded on a silica column and eluted immediately with CH2Cl2:MeOH 5:1 > 3:1. The combined fractions was evaporated and redissolved in CH2Cl2 and filtered. Filtrate was dried to give 9c as light brown solid (yield: 256 mg, 39%). For biological applications purpose 9c was further purified by RP-HPLC to remove a trace of impurity present after purification by silica column chromatography. Thus the counter anion PF6− of 9c changed to CF3COO− and the compound was designated as 9c′. Data for 9c: Rf = 0.51 (silica gel, CH2Cl2:MeOH 4:1). tR (RP-HPLC) = 16.8 min. 1H NMR (250 MHz, CD2Cl2): δ (ppm) 2.54 (m, 2H, Ph–CO–CH2–CH2), 2.75 (min) and 2.82 (maj) (rotamers, m, 2H, CH2–CH2–triazole ring), 3.03 (m, 2H, CH2–CH2–triazole ring), 3.27 (m, 2H, PhCO–CH2–CH2), 3.34–3.56 (m, 4H, NH–CH2–CH2–N and NH–CH2–CH2–N), 4.15 (maj) and 4.32 (min) (rotamers, s, 2H, N–CH2–CO), 4.68 (min) and 4.71 (maj) (rotamers, s, 2H, N–CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 4.87–5.18 (rotamers, m, 4H, 2 × N–CH2–Py), 5.42 (maj) and 5.45, 5.20 (min) (rotamers, s, 2H, triazole–CH2–Ph), 6.52, 6.78 (min) and 6.99 (maj) (rotamers, 1H, NHCO), 7.19–7.50 (m, 13H, 1 × triazole ring H and 12 × aromatic H), 7.56–7.69 (m, 4H, aromatic H), 7.82–7.95 (m, 4H, aromatic H), 8.76 (m, 2H, aromatic H), 9.43 (maj) and 9.50, 9.60, 10.13 (min) (rotamers, NHCO). 13C NMR (62.9 MHz, CD2Cl2): δ (ppm) 20.9 (min) and 21.1 (maj) (rotamers, CH2–CH2–triazole ring), 29.6 (maj) and 39.9 (min) (rotamers, PhCO–CH2–CH2), 31.6 (maj) and 31.9 (min) (rotamers, CH2–CH2–triazole ring), 33.6 (maj) and 33.9 (min) (rotamers, PhCO–CH2–CH2), 37.9 (maj) and 38.3 (min) (rotamers, NH–CH2–CH2–N), 49.1 (NH–CH2–CH2–N), 51.8 (N–CH2–CO), 52.1 (Ph–CH2–triazole ring, overlaps with solvent residual signal), 59.1 (maj) and 59.2 (min) (rotamers, N–CH2–CC), 68.1, 68.3 (maj) and 72.2, 72.4 (min) (rotamers, 2 × N–CH2–Py), 80.8 (CC–CH2–N), 90.3 (N–CH2–CC), 116.1 (maj) and 116.3 (min) (rotamers, aromatic C), 119.5, 120.1 (min) and 119.7 (maj) (rotamers, aromatic C), 121.6 (min) and 121.9 (maj) (rotamers, CH triazole ring), 123.9 (aromatic C), 124.2 (aromatic C), 125.4, 125.6 (min) and 125.8, 125.9 (maj) (rotamers, 2 × aromatic C), 127.8 (min) and 127.9 (maj) (rotamers, aromatic C), 128.4 (min) and 128.5 (maj) (rotamers, aromatic C), 128.9 (aromatic C), 132.7 (aromatic C), 133.1 (aromatic C), 135.2 (aromatic C), 136.6 (aromatic C), 139.3 (min) and 139.6 (maj) (rotamers, aromatic C), 140.5 (aromatic C), 146.7 (maj) and 147.1 (min) (rotamers, C triazole ring), 151.4 (min) and 151.6 (maj) (rotamers, aromatic carbon), 159.3 (min) and 159.6 (maj) (rotamers, aromatic C), 167.1, 167.9, 168.1 (min) and 168.4 (maj) (rotamers, CONH), 172.4 (CONH), 173.4 (maj) and 173.6 (min) (rotamers, CH2CON), 194.9 (min) and 195.3 (maj) (rotamers, Re–CO), 198.8 (min) and 199.2 (maj) (rotamers, Ph–CO). IR bands (ν): 2032s, 1913s, 1670m (br) cm−1. ESI-MS (pos. detection mode): m/z (%): 1071.98 (100) [M-PF6]+.
C), 4.87–5.18 (rotamers, m, 4H, 2 × N–CH2–Py), 5.42 (maj) and 5.45, 5.20 (min) (rotamers, s, 2H, triazole–CH2–Ph), 6.52, 6.78 (min) and 6.99 (maj) (rotamers, 1H, NHCO), 7.19–7.50 (m, 13H, 1 × triazole ring H and 12 × aromatic H), 7.56–7.69 (m, 4H, aromatic H), 7.82–7.95 (m, 4H, aromatic H), 8.76 (m, 2H, aromatic H), 9.43 (maj) and 9.50, 9.60, 10.13 (min) (rotamers, NHCO). 13C NMR (62.9 MHz, CD2Cl2): δ (ppm) 20.9 (min) and 21.1 (maj) (rotamers, CH2–CH2–triazole ring), 29.6 (maj) and 39.9 (min) (rotamers, PhCO–CH2–CH2), 31.6 (maj) and 31.9 (min) (rotamers, CH2–CH2–triazole ring), 33.6 (maj) and 33.9 (min) (rotamers, PhCO–CH2–CH2), 37.9 (maj) and 38.3 (min) (rotamers, NH–CH2–CH2–N), 49.1 (NH–CH2–CH2–N), 51.8 (N–CH2–CO), 52.1 (Ph–CH2–triazole ring, overlaps with solvent residual signal), 59.1 (maj) and 59.2 (min) (rotamers, N–CH2–CC), 68.1, 68.3 (maj) and 72.2, 72.4 (min) (rotamers, 2 × N–CH2–Py), 80.8 (CC–CH2–N), 90.3 (N–CH2–CC), 116.1 (maj) and 116.3 (min) (rotamers, aromatic C), 119.5, 120.1 (min) and 119.7 (maj) (rotamers, aromatic C), 121.6 (min) and 121.9 (maj) (rotamers, CH triazole ring), 123.9 (aromatic C), 124.2 (aromatic C), 125.4, 125.6 (min) and 125.8, 125.9 (maj) (rotamers, 2 × aromatic C), 127.8 (min) and 127.9 (maj) (rotamers, aromatic C), 128.4 (min) and 128.5 (maj) (rotamers, aromatic C), 128.9 (aromatic C), 132.7 (aromatic C), 133.1 (aromatic C), 135.2 (aromatic C), 136.6 (aromatic C), 139.3 (min) and 139.6 (maj) (rotamers, aromatic C), 140.5 (aromatic C), 146.7 (maj) and 147.1 (min) (rotamers, C triazole ring), 151.4 (min) and 151.6 (maj) (rotamers, aromatic carbon), 159.3 (min) and 159.6 (maj) (rotamers, aromatic C), 167.1, 167.9, 168.1 (min) and 168.4 (maj) (rotamers, CONH), 172.4 (CONH), 173.4 (maj) and 173.6 (min) (rotamers, CH2CON), 194.9 (min) and 195.3 (maj) (rotamers, Re–CO), 198.8 (min) and 199.2 (maj) (rotamers, Ph–CO). IR bands (ν): 2032s, 1913s, 1670m (br) cm−1. ESI-MS (pos. detection mode): m/z (%): 1071.98 (100) [M-PF6]+.
Minimal inhibitory concentration (MIC)
The minimal inhibitory concentrations (MIC) were tested against Escherichia coli DSM 30083, Acinetobacter baumannii DSM 30007, Pseudomonas aeruginosa DSM 50071, Bacillus subtilis DSM 402, Staphylococcus aureus DSM 20231, and Staphylococcus aereus ATCC 43300 (MRSA) in a microtiter plate assay according to CLSI guidelines.52E. coli, A. baumannii, S. aureus, and B. subtilis were grown in Mueller Hinton broth, P. aeruginosa in cation adjusted Mueller Hinton II. Compounds were dissolved in DMSO to give 10 mg mL−1 stock solutions. Serial dilution in culture media was carried out automatically with the Tecan Freedom Evo 75 liquid handling workstation (Tecan, Männedorf, Switzerland) from 512 to 0.5 μg mL−1. Compound dilutions were inoculated with 105 bacteria per mL taken from late exponential cultures grown in the same media in a total volume of 200 μL per well. Cells were incubated for 16 hours at 37 °C. The lowest compound concentration inhibiting visible bacterial growth is reported as MIC.Mammalian cell culture and cytotoxicity test
Cytotoxicity studies were performed on three different cell lines by a fluorometric cell viability assay using Resazurin (Promocell GmbH). Human cervical carcinoma cells (HeLa) were cultured in DMEM (Gibco) supplemented with 5% fetal calf serum (FCS, Gibco), 100 U mL−1 penicillin, 100 μg mL−1 streptomycin at 37 °C and 5% CO2. The normal human lung fibroblast MRC-5 cell line was maintained in F-10 medium (Gibco) supplemented with 10% FCS (Gibco), penicillin (100 U mL−1), and streptomycin (100 μg mL−1). The human hepatomacarcinoma (HepG2) cells were cultured in DMEM (Gibco) supplemented with 10% fetal calf serum (FCS, Gibco), 100 U mL−1 penicillin, 100 μg mL−1 streptomycin at 37 °C and 5% CO2.One day before treatment, cells were seeded in triplicate in 96-well plates at a density of 4 × 103 cells per well for HeLa and HepG2, and 7 × 103 for MRC-5 in 100 μL growth medium. Compounds were dissolved in DMSO to give 10 mg mL−1 stock solutions, serial dilutions were prepared using respective cell culture media. Upon treating cells with increasing concentrations of respective compounds for 48 h, the medium was removed, and 100 μL fresh culture medium containing Resazurin (0.2 mg mL−1 final concentration) was added. After 4 h of incubation at 37 °C, fluorescence of the highly red fluorescent product resorufin was quantified at 590 nm emission with 540 nm excitation wavelength in a SpectraMax M5 microplate Reader.
GFP-MinD delocalization assay
B. subtilis 1981 GFP-MinD40 was grown in Belitzky Minimal Medium (BMM)53 until early logarithmic phase. The main culture was subdivided and aliquots were treated with 8 μg mL−1 (2× MIC) 9a, 9b, and 9c′, respectively, or left untreated as control. Nisin was used as positive control at a concentration of 0.75 μg mL−1. After 15 minutes of antibiotic stress, cells were microscopically examined in fluorescent mode as described previously.54BacLight™ membrane disruption assay
BacLight staining (LIVE/DEAD BacLight Bacterial Viability Kit, Invitrogen, Carlsbad, CA, USA) was performed following the manufacturer's instructions. B. subtilis 168 (ref. 55) was grown and stressed with antibiotics as described above. After 15 minutes of antibiotic exposure, cells were stained with a 1:1 mixture of SYTO 9 and propidium iodide (1 μL per mL cells) for 15 minutes, washed twice in 100 mM Tris/1 mM EDTA, pH 7.5, resuspended in the same buffer and subjected to fluorescence microscopy.
Cell wall integrity assay
B. subtilis 168 was grown and stressed with antibiotics as described above. After 15 minutes of antibiotic treatment, 200 μL of culture were withdrawn and immediately fixed in 1 mL of a 1:3 mixture of acetic acid and methanol. 5 μL of fixed bacteria were immobilized in agarose and subjected to light microscopy as described previously.54
Solubility in media
All compounds were dissolved as 10 mg mL−1 stock solutions in DMSO, and first dilutions were prepared with sterile double-distilled water. Concentration series of the compounds ranging from 1 μg mL−1 to 1 mg mL−1 were prepared in either BMM or DMEM (diluted from stock solutions as described above). After 10 min incubation at room temperature, samples were centrifuged at 16100 × g for 10 min (room temperature). Precipitated compounds were visible as pellets. Samples without visible precipitation were mixed again and checked for microprecipitates by light microscopy (Olympus BX51 equipped with a U-UCD8 condenser and a UPlanSApo 100XO objective). The highest compound concentration not leading to visible precipitates was defined as maximum solubility. Experiments were performed independently as duplicates.
Acknowledgements
Financial support from the International Max Planck Research School for Chemical Biology (fellowship to MP), the Research Department Interfacial Systems Chemistry at Ruhr University Bochum (NMN, JEB), the DFG (NMN), the State of North Rhine-Westphalia (NRW), Germany and the European Union, European Regional Development Fund, "Investing in your future" (JEB), the Swiss National Science Foundation (Professorship to GG, grant number PP00P2_133568), the University of Zurich (GG), the Stiftung für Wissenschaftliche Forschung of the University of Zurich (GG), and the COST Action CM1105 (NMN and GG) is gratefully acknowledged. This work is also supported by the Cluster of Excellence RESOLV (EXC 1069) funded by the Deutsche Forschungsgemeinschaft. The authors thank PD Dr Stefano Ferrari for access to cell culture laboratories.References
- G. Taubes, Science, 2008, 321, 356–361 CrossRef CAS PubMed
.
- S. Saxena and C. Gomber, Cent. Eur. J. Med., 2010, 5, 12–29 CrossRef CAS PubMed
- R. M. Klevens, M. A. Morrison, J. Nadle, S. Petit, K. Gershman, S. Ray, L. H. Harrison, R. Lynfield, G. Dumyati, J. M. Townes, A. S. Craig, E. R. Zell, G. E. Fosheim, L. K. McDougal, R. B. Carey and S. K. Fridkin, JAMA, J. Am. Med. Assoc., 2007, 298, 1763–1771 CrossRef CAS PubMed
-
ECDC/EMEA Joint Working Group, The Bacterial Challenge, The Publications Office of the European Union, 2009, Stockholm Search PubMed
- J. E. Bandow and N. Metzler-Nolte, ChemBioChem, 2009, 10, 2847–2850 CrossRef CAS PubMed
- P. Kloss, L. Xiong, D. L. Shinabarger and A. S. Mankin, J. Mol. Biol., 1999, 294, 93–101 CrossRef CAS PubMed
- E. P. Abraham, E. Chain, C. M. Fletcher, A. D. Gardner, N. G. Heatley, M. A. Jennings and H. W. Florey, Lancet, 1941, 238, 177–189 CrossRef
- M. Wenzel and J. E. Bandow, Proteomics, 2011, 11, 3256–3268 CrossRef CAS PubMed
- M. S. Butler, M. A. Blaskovich and M. A. Cooper, J. Antibiot., 2013, 66, 571–591 CrossRef CAS PubMed
- H. Brötz-Oesterhelt and N. A. Brunner, Curr. Opin. Pharmacol., 2008, 8, 564–573 CrossRef PubMed
-
G. Jaouen and N. Metzler-Nolte, in Topics in Organometallic Chemistry, Springer, Heidelberg, Germany, 1st edn, 2010, vol. 32 Search PubMed
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed
-
C. Biot and D. Dive, in Medicinal Organometallic Chemistry (Topics in Organometallic Chemistry), ed. G. Jaouen and N. Metzler-Nolte, Springer, Heidelberg, Germany, 2010, vol. 32, ch. 7, pp. 155–193 Search PubMed
- C. Biot, W. Castro, C. Y. Botte and M. Navarro, Dalton Trans., 2012, 6321–6580 Search PubMed
- F. Dubar, T. J. Egan, B. Pradines, D. Kuter, K. K. Ncokazi, D. Forge, J.-F. Paul, C. Pierrot, H. Kalamou, J. Khalife, E. Buisine, C. Rogier, H. Vezin, I. Forfar, C. Slomianny, X. Trivelli, S. Kapishnikov, L. Leiserowitz, D. Dive and C. Biot, ACS Chem. Biol., 2011, 6, 275–287 CrossRef CAS PubMed
- N. C. Lloyd, H. W. Morgan, B. K. Nicholson and R. S. Ronimus, Angew. Chem., Int. Ed., 2005, 44, 941–944 CrossRef CAS PubMed
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC
- M. Patra, G. Gasser and N. Metzler-Nolte, Dalton Trans., 2012, 41, 6350–6358 RSC
- K. Strebhard and A. Ullrich, Nat. Rev. Cancer, 2008, 8, 473–480 CrossRef PubMed
-
S. Gibaud and G. Jaouen, in Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springer-Verlag, Berlin Heidelberg, 2010, pp. 1–20, and references therein Search PubMed
- W. J. Youngs, A. R. Knapp, P. O. Wagersa and C. A. Tessiera, Dalton Trans., 2012, 327–336 RSC
- K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed
- C. L. Cannon, L. A. Hogue, R. K. Vajravelu, G. H. Capps, A. Ibricevic, K. M. Hindi, A. Kascatan-Nebioglu, M. J. Walter, S. L. Brody and W. J. Youngs, Antimicrob. Agents Chemother., 2009, 53, 3285–3293 CrossRef CAS PubMed
- M. Patra, G. Gasser, A. Pinto, K. Merz, I. Ott, J. E. Bandow and N. Metzler-Nolte, ChemMedChem, 2009, 4, 1930–1938 CrossRef CAS PubMed
- M. Patra, G. Gasser, M. Wenzel, K. Merz, J. E. Bandow and N. Metzler-Nolte, Organometallics, 2012, 31, 5760–5771 CrossRef CAS
- M. Patra, G. Gasser, M. Wenzel, K. Merz, J. E. Bandow and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2011, 3295–3302 CrossRef CAS
- J. T. Chantson, M. V. V. Falzacappa, S. Crovella and N. Metzler-Nolte, ChemMedChem, 2006, 1, 1268–1274 CrossRef CAS PubMed
- J. T. Chantson, M. V. Verga-Falzacappa, S. Crovella and N. Metzler-Nolte, J. Organomet. Chem., 2005, 690, 4564–4572 CrossRef CAS PubMed
- H. B. Albada, P. Prochnow, S. Bobersky, J. E. Bandow and N. Metzler-Nolte, Chem. Sci., 2014, 5, 4453–4459 RSC
- H. B. Albada, A.-I. Chiriac, M. Wenzel, M. Penkova, J. E. Bandow, H.-G. Sahl and N. Metzler-Nolte, Beilstein J. Org. Chem., 2012, 8, 1753–1764 CrossRef CAS PubMed
- M. Wenzel, A. I. Chiriac, A. Otto, D. Zweytick, C. May, C. Schumacher, R. Gust, H. B. Albada, M. Penkova, U. Krämer, R. Erdmann, N. Metzler-Nolte, S. K. Straus, E. Bremer, D. Becher, H. Brötz-Oesterhelt, H.-G. Sahl and J. E. Bandow, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1409–E1418 CrossRef CAS PubMed
- M. Wenzel, M. Patra, C. H. R. Senges, J. J. Stepanek, A. Pinto, P. Prochnow, I. Ott, N. Metzler-Nolte and J. E. Bandow, ACS Chem. Biol., 2013, 8, 1442–1450 CrossRef CAS PubMed
- M. Patra, G. Gasser, D. Bobukhov, K. Merz, A. V. Shtemenko and N. Metzler-Nolte, Dalton Trans., 2010, 5617–5619 RSC
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC
- G. Gasser, N. Hüsken, S. D. Köster and N. Metzler-Nolte, Chem. Commun., 2008, 3675–3677 RSC
- A. Hess and N. Metzler-Nolte, Chem. Commun., 1999, 885–886 RSC
- H. W. P. N'Dongo, I. Neundorf, K. Merz and U. Schatzschneider, J. Inorg. Biochem., 2008, 102, 2114–2119 CrossRef PubMed
- R. Bauer and L. M. Dicks, Int. J. Food. Microbiol., 2005, 101, 201–216 CrossRef CAS PubMed
- H. Strahl and L. W. Hamoen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12281–12286 CrossRef CAS PubMed
- V. Teixeira, M. J. Feio and M. Bastos, Prog. Lipid Res., 2012, 51, 149–177 CrossRef CAS PubMed
- T. Schneider, T. Kruse, R. Wimmer, I. Wiedemann, V. Sass, U. Pag, A. Jansen, A. K. Nielsen, P. H. Mygind, D. S. Raventós, S. Neve, B. Ravn, A. M. J. J. Bonvin, L. D. Maria, A. S. Andersen, L. K. Gammelgaard, H.-G. Sahl and H.-H. Kristensen, Science, 2010, 328, 1168–1172 CrossRef CAS PubMed
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC
- F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12–21 CrossRef CAS PubMed
- K. K.-W. Lo, A. W.-T. Choi and W. H.-T. Law, Dalton Trans., 2012, 6021–6047 RSC
- M. Patra and G. Gasser, ChemBioChem, 2012, 13, 1232–1252 CrossRef CAS PubMed
- I. Kitanovic, S. Can, H. Alborzina, A. Kitanovic, V. Pierroz, A. Leonidvoa, A. Pinto, B. Spingler, S. Ferrari, R. Molteni, A. Steffen, N. Metzler-Nolte, S. Wölfl and G. Gasser, Chem.–Eur. J., 2014, 20, 2496–2507 CrossRef CAS PubMed
- A. Leonidova, V. Pierroz, R. Rubbiani, Y. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2014, 5, 4044–4056 RSC
- A. Leonidova, V. Pierroz, L. A. Adams, N. Barlow, S. Ferrari, B. Graham and G. Gasser, ACS Med. Chem. Lett., 2014, 5, 809–814 CrossRef CAS PubMed
- A. Leonidova and G. Gasser, ACS Chem. Biol., 2014 DOI:10.1021/cb500528c
- D. G. Cabrera, B. D. Koivisto and D. A. Leigh, Chem. Commun., 2007, 4218–4220 RSC
- S. Top and G. Jaouen, J. Org. Chem., 1981, 46, 78–82 CrossRef CAS
- J. Stülke, R. Hanschke and M. Hecker, J. Gen. Microbiol., 1993, 139, 2041–2045 CrossRef PubMed
- M. Wenzel, B. Kohl, D. Münch, N. Raatschen, H. B. Albada, L. W. Hamoen, N. Metzler-Nolte, H. G. Sahl and J. E. Bandow, Antimicrob. Agents Chemother., 2012, 56, 5749–5757 CrossRef CAS PubMed
- C. Agnostopoulos and J. Spizizen, J. Bacteriol., 1961, 81, 741–746 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data of all newly synthesized compounds, HPLC chromatograms and ESI mass spectra of 9a, 9b and 9c′ and 10, copies of 1H and 13C NMR spectra for new compounds. See DOI: 10.1039/c4sc02709d |
| This journal is © The Royal Society of Chemistry 2015 |