
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective α-amination enabled by a BINAM-derived phase-transfer catalyst†
H. M.
Nelson‡
,
J. S.
Patel‡
,
H. P.
Shunatona
and
F. D.
Toste
*
Department of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: fdtoste@berkeley.edu
First published on 2nd October 2014
Abstract
Chiral anion phase-transfer of aryldiazonium cations was utilized to achieve highly enantioselective α-amination of carbonyl compounds. A broad scope of indanone- and benzosuberone-derived substrates was amenable to this strategy. Critical to obtaining high levels of enantioselectivity was the use of BINAM-derived phosphoric acids. The utility of this transformation was demonstrated through facile conversion of diazene products to valuable α-amino acid derivatives.
Chiral amines are attractive targets for enantioselective methodology due to their prominent representation amongst bioactive molecules and chiral catalysts.1 Particularly attractive are α-amino acids, which are utilized directly as building blocks for pharmaceuticals, natural products, agrochemicals, and as precursors for commonly employed chiral ligands.1 Several methods exist that provide access to enantioenriched, protected amines.2 The use of dialkyl azodicarboxylates as masked electrophilic nitrogen sources in auxiliary-directed,3 organocatalytic4 and transition metal catalyzed5 amination reactions is prevalent. These methodologies furnish highly enantioenriched hydrazines that can be converted to amines after multiple chemical operations.
We hypothesized that the use of aryldiazonium cations as electrophilic nitrogen sources within a chiral anion phase-transfer (CAPT) manifold could provide a complementary enantioselective amination method.6 In this scenario, chiral anion phase-transfer of an insoluble aryldiazonium cation would generate a soluble N-electrophilic, chiral diazonium ion pair. This species could react with nucleophilic carbonyl derivatives to provide enantioenriched α-diazenated compounds (Fig. 1a and b). Importantly, this methodology would complement established α-amination methodologies in several ways. Firstly, well-established methods for reductive cleavage of azo compounds such as 2 would provide facile access to unprotected α-amino acid derivatives (3).7 Secondly, straightforward preparation of complex aryldiazonium cations from readily available anilines would facilitate development of application-specific amination reagents with tunable reactivity.8 This manuscript details the successful execution of this hypothesis through the first examples of catalytic, enantioselective α-amination using aryldiazonium cations. This advance is enabled by the preparation of BINAM-derived chiral phosphoric acids (BDPAs) that proved critical to obtaining high levels of enantioinduction.
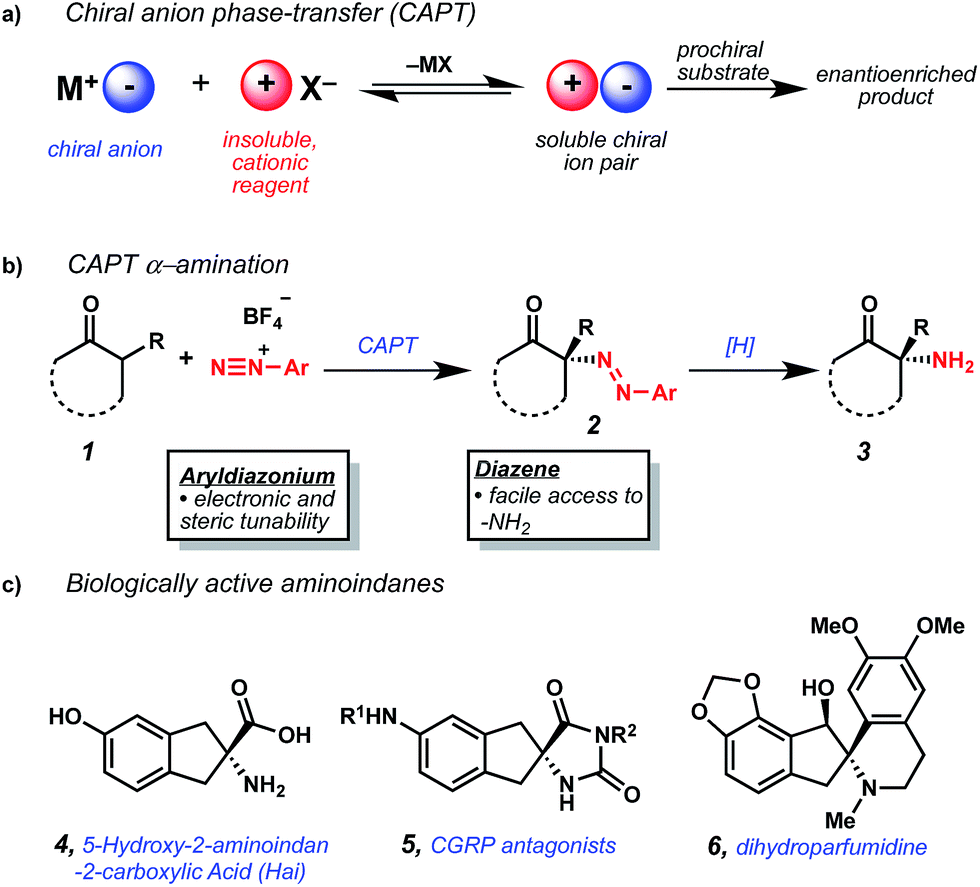 | ||
| Fig. 1 (a) Chiral anion phase-transfer concept. (b) Application to α-amination. (c) Biologically active amino-indanone derivatives. | ||
As a model system, we were attracted to indanone-derived β-ketoesters due to their potential for elaboration into conformationally constrained tyrosine analogues (CCTAs) such as 5-hydroxy-2-aminoindan-2-carboxylic acid (Hai) (4),9 spirohydantoin-derived CGRP antagonists investigated for treatment of migraine headaches (5),10 and amino-indane natural products such as dihydroparfumidine11 (6) (Fig. 1c). We initially found that exposure of β-ketoester 17 to TCyP (7), Na3PO4, and PhN2BF4 provided diazene 18 with excellent conversion, albeit with low enantioselectivity (Table 1).12
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Solv. | Base | Conv.b (%) | eec (%) |
| a Conditions: 17 (1 equiv.), cat. (13–16) (5 mol%), base (6 equiv.), PhN2BF4 (1.2 equiv.), solvent (0.025 M), rt, 2–24 h. b Conversion based on consumption of starting material as determined by 1H NMR. c Determined by chiral phase HPLC. | |||||
| 1 | 7 | Hexanes | Na3PO4 | >95 | 9 |
| 2 | 8 | Hexanes | Na3PO4 | >95 | 7 |
| 3 | 9 | Hexanes | Na3PO4 | >95 | 0 |
| 4 | 10 | Hexanes | Na3PO4 | >95 | 2 |
| 5 | 11 | Hexanes | Na3PO4 | >95 | 7 |
| 6 | 12 | Hexanes | Na3PO4 | >95 | 4 |
| 7 | 7 | Hexanes | NaHCO3 | >95 | 21 |
| 8 | 7 | Hexanes | NaH2PO4 | >95 | 34 |
| 9 | 13 | Hexanes | NaH2PO4 | >95 | −7 |
| 10 | 14 | Hexanes | NaH2PO4 | >95 | 4 |
| 11 | 15 | Hexanes | NaH2PO4 | >95 | 5 |
| 12 | 16 | Hexanes | NaH2PO4 | >95 | 87 |
| 13 | 16 | 2-MeTHF | NaH2PO4 | >95 | 10 |
| 14 | 16 | Toluene | NaH2PO4 | >95 | 40 |
| 15 | 16 | Cyclohexane | NaH2PO4 | >95 | 90 |
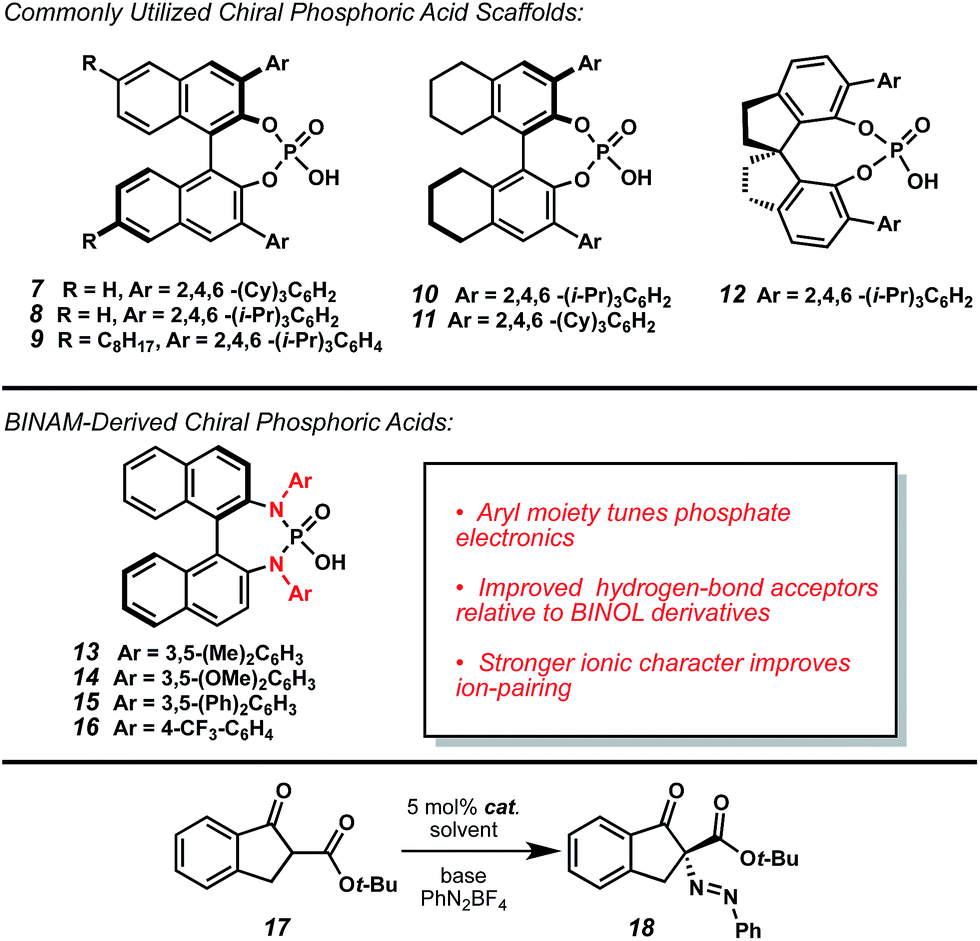
With this encouraging result in hand, a screen of CAPT catalysts and inorganic bases was undertaken (Table 1). Examination of the commonly employed chiral phosphate scaffolds did not identify a catalyst capable of providing improved enantioselectivity (entries 1–6); however, linear screening carried out with TCyP (7) proved fruitful, as use of weaker bases, such as NaH2PO4, increased enantioselectivity to 34% ee while maintaining excellent conversion (entry 7, 8).
Challenged by low levels of enantioselectivity using common chiral phosphate scaffolds, we undertook efforts to design novel CAPT catalysts. We were drawn to the BINAM-derived phosphoric acids (BDPAs, Table 1, 13–16) first prepared by Ishihara and coworkers,13 as they offer two potential improvements over BINOL-derived phosphates (7–11): first, we hypothesized that the nitrogen lone pairs would improve hydrogen-bonding interactions with the substrates relative to traditional chiral phosphoric acid catalysts (7–12).14 Second, increased resonance donation from the nitrogens could improve ion-pairing with the diazonium cation. Furthermore, this catalyst system would be highly modular, allowing for variation of both the ionic character and the hydrogen-bonding strength via synthetic modulation of the N-aryl groups.15
A small library of BDPAs was prepared (13–16). Upon examination of these catalysts under our diazenation conditions (Table 1, entries 9–12), we were pleased to find that electron-poor BDPA 16 provided the diazenated product (18) in excellent conversion and 87% ee (entry 12). Fine-tuning of solvent (entries 13–15) identified cyclohexane as optimal, yielding indanone 18 in 90% ee (entry 8).
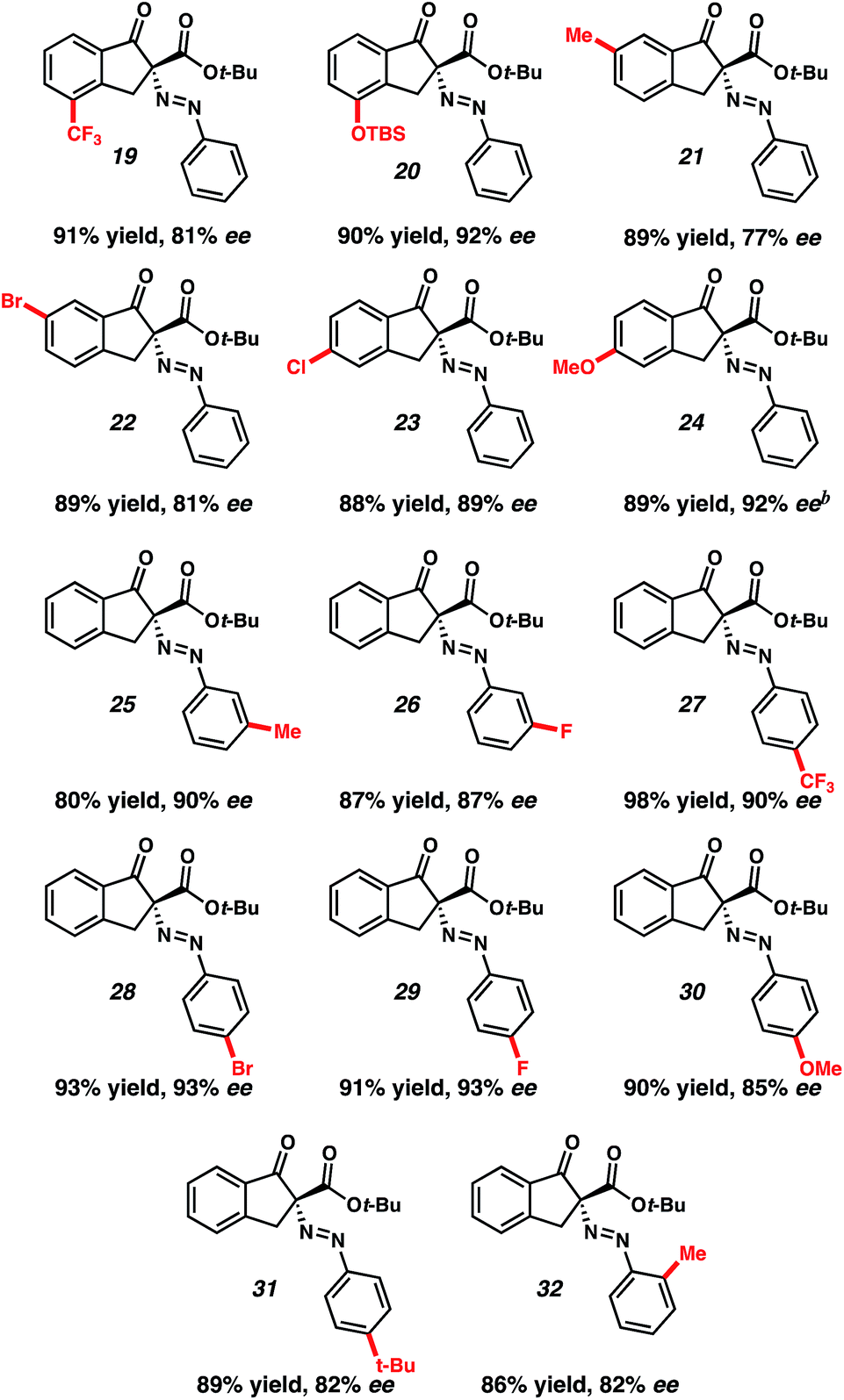
With optimized reaction conditions in hand, we investigated the scope of this reactivity. Indanones with electron-rich (20, 21, 24, Table 2) and electron-poor (19, 22, 23) substitution at the −4, −5, and −6 positions provided their corresponding diazenes in good yields and enantioselectivities. Importantly, both protected phenol and halogenated derivatives (20 and 22–24) were competent under the reaction conditions, permitting derivatization of the reaction products. Additionally, substitution of the diazonium aryl group with electron-poor, as well as electron-rich, groups at -ortho, -meta, and -para position provided excellent yields and good to excellent enantioselectivities (Table 2, 25–32).
Additional ketone derivatives were amenable to CAPT diazenation with slight modification of the reaction conditions (Scheme 1). Benzosuberone derivative 33 was a suitable nucleophile, affording diazene 34 in 96% ee and 78% yield. Use of β-ketoamide 35 provided diazene 36 in 90% ee and 91% yield. We were pleased to find that nonstabilized enamide 37 underwent enantioselective C–N bond formation, providing imine 38 in 80% ee and 59% yield.
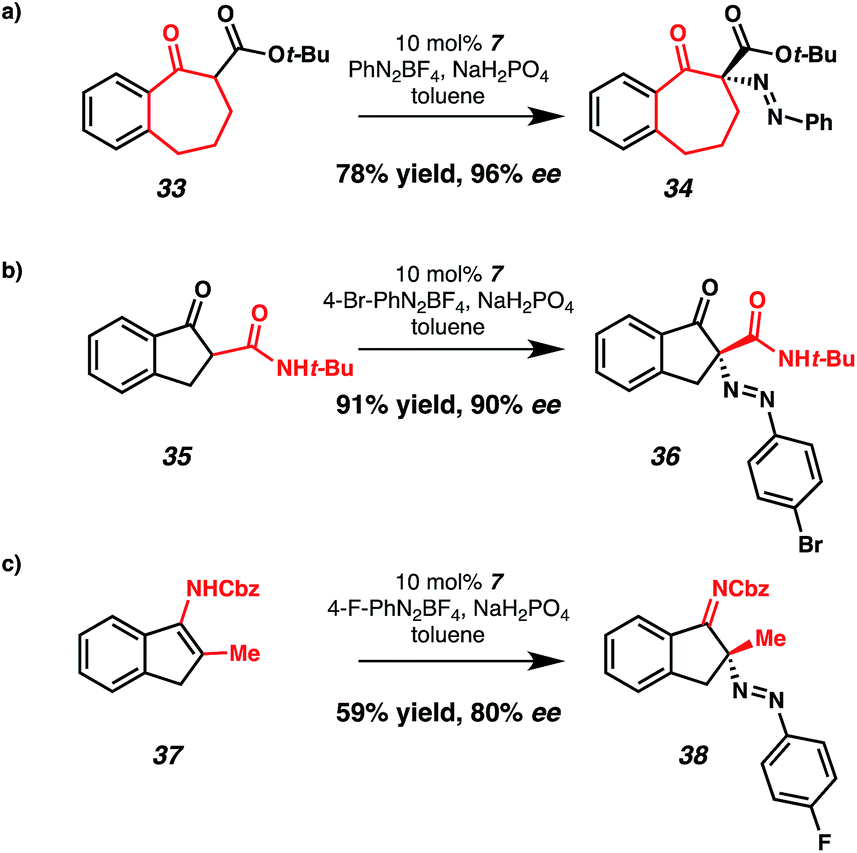 | ||
| Scheme 1 Diazenation of additional substrates. | ||
We were pleased to find that hydrogenation of indanone 18 and benzosuberone 34 under standard conditions smoothly formed their respective β-hydroxy amino acid derivatives with excellent diastereoselectivity and without loss of enantioenrichment (Scheme 2, 39 and 40). Additionally, compound 24 was reduced to protected CCTA 41, a synthetic precursor of Hai (4) using standard heterogeneous Pd/C conditions followed by homogeneous reduction with polymethylhydrosiloxane (PHMS) and PdCl2 (Scheme 2c).16 Importantly, enantioenriched compounds akin to amine 41 (Scheme 1c) have been previously prepared via classical resolution in 9 steps.9b Furthermore, as CCTAs such as Hai (4) are utilized for study of protein conformation,9 we envisioned that 15N-labeling would be of great utility, and a powerful application of our methodology. Towards this end, isotopically enriched amino indanone 42 was prepared in a three-step sequence from inexpensive Na15NO2. It is noteworthy that the use of an azodicarboxylate electrophile for this application would require two equivalents of isotopically enriched nitrogen.
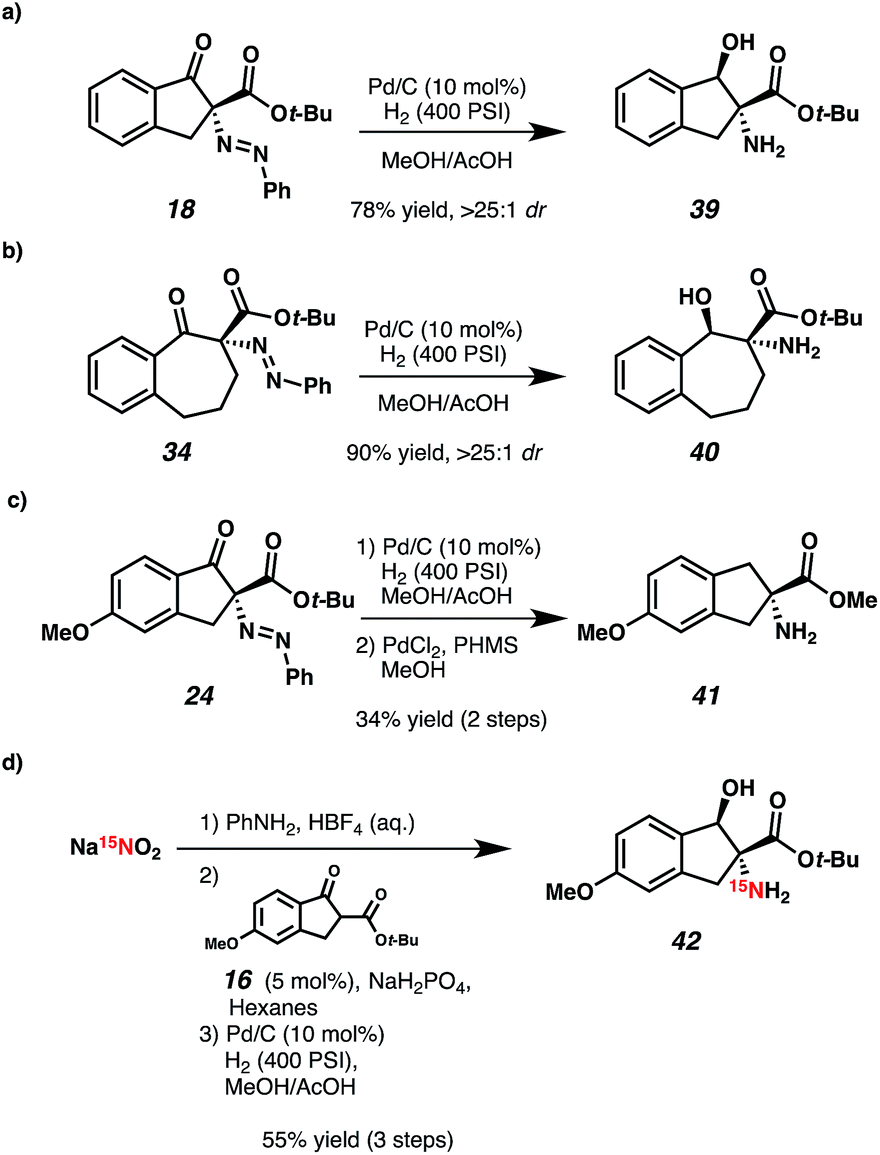 | ||
| Scheme 2 Applications of diazene products. | ||
Conclusions
In closing, we have developed a method for the enantioselective α-diazenation of enolate derivatives. This work was enabled by the development of novel BDPAs. The presented methodology possesses a broad scope, allowing for diazenation of diverse nucleophiles. As an application of our methodology, several diazenes were directly reduced to provide amino acid derivatives. Furthermore, facile 15N-labeling was demonstrated through preparation of protected amino acid 42.Acknowledgements
We gratefully acknowledge NIHGMS (R01 GM104534) for financial support. H.M.N. would like to acknowledge the UNCF and Merck for generous funding. We would also like to thank Dr Matthew Winston for useful discussion regarding 15N labeling and acknowledge the College of Chemistry CheXray (NIH Shared Instrumentation Grant S10-RR027172) and Antonio DiPasquale for X-ray crystallographic data.Notes and references
- (a) T. C. Nugent, Chiral amine synthesis: methods, developments and applications, John Wiley & Sons, 2011 Search PubMed; (b) M. T. Reetz, Angew. Chem., Int. Ed., 1991, 30, 1531–1546 CrossRef.
- (a) E. Erdik, Tetrahedron, 2004, 60, 8747–8782 CrossRef CAS; (b) C. Najera and J. Sansano, Chem. Rev., 2007, 107, 4584–4671 CrossRef CAS PubMed; (c) J. M. Janey, Angew. Chem., Int. Ed., 2005, 44, 4292–4300 CrossRef CAS PubMed.
- (a) L. A. Trimble and J. C. Vederas, J. Am. Chem. Soc., 1986, 108, 6397–6399 CrossRef CAS; (b) C. Gennari, L. Colombo and G. Bertolini, J. Am. Chem. Soc., 1986, 108, 6394–6395 CrossRef CAS; (c) D. A. Evans, T. C. Britton, R. L. Dorow and J. F. Dellaria, Tetrahedron, 1988, 44, 5525–5540 CrossRef CAS; (d) D. A. Evans, T. C. Britton, R. L. Dorow and J. F. Dellaria, J. Am. Chem. Soc., 1986, 108, 6395–6397 CrossRef CAS.
- (a) A. Bøgevig, K. Juhl, N. Kumaragurubaran, W. Zhuang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2002, 41, 1790–1793 CrossRef; (b) B. List, J. Am. Chem. Soc., 2002, 124, 5656–5657 CrossRef CAS PubMed; (c) H. Konishi, T. Y. Lam, J. P. Malerich and V. H. Rawal, Org. Lett., 2010, 12, 2028–2031 CrossRef CAS PubMed; (d) M. Terada, M. Nakano and H. Ube, J. Am. Chem. Soc., 2006, 128, 16044–16045 CrossRef CAS PubMed; (e) R. He, X. Wang, T. Hashimoto and K. Maruoka, Angew. Chem., Int. Ed., 2008, 47, 9466–9468 CrossRef CAS PubMed; (f) C. L. Zhu, F. G. Zhang, W. Meng, J. Nie, D. Cahard and J. A. Ma, Angew. Chem., Int. Ed., 2011, 50, 5869–5872 CrossRef CAS PubMed; (g) D. G. Blackmond, A. Moran, M. Hughes and A. Armstrong, J. Am. Chem. Soc., 2010, 132, 7598–7599 CrossRef CAS PubMed; (h) C. Liu, Q. Zhu, K. W. Huang and Y. Lu, Org. Lett., 2011, 13, 2638–2641 CrossRef CAS PubMed; (i) T. Y. Liu, H. L. Cui, Y. Zhang, K. Jiang, W. Du, Z. Q. He and Y. C. Chen, Org. Lett., 2007, 9, 3671–3674 CrossRef CAS PubMed.
- (a) D. A. Evans and S. G. Nelson, J. Am. Chem. Soc., 1997, 119, 6452–6453 CrossRef CAS; (b) G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521–11524 CrossRef CAS PubMed; (c) K. Juhl and K. A. Jørgensen, J. Am. Chem. Soc., 2002, 124, 2420–2421 CrossRef CAS PubMed; (d) T. Mashiko, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 14990–14999 CrossRef CAS PubMed.
- (a) H. M. Nelson, S. H. Reisberg, H. P. Shunatona, J. S. Patel and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 5600–5603 CrossRef CAS PubMed; (b) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed.
- H. M. Pinheiro, E. Touraud and O. Thomas, Dyes Pigm., 2004, 61, 121–139 CrossRef CAS.
- (a) A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622–4643 CAS; (b) H. Bonin, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2011, 353, 3063–3084 CrossRef CAS.
- (a) R. M. Pinder, B. H. Butcher, D. A. Buxton and D. J. Howells, J. Med. Chem., 1971, 14, 892–893 CrossRef CAS PubMed; (b) F. N. Murigi, G. S. Nichol and E. A. Mash, J. Org. Chem., 2010, 75, 1293–1296 CrossRef CAS PubMed; (c) H. I. Mosberg, A. L. Lomize, C. Wang, H. Kroona, D. L. Heyl, K. Sobczyk-Kojiro, W. Ma, C. Mousigian and F. Porreca, J. Med. Chem., 1994, 37, 4371–4383 CrossRef CAS PubMed; (d) T. Deeks, P. A. Crooks and R. D. Waigh, J. Med. Chem., 1983, 26, 762–765 CrossRef CAS PubMed; (e) M. T. Cox, Opiate Receptor Antagonists, US Pat., 4464358 A, 7 August 1984.
- (a) M. R. Wood, K. M. Schirripa, J. J. Kim, R. A. Bednar, J. F. Fay, J. G. Bruno, E. L. Moore, S. D. Mosser, S. Roller, C. A. Salvatore, J. P. Vacca and H. G. Selnick, Bioorg. Med. Chem. Lett., 2010, 20, 6827–6830 CrossRef CAS PubMed; (b) C. A. Stump, I. M. Bell, R. A. Bednar, J. F. Fay, S. N. Gallicchio, J. C. Hershey, R. Jelley, C. Kreatsoulas, E. L. Moore, S. D. Mosser, A. G. Quigley, S. A. Roller, C. A. Salvatore, S. S. Sharik, C. R. Theberge, C. B. Zartman, S. A. Kane, S. L. Graham, H. G. Selnick, J. P. Vacca and T. M. Williams, Bioorg. Med. Chem. Lett., 2010, 20, 2572–2576 CrossRef CAS PubMed; (c) I. M. Bell and H. G. Selnick, Monocyclic Amide CGRP Receptor Antagonists, US Pat., 172,205, 14 July 2011.
- Z. H. Mardirossian, H. G. Kiryakovt, J. P. Ruder and D. B. MacLean, Phytochemistry, 1983, 22, 759–761 CrossRef CAS.
- The connectivity and absolute stereochemistry of 18 was confirmed by X-ray crystallography. See the ESI† for diffraction data.
- (a) M. Hatano, T. Ikeno, T. Matsumura, S. Torii and K. Ishihara, Adv. Synth. Catal., 2008, 350, 1776–1780 CrossRef CAS; (b) M. Terada, K. Sorimachi and F. Uraguchi, Synlett, 2006, 1, 133–136 CrossRef.
- Hydrogen-bonding of chiral anion phase-transfer catalysts to substrates has been envoked in several transition state models, see: (a) X. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225–5228 CrossRef CAS PubMed; (b) R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268–1271 CrossRef CAS PubMed; (c) J. Wu, Y.-M. Wang, A. Drljevic, V. Rauniyar, R. J. Phipps and F. D. Toste, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13729–13733 CrossRef CAS PubMed.
- See the ESI† for the syntheses of BDPAs.
- H. Wang, L. Li, X.-F. Bai, J.-Y. Shang, K.-F. Yang and L.-W. Xu, Adv. Synth. Catal., 2013, 355, 341–347 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures and characterization data. CCDC 1025135. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02494j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |