
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into hydroacylation with non-chelating aldehydes†
Stephen K.
Murphy
ab,
Achim
Bruch
a and
Vy M.
Dong
*a
aDepartment of Chemistry, University of California, Irvine, California 92697-2025, USA. E-mail: dongv@uci.edu
bDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada
First published on 22nd September 2014
Abstract
The combination of a small-bite-angle diphosphine bis(dicyclohexylphosphino)methane (dcpm) and [Rh(cod)OMe]2 catalyses the hydroacylation of 2-vinylphenols with a wide range of non-chelating aldehydes. Here we present a detailed experimental study that elucidates the factors contributing to the broad aldehyde scope and high reactivity. A variety of catalytically relevant intermediates were isolated and a [Rh(dcpm)(vinylphenolate)] complex was identified as the major catalytically relevant species. A variety of off-cycle intermediates were also identified that can re-enter the catalytic cycle by substrate- or 1,5-cyclooctadiene-mediated pathways. Saturation kinetics with respect to the 2-vinylphenol were observed, and this may contribute to the high selectivity for hydroacylation over aldehyde decarbonylation. A series of deuterium labelling experiments and Hammett studies support the oxidative addition of Rh to the aldehyde C–H bond as an irreversible and turnover-limiting step. The small bite angle of dcpm is crucial for lowering the barrier of this step and providing excellent reactivity with a variety of aldehydes.
Introduction
Central challenges in Rh-catalysed olefin hydroacylation are to promote aldehyde C–H activation while preventing decarbonylation, and to control the regioselectivity of C–C bond formation (Fig. 1).1 Most intermolecular hydroacylations are inherently linear-selective and rely on chelating aldehydes such as salicylaldehydes,2 2-aminobenzaldehydes,3 β-sulphur aldehydes,4 or 2-pyridylaldimines5 to promote C–H activation and suppress decarbonylation. Brookhart, Tanaka, and Jun have made breakthroughs in the field by developing linear-selective protocols with non-chelating aldehydes.6 Alternatively, Co catalysts,7 Ru hydrides8 and N-heterocyclic carbenes9 have emerged as promising catalysts for the linear-selective coupling of aldehydes with α,β-unsaturated carbonyl compounds, 1,3-dienes, cyclopropenes, and some styrenes. Developing methods that provide the alternative branched products without relying on chelating aldehydes would enable access to unique chemical space by hydroacylation.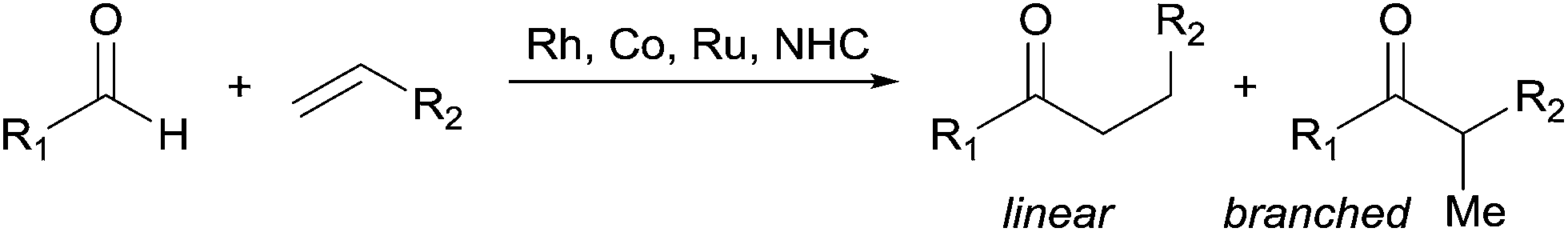 | ||
| Fig. 1 Intermolecular olefin hydroacylation. | ||
We recently reported a strategy for branched-selective hydroacylation with an aldehyde scope that is among the most broad reported to date (Fig. 2).10 Alkyl, alkenyl, and aryl aldehydes were coupled with 2-vinylphenols in generally >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 branched-to-linear (b:l) selectivity and excellent yields. We hypothesized that the phenolic group on the olefin promotes the branched regioselectivity in analogy to our double-chelating hydroacylations with salicylaldehydes.2c–e A [Rh(cod)OMe]2 catalyst in combination with a small-bite-angle diphosphine, bis(dicyclohexylphosphino)methane (dcpm), was crucial for reactivity. This method enabled short syntheses of four neolignan natural products using hydroacylation in combination with an acid-catalysed cyclocondensation. Herein we report a mechanistic study that sheds light on the catalyst resting states of this transformation, the rate-limiting step, and the properties of the substrates and ligands that enable reactivity with non-chelating aldehydes.
:1 branched-to-linear (b:l) selectivity and excellent yields. We hypothesized that the phenolic group on the olefin promotes the branched regioselectivity in analogy to our double-chelating hydroacylations with salicylaldehydes.2c–e A [Rh(cod)OMe]2 catalyst in combination with a small-bite-angle diphosphine, bis(dicyclohexylphosphino)methane (dcpm), was crucial for reactivity. This method enabled short syntheses of four neolignan natural products using hydroacylation in combination with an acid-catalysed cyclocondensation. Herein we report a mechanistic study that sheds light on the catalyst resting states of this transformation, the rate-limiting step, and the properties of the substrates and ligands that enable reactivity with non-chelating aldehydes.
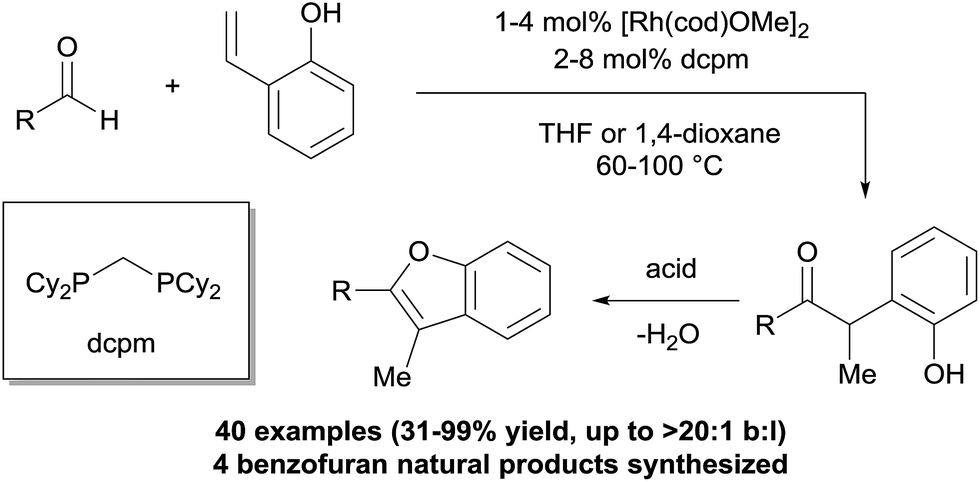 | ||
| Fig. 2 Intermolecular 2-vinylphenol hydroacylation and cyclocondensation to access benzofuran natural products. | ||
Results and discussion
A. Assignment of catalyst resting states
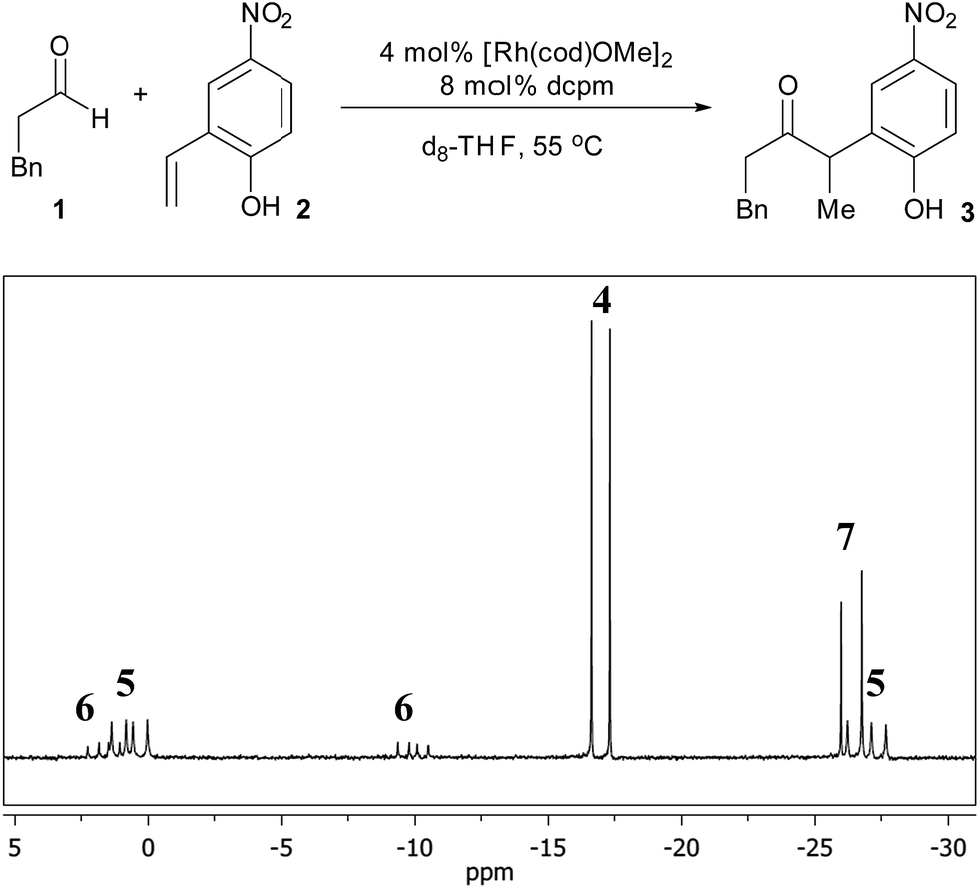
To identify potential reaction intermediates, we monitored the 31P NMR of a catalytic hydroacylation involving hydrocinnamaldehyde (1) and 4-nitro-2-vinylphenol (2) (Table 1). At 12% conversion, four different phosphorus-containing complexes (4, 5, 6 and 7) were detected in an approximately 3:3:1:1 ratio, respectively, based on integration of the 31P NMR signals. Over the course of catalysis, complexes 5 and 6 decreased in concentration while only complexes 4 and 7 remained at the end of the reaction.
|
|
|||
|---|---|---|---|
| Complex | δ (ppm) | J Rh–P (Hz) | J P–P (Hz) |
| 4 | −17.0 | 111 | — |
| 5 | 0.7, −26.9 | 125, 148 | 92 |
| 6 | 1.7, −9.9 | 126, 117 | 69 |
| 7 | −26.4 | 126 | — |
The major complex 4 was prepared by reacting [Rh(cod)OMe]2 with dcpm and 4-nitro-2-vinylphenol in a 1:2:2 molar ratio in THF (Fig. 3, top). A cationic [Rh(dcpm)2]+ fragment was identified by ESI+ MS and by comparison of the NMR data with that of [Rh(dcpm)2]BF4 (8). The synthesis of 4 was accompanied by quantitative formation of 1,5-cyclooctadiene, MeOH and another Rh complex 9, containing a coordinated vinylphenolate anion as judged by 1H NMR analysis. The aromatic protons of the vinylphenolate were shifted upfield and the vinylic peaks appeared between 2 and 4 ppm—within the range of coordinated olefins. The 13C NMR showed coupling between the vinylic carbon atoms and the Rh center (JRh–C = 11 and 15 Hz). Based on this data and the ESI− MS, we assigned this complex as an anionic 2:1 vinylphenolate-to-Rh complex 9. To support our assignment of anion 9, the [(18-crown-6)K][Rh(nitrovinylphenolate)2] salt 10 was synthesized by treating two equivalents of 4-nitro-2-vinylphenol with [Rh(cod)OMe]2, t-BuOK and 18-crown-6 (Fig. 3, bottom). The solid state structure of salt 10 (Fig. 3, right) contains two vinylphenolate anions bound to a Rh atom in a cis orientation, where the phenolate oxygen atoms bind to the potassium centre. Only small differences in chemical shift were noted between 9 and 10, which are likely due to potassium coordination in 10. Additionally, small shoulder peaks were noted in the 1H NMR spectrum of 9, which are likely a result of geometrical isomers.
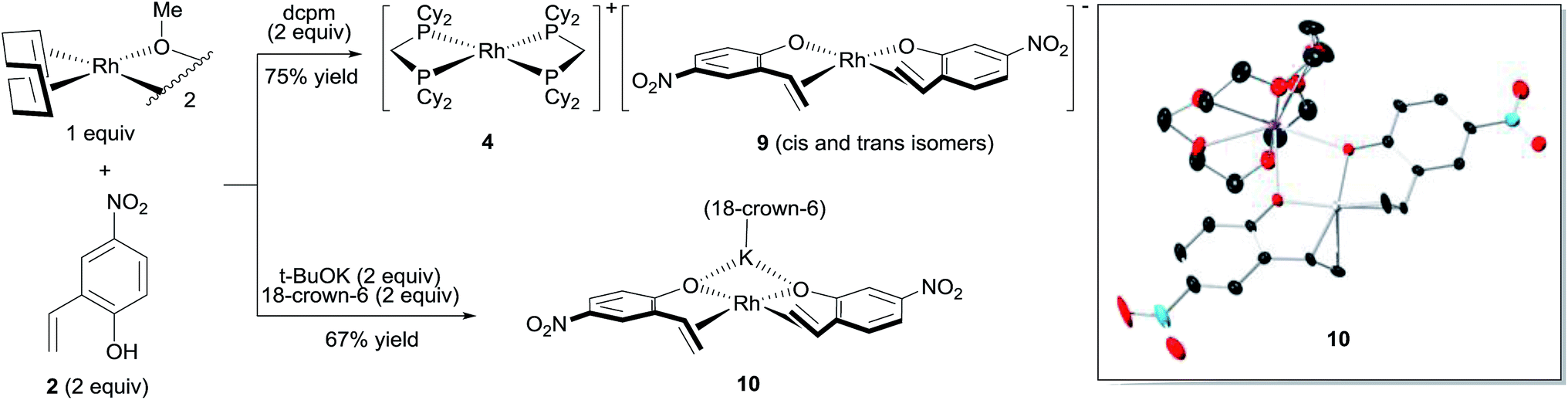 | ||
| Fig. 3 Synthesis of [Rh(vinylphenolate)2]− complexes. | ||
Given that the double salt of 4 and 9 accounts for nearly 60% of the Rh during catalysis, the reactivity of these species strongly correlates with the activity of the overall catalyst system. We thus decided to examine the reactivity of each ion independently by studying complexes 8 ([Rh(dcpm)2]BF4) and 10 [(18-crown-6)K][Rh(nitrovinylphenolate)2] (Table 2). While [Rh(diphosphine)2]+ complexes catalyse the cyclization of 4-pentenals at elevated temperatures,11 complex 8 did not catalyse vinylphenol hydroacylation even in the presence of base (t-BuOK, entries 2 and 3). Salt 10 also did not catalyse hydroacylation (entry 4), but it could be activated by dcpm to give an initial turnover frequency (TOFinit) four times lower than the optimized reaction (entries 1 and 5, 15 h−1versus 4 h−1, respectively). Although this rate is low, the observation of catalysis supports the intermediacy of phosphine-ligated species and suggests that 4 and 9 are not directly on the catalytic cycle. The combination of 8, 10, and 1,5-cyclooctadiene resulted in slow catalysis (entries 6 and 7, approx. 50% conv. after 24 hours). Apparently, the 1,5-cyclooctadiene converts the double salt back to a catalytically active species, although this effect is likely smaller than the inhibitory effect of 1,5-cyclooctadiene that occurs due to binding of the active catalyst (vide infra).
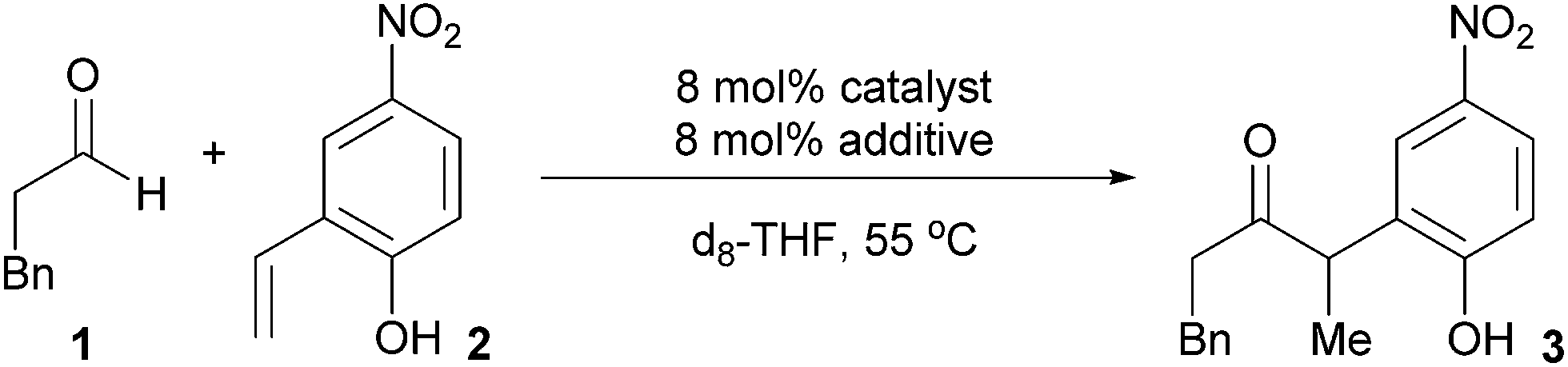
Complex 7 is the next most abundant Rh complex during catalysis, and was identified as a cationic [Rh(dcpm)(cod)]+ fragment by comparison of the 31P NMR spectrum to that of [Rh(dcpm)(cod)]BF4 (11). This latter complex is a kinetically competent catalyst in the presence of t-BuOK and provides a similar initial turnover frequency to the optimized conditions (18 h−1versus 15 h−1, respectively).12 When a suspension of 4-nitro-2-vinylphenol and t-BuOK was vigorously stirred with complex 11, a nearly quantitative exchange reaction occurred to generate substrate-bound 5 (Fig. 4). In the presence of excess aldehyde, the olefin ligand of complex 5 exchanges with the aldehyde to generate intermediate 6, which is observed during catalysis. Complex 6 is only observed transiently and undergoes hydroacylation and a substitution reaction at room temperature to form cation 7 and anion 3−, which aggregates with excess phenols as judged by ESI− MS.
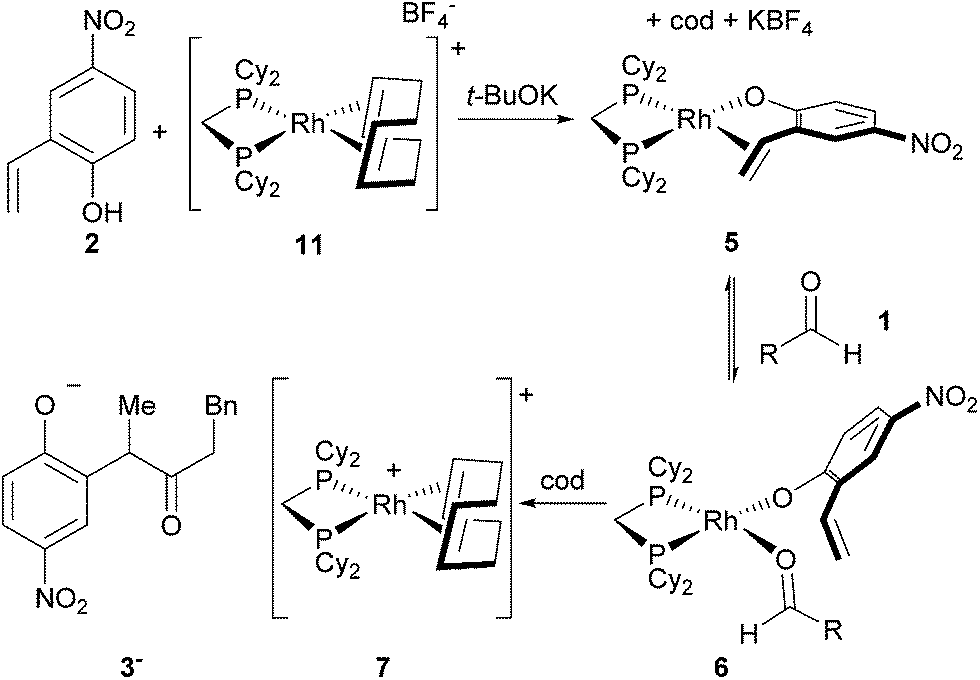 | ||
| Fig. 4 Formation of the olefin-bound complex and its behaviour in the presence of excess aldehyde (R = CH2Bn). | ||
Our investigation of catalyst resting states reveals that 5 and 6 are directly on the catalytic cycle, while a variety of off-cycle intermediates can re-enter the cycle via pathways mediated by 1,5-cyclooctadiene or 2-vinylphenol. The strong coordination of the 2-vinylphenol compared to the aldehyde is likely a key to promoting hydroacylation over competitive decarbonylation. Of the observed Rh complexes, oxidative addition would only be possible from 6, which already contains the O-coordinated 2-vinylphenolate moiety. Formation of the acyl-Rh-hydride would likely be followed by rapid coordination of the olefin to generate a coordinatively saturated intermediate that is stable towards decarbonylation,13 thus providing the desired chemoselectivity.
B. Kinetic analysis using in situ NMR monitoring of reaction progress
To identify kinetic parameters, we studied the hydroacylation of 4-nitro-2-vinylphenol under catalytic conditions with various amounts of hydrocinnamaldehyde (Fig. 5). The coupling reaction with 1.5 equivalents of hydrocinnamaldehyde follows a curved reaction profile with an initial turnover frequency of 15 h−1 and no induction period. Decreasing the aldehyde concentration by half decreases the initial turnover frequency by half (8 h−1), a result that suggests a first order dependence of the rate on aldehyde concentration. Using a large excess of aldehyde (8 equiv) yields a reaction profile that is linear up to 80% conversion with an initial turnover frequency of only 23 h−1. Analysing the 31P spectrum under these highly concentrated conditions reveals that the equilibrium between 5 and 6 is completely shifted toward 6. The formation of 6, which contains both a coordinated aldehyde and phenolate, leads to saturation kinetics with respect to both substrates. Under typical catalytic conditions, however, this kinetic data and the assignment of catalyst resting states supports a first order dependence of the rate on aldehyde concentration and a zeroth order dependence on 2-vinylphenol concentration. These results corroborate the concentration-dependent selectivity for hydroacylation over competitive aldol dehydration (Fig. 6).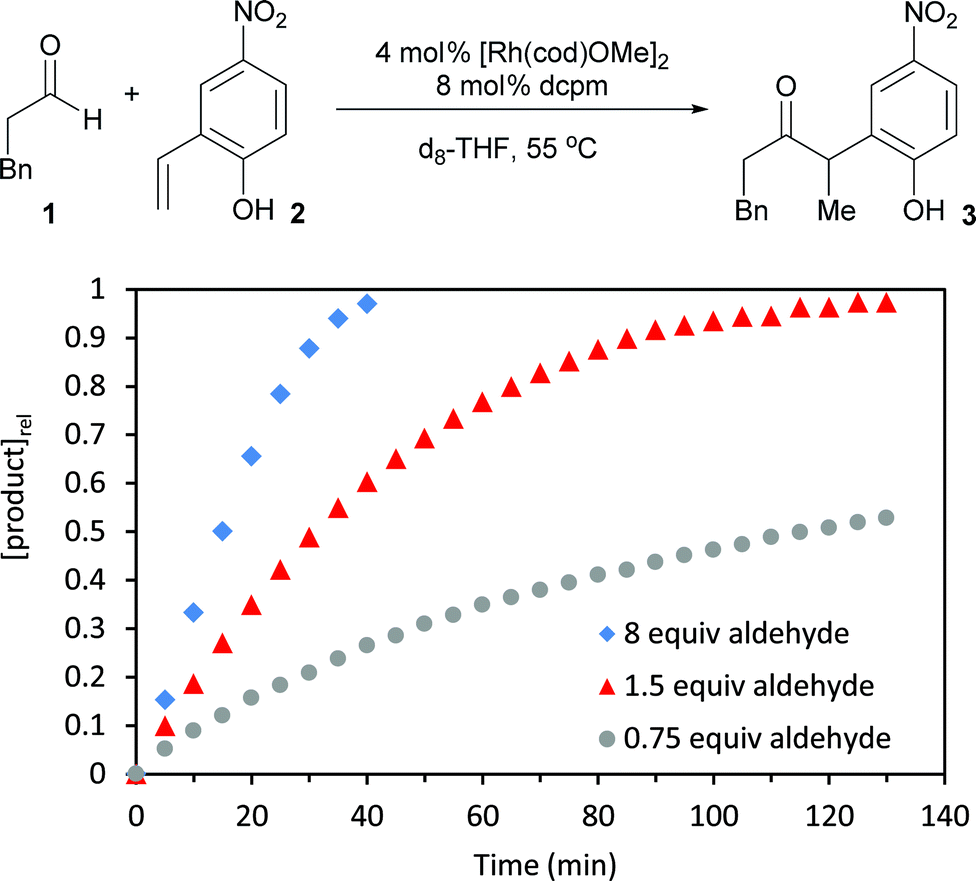 | ||
| Fig. 5 Kinetic data for hydroacylation with various amounts of hydrocinnamaldehyde. | ||
 | ||
| Fig. 6 Concentration-dependent chemoselectivity for hydroacylation over aldol dehydration. | ||
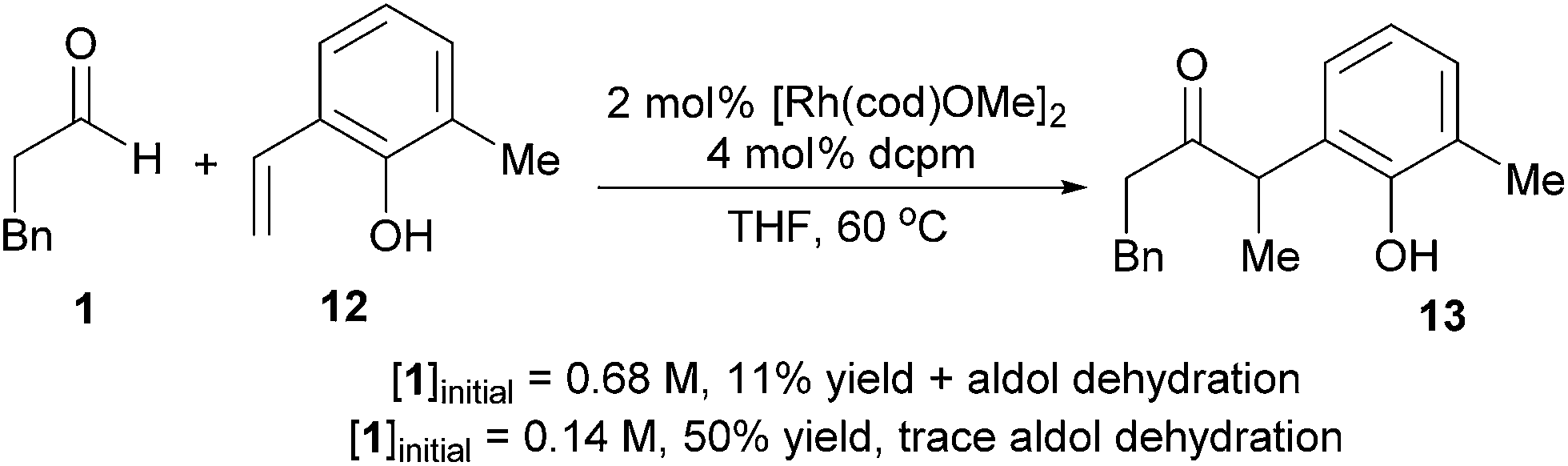
The substrate 6-methyl-2-vinylphenol (12) undergoes slow hydroacylation due to its steric bulk, which allows the aldehyde to be consumed by dimerization. However, greater chemoselectivity for hydroacylation over aldol dehydration was observed upon fivefold dilution. Diluting the reaction slows down aldol dehydration (second order in aldehyde) more dramatically than vinylphenol hydroacylation (first order in aldehyde, zeroth order in vinylphenol).
C. Deuterium labelling studies
We probed the turnover-limiting step of the reaction by tracking the deuterium label for hydroacylation with deuterated 2-naphthaldehyde (Table 3, entry 1). This substrate coupled with 4-nitro-2-vinylphenol in 78% yield and 12:1 b:l selectivity. 1H and 2D NMR analysis revealed that deuterium was incorporated into only the β-positions of the resulting ketones. Analysis of the products by mass spectrometry revealed that they were exclusively monodeuterated. Analogous results were obtained for the reaction of deuterated hydrocinnamaldehyde with 4-nitro-2-vinylphenol (Table 3, entry 2). The lack of deuterium scrambling and multiply-deuterated products rules out reductive elimination as the turnover-limiting step and suggests that at least one of the earlier steps in the catalytic cycle is irreversible.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligandd | R | Time (h) | Yield (%) | b:l |
|
a b:l ratios were determined by NMR analysis of the crude reaction mixtures.
b Entry 1: 1,4-dioxane, 100 °C. Entries 2–5: THF, 60 °C.
c Deuterium content measured by 1H and 2D NMR, and ESI MS.
d dcpe = 1,2-bis(dicyclohexylphosphino)ethane; dcpp = 1,3-bis(dicyclohexylphosphino)propane; dcpb = 1,4-bis(dicyclohexylphosphino)butane.
|
|||||
| 1 | dcpm | 2-nap | 24 | 78 | 12:1 |
| 2 | dcpm | CH2Bn | 2 | 90 | >20:1 |
| 3 | dcpe | CH2Bn | 12 | 45 | >20:1 |
| 4 | dcpp | CH2Bn | 12 | 41 | >20:1 |
| 5 | dcpb | CH2Bn | 12 | 35 | >20:1 |
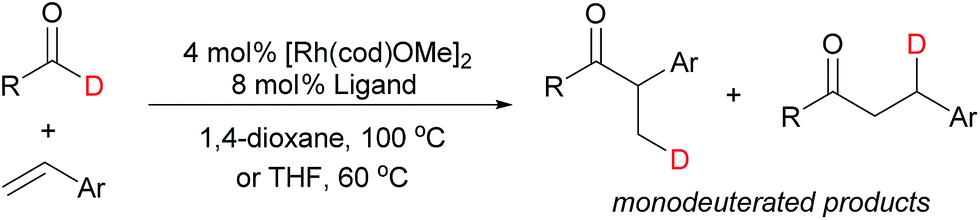
This observation contrasts almost all other olefin hydroacylation studies where extensive deuterium-scrambling is observed as a result of rate-limiting reductive elimination.14,15 When larger-bite-angle diphosphines were used in the test for deuterium scrambling (Table 3, entries 3–5), the reaction rates were significantly reduced, deuterium scrambling still did not occur, and only monodeuterated products were identified. These results suggest that reductive elimination is not rate-determining for this substrate combination, regardless of ligand bite angle. Accordingly, the fact that smaller-bite-angle diphosphines provide faster rates can be attributed to their abilities to promote earlier steps in the catalytic cycle (vide infra).
To further probe the turnover-limiting step, we measured kinetic isotope effects (KIE) via intermolecular competition experiments of deuterated aldehydes with their protio analogues.16 With 4-nitro-2-vinylphenol as the coupling partner, a KIE of 2.5 ± 0.2 was observed for the reaction with hydrocinnamaldehyde and 2.4 ± 0.2 for the reaction with 2-naphthaldehyde. The similar magnitudes of the KIE values indicate that these reactions proceed by similar mechanisms. We recently reported a KIE of 2.6 for an intermolecular ketone hydroacylation with non-chelating aldehydes, in which oxidative addition of Rh to the aldehyde C–H bond was rate limiting.17 Madsen reported a similar value of 2.85 for the oxidative addition of aldehydes to Rh(dppp)+.18 Lower KIE values of 1.4–1.7 are associated with hydroacylation reactions that involve rate-limiting migratory insertion or reductive elimination.19,4e The large primary KIEs observed in the present case suggest that irreversible oxidative addition is likely the turnover-limiting step.
D. Hammett studies
We conducted a Hammett study by reacting different para-substituted benzaldehydes with 4-nitro-2-vinylphenol (Fig. 7).20,21 Aldehydes with electron-withdrawing substituents reacted faster, while those with electron-donating groups reacted slower. A positive ρ value of 0.79 was measured, which indicates a build-up of negative charge in the transition state of the turnover-limiting step. This description is consistent with the transition state for oxidative addition of Rh to the aldehyde C–H bond.22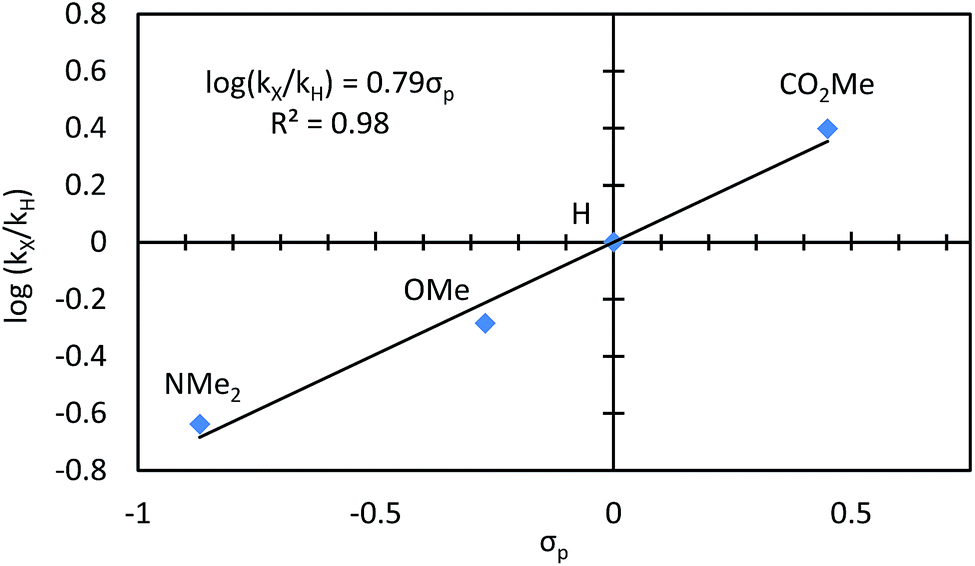 | ||
| Fig. 7 Hammett study for para-substituted benzaldehydes. | ||
E. Rationale for the ligand effects
Having established the catalyst resting states and the turnover-limiting step, we conclude that the rate of 2-vinylphenol hydroacylation is dictated predominantly by the rate of oxidative addition. Thus, the ligand dcpm likely accelerates this key step relative to its wide-bite-angle counterparts. To better understand the steric component of this effect,23 we examined the reactivity of the ligand (tBu)2PCH2P(tBu)(Me), which has a larger steric profile than dcpm but similar electronic properties and bite angle. This ligand did not provide an active catalyst. We thus propose that the small size of dcpm reduces steric repulsion in the transition state for oxidative addition. A pronounced electronic effect is also apparent, based on the comparison of dcpm with dppm (bis(diphenylphosphino)methane): faster rates were observed with the more basic disphosphine dcpm, despite its increased steric bulk. Our results contrast findings on the addition of H2 to Rh(diphosphine)2+ complexes, and the hydroacylation of alkenes with β-sulfur-substituted aldehydes. In the first case, wider bite angles favoured oxidative addition as a result of an electronic bite-angle effect;24 and in the latter case, small-bite-angle diphosphines promoted a rate-limiting reductive elimination.4eF. Proposed mechanism
On the basis of our experimental evidence and literature precedence, we propose the mechanism shown in Fig. 8. The catalyst precursor [Rh(cod)OMe]2 reacts with dcpm and vinylphenol 2 to form either the catalytically inactive double salt of 4 and 9 or the major catalyst resting state 5. Exchange of the olefin ligand for aldehyde 1 leads to intermediate 6. Both complexes 5 and 6 are catalyst resting states and they decrease in concentration as the reaction progresses. Supported by kinetic isotope effects and a Hammett study, we propose that oxidative addition is turnover-limiting. The resulting acyl-RhIII-hydride 14 does not reductively decarbonylate to any measurable extent, but instead undergoes migratory insertion to yield intermediate 15.25 Reductive elimination and displacement of the product 3 with substrate 2 completes the catalytic cycle. Alternatively, 1,5-cyclooctadiene can displace either the vinylphenol (2) or product (3) to form the off-cycle intermediate 7, which increases in concentration during catalysis. The counterion of 7 is a dissociated phenolate anion that can hydrogen bond with excess substrate or product.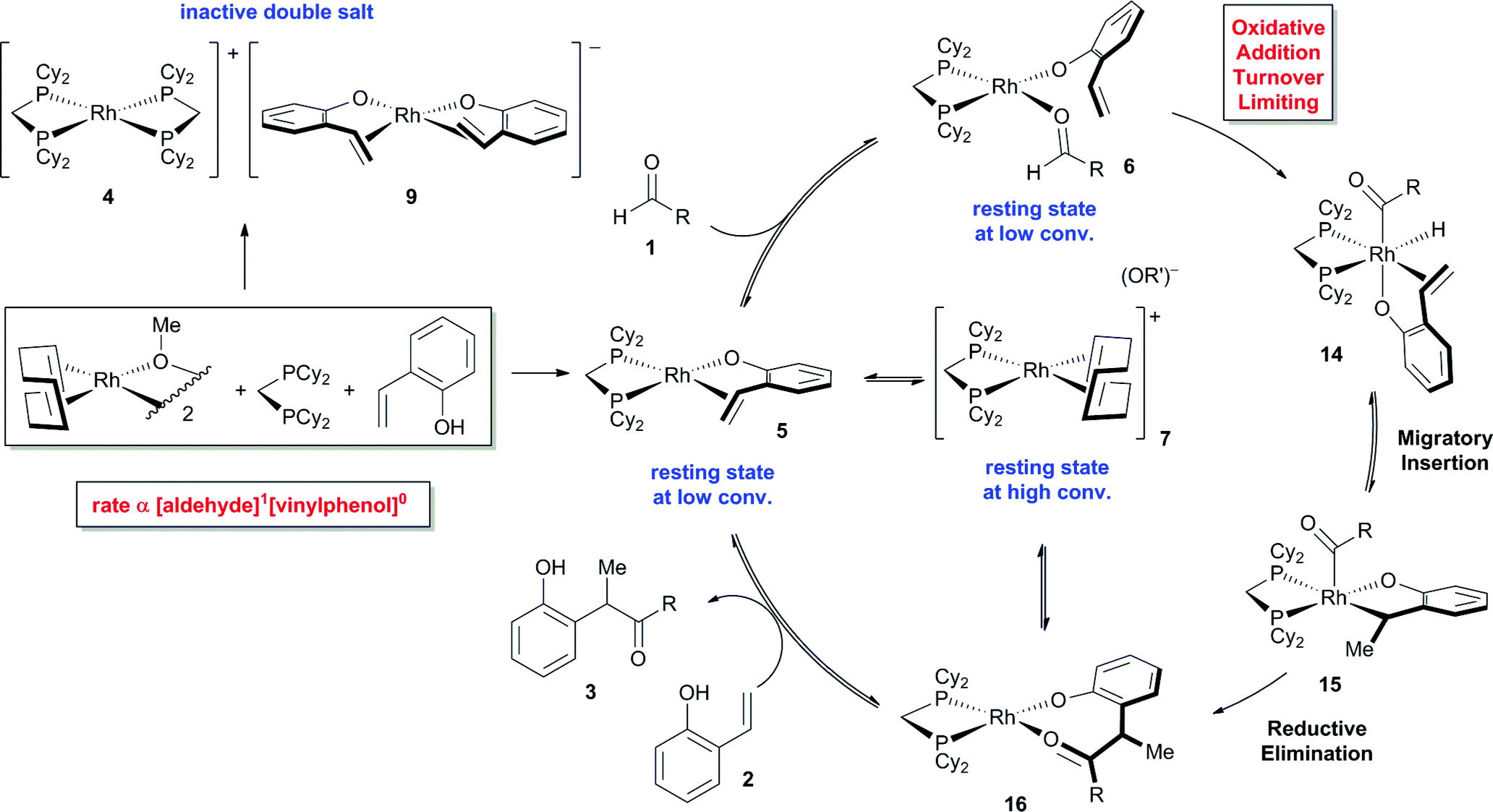 | ||
| Fig. 8 Proposed mechanism for vinylphenol hydroacylation (4-nitro group omitted from the vinylphenol, R = CH2Bn). | ||
Conclusions
We have disclosed a thorough mechanistic study on the branched-selective hydroacylation of 2-vinylphenols with a wide range of aryl, alkenyl, and alkyl aldehydes. Analysis of the catalyst resting states revealed that most of the catalyst is sequestered as an inactive double salt of Rh, while the active catalyst consists of a Rh(dcpm)(vinylphenolate) fragment. Non-directed oxidative addition of Rh to the aldehyde C–H bond occurs even at room temperature, followed by rapid hydroacylation. This strong binding of the vinylphenolate is likely a key to promoting hydroacylation over competitive aldehyde decarbonylation and aldol dehydration. KIE measurements and a Hammett study support oxidative addition as the turnover-limiting step. The high reactivity of neutral [Rh(X)(dcpm)] fragments likely arises from the electron-rich character of the complex, and small bite angle of the diphosphine, which promotes oxidative addition. In contrast, previous studies that use chelating aldehydes involve a fast and reversible C–H bond activation. This difference in mechanism highlights the challenge of non-directed aldehyde activation. Given the mild conditions of this transformation, [Rh(X)(dcpm)] fragments hold promise for future hydroacylations with non-chelating aldehydes.Acknowledgements
We thank the National Institutes of Health (GM105938) for funding. S.K.M. is grateful for a Canada Graduate Scholarship (CGS), and A.B. is grateful for a fellowship within the Postdoc-Programme of the German Academic Exchange Service (DAAD). We thank Dr Joseph W. Ziller and Jordan Corbey for X-ray crystallographic analysis.Notes and references
- For a review on hydroacylation, see: M. C. Willis, Chem. Rev., 2010, 110, 725 CrossRef CAS PubMed.
- (a) M. Tanaka, M. Imai, Y. Yamamoto, K. Tanaka, M. Shimowatari, S. Nagumo, N. Kawahara and H. Suemune, Org. Lett., 2003, 5, 1365 CrossRef CAS PubMed; (b) M. Imai, M. Tanaka, K. Tanaka, Y. Yamamoto, N. Imai-Ogata, M. Shimowatari, S. Nagumo, N. Kawahara and H. Suemune, J. Org. Chem., 2004, 69, 1144 CrossRef CAS PubMed; (c) M. M. Coulter, K. G. M. Kou, B. Galligan and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16330 CrossRef CAS PubMed; (d) S. K. Murphy, M. M. Coulter and V. M. Dong, Chem. Sci., 2012, 3, 355 RSC; (e) S. K. Murphy, D. A. Petrone, M. M. Coulter and V. M. Dong, Org. Lett., 2011, 13, 6216 CrossRef CAS PubMed; (f) K. Kokubo, K. Matsumasa, M. Miura and M. Nomura, J. Org. Chem., 1997, 62, 4564 CrossRef CAS; (g) K. Kokubo, K. Matsumasa, Y. Nishinaka, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1999, 72, 303 CrossRef CAS; (h) R. T. Stemmler and C. Bolm, Adv. Synth. Catal., 2007, 349, 1185 CrossRef CAS; (i) Y. Inui, M. Tanaka, M. Imai, K. Tanaka and H. Suemune, Chem. Pharm. Bull., 2009, 57, 1158 CrossRef CAS; (j) D. H. T. Phan, K. G. M. Kou and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16354 CrossRef CAS PubMed; (k) M. von Delius, C. M. Le and V. M. Dong, J. Am. Chem. Soc., 2012, 134, 15022 CrossRef CAS PubMed.
- M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 13280 CrossRef CAS PubMed.
- For hydroacylation with β-sulfur substituted aldehydes, see: (a) M. C. Willis, S. J. McNally and P. J. Beswick, Angew. Chem., Int. Ed., 2004, 43, 340 CrossRef CAS PubMed; (b) G. L. Moxham, H. E. Randell-Sly, S. K. Brayshaw, R. L. Woodward, A. S. Weller and M. C. Willis, Angew. Chem., Int. Ed., 2006, 45, 7618 CrossRef CAS PubMed; (c) G. L. Moxham, H. E. Randell-Sly, S. K. Brayshaw, A. S. Weller and M. C. Willis, Chem. – Eur. J., 2008, 14, 8383 CrossRef CAS PubMed; (d) J. D. Osborne and M. C. Willis, Chem. Commun., 2008, 5025 RSC; (e) A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885 CrossRef CAS PubMed; (f) I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779 CrossRef CAS.
- For (2-pyridyl)aldimines, see: (a) J. W. Suggs, J. Am. Chem. Soc., 1979, 101, 489 CrossRef CAS; (b) C.-H. Jun, J.-B. Kang and J.-Y. Kim, J. Organomet. Chem., 1993, 458, 193 CrossRef CAS; (c) C.-H. Jun, J.-S. Han, J.-B. Kang and J.-Y. Kim, Tetrahedron Lett., 1993, 34, 6431 CrossRef CAS; (d) C.-H. Jun, J.-S. Han, J.-B. Kang and S.-I. Kim, J. Organomet. Chem., 1994, 474, 183 CrossRef CAS; (e) M. C. Willis and S. Sapmaz, Chem. Commun., 2001, 2558 RSC.
- (a) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (b) K. Tanaka, Y. Shibata, T. Suda, Y. Hagiwara and M. Hirano, Org. Lett., 2007, 9, 1215 CrossRef CAS PubMed; (c) Y. Shibata and K. Tanaka, J. Am. Chem. Soc., 2009, 131, 12552 CrossRef CAS PubMed; (d) A. H. Roy, C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 2082 CrossRef CAS PubMed; (e) C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 1997, 119, 3165 CrossRef CAS; (f) C. P. Lenges, P. S. White and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 6965 CrossRef CAS.
- Q. A. Chen, D. K. Kim and V. M. Dong, J. Am. Chem. Soc., 2014, 136, 3772 CrossRef CAS PubMed.
- For branched-selective hydroacylation using complementary Ru-hydride catalysis, see: (a) T. Fukuyama, T. Doi, S. Minamino, S. Omura and I. Ryu, Angew. Chem., Int. Ed., 2007, 46, 5559 CrossRef CAS PubMed; (b) S. Omura, T. Fukuyama, J. Horiguchi, Y. Murakami and I. Ryu, J. Am. Chem. Soc., 2008, 130, 14094 CrossRef CAS PubMed; (c) F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14120 CrossRef CAS PubMed; (d) For a review of hydroacylation methods that proceed by a mechanism other than aldehyde C–H bond activation, see: J. C. Leung and M. J. Krische, Chem. Sci., 2012, 3, 2202 RSC.
- For styrenes, see: (a) M. Schedler, D.-S. Wang and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 2585 CrossRef CAS PubMed; (b) For cyclopropenes, see: X. Bugaut, F. Liu and F. Glorius, J. Am. Chem. Soc., 2011, 133, 8130 CrossRef CAS PubMed; (c) For selected examples of the intermolecular Stetter reaction with α,β-unsaturated carbonyl compounds, see: D. Enders, J. Han and A. Henseler, Chem. Commun., 2008, 3989 RSC; (d) D. Enders and J. Han, Synthesis, 2008, 3864 CrossRef CAS PubMed; (e) Q. Liu, S. Perreault and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14066 CrossRef CAS PubMed; (f) Q. Liu and T. Rovis, Org. Lett., 2009, 11, 2856 CrossRef CAS PubMed; (g) D. A. DiRocco, K. M. Oberg, D. M. Dalton and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10872 CrossRef CAS PubMed; (h) T. Jousseaume, N. E. Wurz and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1410 CrossRef CAS PubMed.
- S. K. Murphy, A. Bruch and V. M. Dong, Angew. Chem., Int. Ed., 2014, 53, 2455 CrossRef CAS PubMed.
- (a) B. R. James and C. G. Young, J. Chem. Soc., Chem. Commun., 1983, 1215 RSC; (b) B. R. James and C. G. Young, J. Organomet. Chem., 1985, 285, 321 CrossRef CAS.
- We expected catalyst 11 to exhibit a faster rate than the optimized reaction because none of the Rh would be sequestered as the double salt of 4 and 9. The fact that 11 provides only a slightly higher initial TOF than the optimized reaction is likely a result of the low solubility of the t-BuOK–vinylphenol mixture and the lack of stirring during NMR analysis.
- (a) D. Milstein, Organometallics, 1982, 1, 1549 CrossRef CAS; (b) D. Milstein, J. Chem. Soc., Chem. Commun., 1982, 1357 RSC.
- For examples of hydroacylation where deuterium scrambling is observed as a result of rate-limiting reductive elimination, see: (a) R. E. Campbell, C. F. Lochow, K. P. Vora and R. G. Miller, J. Am. Chem. Soc., 1980, 102, 5824 CrossRef CAS; (b) R. E. Campbell and R. G. Miller, J. Organomet. Chem., 1980, 186, C27 CrossRef CAS; (c) D. P. Fairlie and B. Bosnich, Organometallics, 1988, 7, 946 CrossRef CAS; (d) I. F. D. Hyatt, H. K. Anderson, A. T. Morehead Jr and A. L. Sargent, Organometallics, 2008, 27, 135 CrossRef CAS; (e) Ref. 6d ; (f) Ref. 2d ; (g) Ref. 2b .
- To our knowledge, there are only two examples of olefin hydroacylation where reductive elimination has been ruled out as the rate-limiting step, see: (a) M. C. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932 CrossRef CAS PubMed; (b) Ref. 2k .
- An intermolecular KIE experiment is appropriate for this system given that C–H bond cleavage is the first step in the catalytic cycle and our observed catalyst resting states ([Rh(dcpm)(vinylphenolate)]) and [Rh(dcpm)(vinylphenolate)(aldehyde)] directly precede aldehyde activation. For a discussion on KIEs in metal catalysis, see: (a) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed; (b) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857 CrossRef PubMed.
- K. G. M. Kou, D. N. Le and V. M. Dong, J. Am. Chem. Soc., 2014, 136, 9471 CrossRef CAS PubMed.
- P. Fristrup, M. Kreis, A. Palmelund, P.-O. Norrby and R. Madsen, J. Am. Chem. Soc., 2008, 130, 5206 CrossRef CAS PubMed.
- Z. Shen, P. K. Dornan, H. A. Khan, T. K. Woo and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 1077 CrossRef CAS PubMed.
- For use of a Hammett plot to study the mechanism of Rh(dppp)+ catalysed aldehyde decarbonylation, see ref. 18.
- In line with our observation of saturation kinetics with respect to the 2-vinylphenol, modifying the 2-vinylphenols at the 3 or 4 position also changes the reaction rate even though the substrate does not appear in the rate equation. More electron rich 2-vinylphenols appear to react slower than electron deficient substrates, but electron rich substrates also lead to unidentified side-products.
- For the effect of electronics on oxidative addition, see: (a) L. Xue and Z. Lin, Chem. Soc. Rev., 2010, 39, 1692 RSC; (b) K. Wang, T. J. Emge, A. S. Goldman, C. Li and S. P. Nolan, Organometallics, 1995, 14, 4929 CrossRef CAS; (c) Ref. 6d .
- (a) For case studies on the steric and electronic components of the bite angles effect of various catalytic reactions, see: Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890 RSC; (b) For a comparison of dppm and (tBu)2PCH2(tBu)2 in CO/ethylene oligomerization, see: E. Drent, J. A. M. Broekhoven and M. J. Doyle, J. Organomet. Chem., 1991, 417, 235 CrossRef CAS; (c) C. Bianchini, H. M. Lee, A. Meli, W. Oberhauser, M. Peruzzini and F. Vizza, Organometallics, 2002, 21, 16 CrossRef CAS.
- D. L. DuBois, D. M. Blake, A. Miedaner, C. J. Curtis, M. R. DuBois, J. A. Franz and J. C. Linehan, Organometallics, 2006, 25, 4414 CrossRef CAS.
- Branched-selective migratory insertion is generally fast and reversible for olefin hydroacylation, whereas linear-selective insertion has a higher barrier, see: (a) Ref. 16b ; (b) Ref. 2d ; (c) Ref. 15g ; (d) for a related example of the hydroformylation reaction, see: C. P. Casey and L. M. Petrovich, J. Am. Chem. Soc., 1995, 117, 6007 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, reaction procedures, characterization data. CCDC 1012849. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02026j |
| This journal is © The Royal Society of Chemistry 2015 |