DOI:
10.1039/C4SC02410A
(Edge Article)
Chem. Sci., 2015,
6, 308-321
Optical and electronic properties of air-stable organoboron compounds with strongly electron-accepting bis(fluoromesityl)boryl groups†
Received
8th August 2014
, Accepted 1st October 2014
First published on 1st October 2014
Abstract
Three compounds with phenyl (1), 4-tert-butylphenyl (2) and 4-N,N-diphenylaminophenyl (3) groups attached to bis(fluoromesityl)boryl ((FMes)2B) through B–C bonds have been prepared. The restricted rotation about the B–C bonds of boron-bonded aryl rings in solution has been studied by variable-temperature 19F NMR spectroscopy, and through-space F–F coupling has been observed for 3 at low temperature. Steric congestion inhibits binding of 1 by Lewis bases DABCO and tBu3P and the activation of H2 in their presence. Photophysical and electrochemical studies have been carried out on 2, 3, and an analogue of 3 containing a bis(mesityl)boryl ((Mes)2B) group, namely 4. Both 2 and 3 show bright emission in nonpolar solvents and in the solid-state, very strong electron-accepting ability as measured by cyclic voltammetry, and good air-stability. In addition, 2 displayed unusually long-lived emission (τ = 2.47 s) in 2-MeTHF at 77 K. The much stronger acceptor strength of (FMes)2B than (Mes)2B leads to significantly red-shifted emission in solution and the solid state, stronger emission solvatochromism, and significantly lower reduction potentials. Theoretical calculations confirm that 2 and 3 tend to form highly twisted excited states with good conjugation between one FMes group and the boron atom, which correlate well with their blue-shifted solid-state emissions and low kr values in solution.
Introduction
In conjugated three-coordinate organoboron compounds, the boron centre can accept electron density into its empty 2pz orbital following photoexcitation.1,2 Using this fascinating property, a large number of organoboron systems have been constructed for various applications, such as nonlinear optics (NLO),3–5 anion sensing,6,7 and organic light-emitting diodes (OLEDs).8 Furthermore, the strong Lewis acidity of three-coordinate boron compounds can be exploited in frustrated Lewis pair (FLP) chemistry.9 Three-coordinate boron is intrinsically sensitive towards nucleophiles and must be inhibited from reaction with water, in particular. The two major strategies that have been pursued to provide organoboron compounds with sufficient air-stability for practical use are steric protection of the boron centre with bulky substituents1a and structural constraint by incorporation of the boron atom in a rigid, planar structure;10 however, the former strategy is often easier to achieve with regards to chemical synthesis. A representative example of the steric protection strategy is the use of the bis(mesityl)boryl ((Mes)2B) group, wherein the boron atom is protected effectively by the ortho-methyl substituents of two mesityl groups, and as such this moiety has been employed extensively as an electron acceptor for constructing air-stable organoboron materials.1 Considering that the electron-accepting ability of the boryl group plays an important role in determining the overall performance of the material, enhancement of this ability is expected to lead to further improvements, such as better electron injecting and transporting properties in OLEDs, lower energy emission, and larger two-photon absorption (TPA) cross sections.11 However, only a few boryl groups with higher electron affinity or Lewis acidity than (Mes)2B have been reported,7d,12 and most of these do not give sufficient steric protection to the boron centre to render it air-stable.6f,12a–d Therefore, we were motivated to develop air-stable organoboron compounds that fully exploit the favourable electronic properties expected of boryl groups more strongly electron-accepting than (Mes)2B.
In previous work, we have investigated theoretically the alternative acceptor group bis(fluoromesityl)boryl ((FMes)2B, FMes = fluoromesityl = 2,4,6-tris(trifluoromethyl)phenyl), which is an analogue of (Mes)2B with the methyl groups replaced by electron-withdrawing CF3 groups.13 On the basis of our computed results on an extensive series of X2B–dithienyl–BX2 derivatives, we predicted that (FMes)2B should be a much stronger acceptor than (Mes)2B, and that it can reasonably be expected to provide similar, or even greater, steric protection to the boron centre due to the larger CF3 groups, which have a similar volume to an ethyl group.14 Furthermore, we showed that (FMes)2B should be almost as strong an electron acceptor group as (C6F5)2B, but with much greater steric encumbrance. We have also reported the synthesis of (FMes)2BF, which we believed could serve as a useful reagent in the synthesis of (FMes)2B-containing compounds.15 Recently, Irle, Yamaguchi and coworkers reported one compound with a (FMes)2B group directly connected to a carbazole unit through a B–N bond.16 The fluorescence spectrum of this compound has a large Stokes shift of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 in cyclohexane (λabs = 369 nm; λem = 603 nm), but it also has a very low fluorescence quantum yield (ΦF) of 0.03 in the same solvent. Unfortunately, the electron-accepting ability of this compound was not evaluated electrochemically; thus, it is difficult to judge fully the improvement of (FMes)2B over its (Mes)2B analogue in this regard. In a related study, the same authors investigated the aforementioned (FMes)2B-containing compound and a second compound, namely (FMes)2B–NPh2, by DFT and TD-DFT methods; they predicted that the absorption of the former should be red-shifted from the latter and that they should both emit from a twisted intramolecular charge transfer (TICT) state, wherein twisting occurs about the B–N bond while preserving overall C2 symmetry.17
500 cm−1 in cyclohexane (λabs = 369 nm; λem = 603 nm), but it also has a very low fluorescence quantum yield (ΦF) of 0.03 in the same solvent. Unfortunately, the electron-accepting ability of this compound was not evaluated electrochemically; thus, it is difficult to judge fully the improvement of (FMes)2B over its (Mes)2B analogue in this regard. In a related study, the same authors investigated the aforementioned (FMes)2B-containing compound and a second compound, namely (FMes)2B–NPh2, by DFT and TD-DFT methods; they predicted that the absorption of the former should be red-shifted from the latter and that they should both emit from a twisted intramolecular charge transfer (TICT) state, wherein twisting occurs about the B–N bond while preserving overall C2 symmetry.17
The strong electron affinity to enhance optoelectronic properties is related to the strong Lewis acidity required in FLP chemistry for promoting small molecule activation. The strongly Lewis acidic (FMes)2B group has been investigated recently in this context.18 The borane (FMes)2BH forms FLPs with tertiary amines that can activate H2 and, subsequently, reduce enamines in near quantitative yield.18a This borane also forms an FLP with diisopropylamine and a classical Lewis pair with diethylamine, both of which can activate CO2 at room temperature.18b Furthermore, (E)-vinylboranes and (Z)-(2-B(FMes)2-vinyl)gold compounds have been synthesised using a DABCO/HB(FMes)2 FLP to activate terminal alkynes.18c
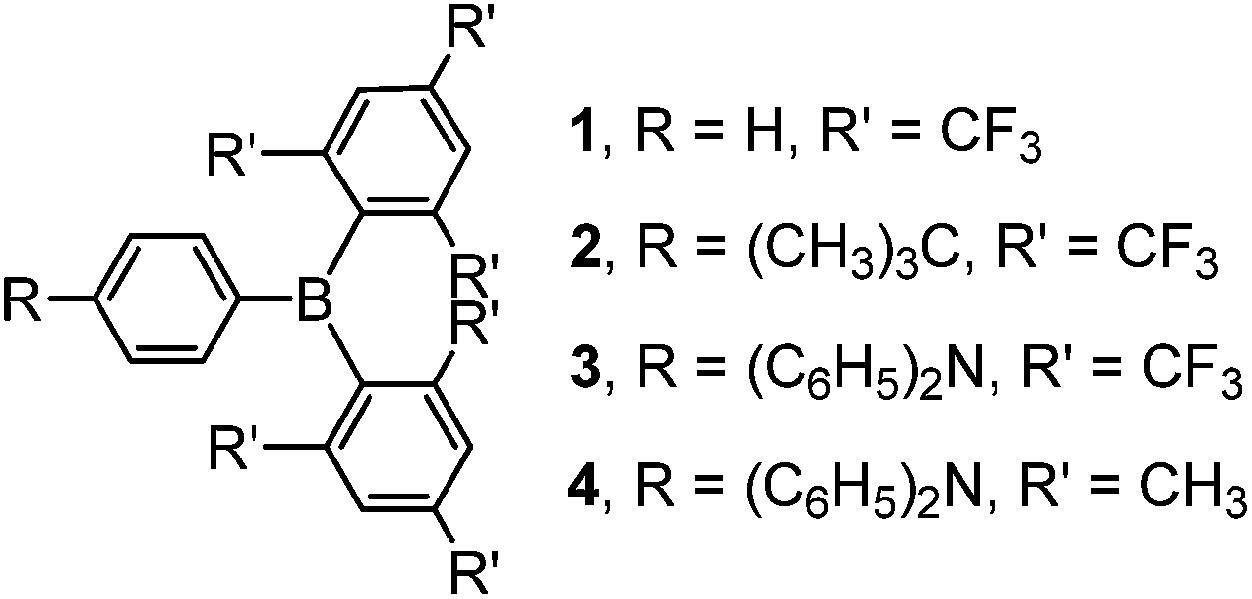
Despite the promising results of these earlier studies, to the best of our knowledge, no three-coordinate boron compound with an aryl ring directly connected to an (FMes)2B moiety through a B–C bond has yet been reported. Thus, we set out to prepare representative examples of this class of compound, namely compounds 1–3 (Scheme 1) with phenyl, 4-tert-butylphenyl and 4-N,N-diphenylaminophenyl groups attached to (FMes)2B through B–C bonds, respectively. Compound 1 was used as an example to study the FLP reactivity, to allow comparison with (FMes)2BH. Compounds 2 and 3 with electron-donating tert-butyl and diphenylamino groups were applied in the photophysical and electrochemical studies. Donor–acceptor organoboron systems often show attractive optoelectronic properties and, therefore, the very different electron-donating abilities of the tert-butyl and diphenylamino groups were used to tune these properties. A known analogue of 3 with a (Mes)2B group (4) (ref. 19) was synthesised to permit direct comparison of the (FMes)2B and (Mes)2B groups, allowing us to evaluate the combined effects of varying the donor and acceptor strength.
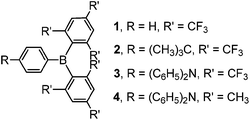 |
| | Scheme 1 Molecular structures of compounds 1–4. | |
Results and discussion
Synthesis
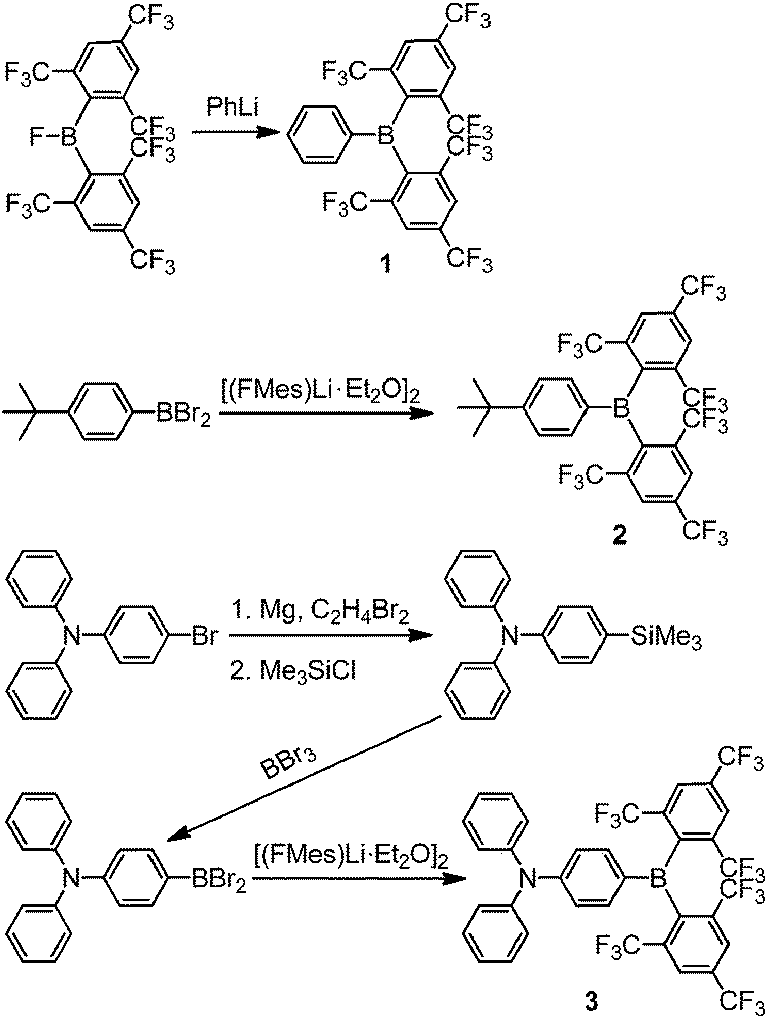
Two different general procedures were employed for the syntheses of 1–3 (Scheme 2). Compound 1 was prepared from the reaction of (FMes)2BF15 with PhLi, whilst 2 and 3 were obtained from the reaction of [(FMes)Li·Et2O]2 (ref. 20) with the corresponding arylboron dibromide and were purified on a silica gel column using standard grade solvents in air, giving an excellent indication of their stability. Further experimental details are given in the ESI.†
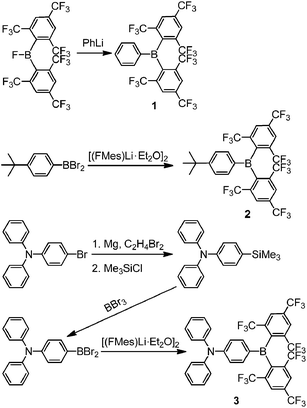 |
| | Scheme 2 Syntheses of compounds 1–3. | |
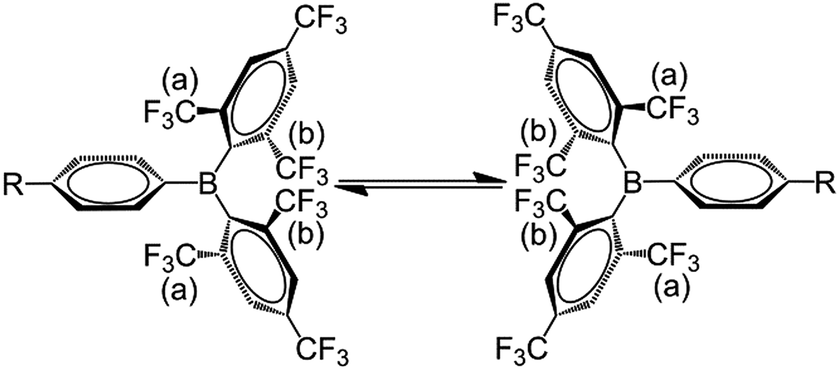
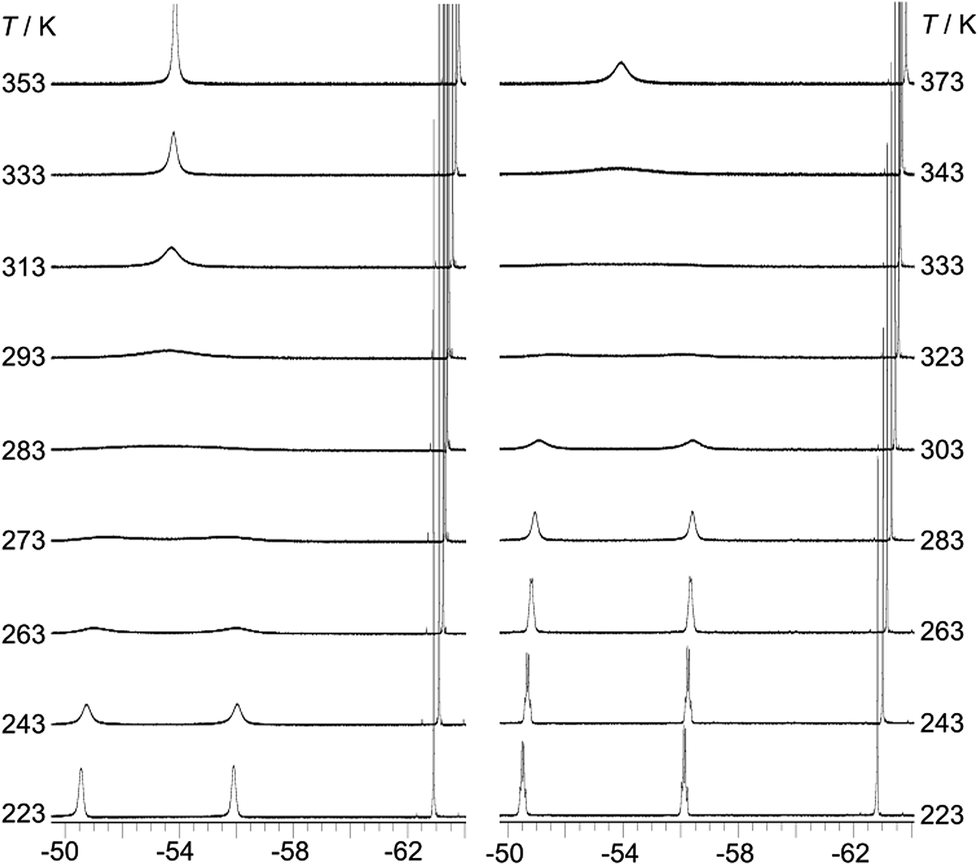
The room temperature 19F{1H} NMR spectra of 1–3 in toluene-d8 show sharp singlets at ca. −63.5 ppm, which can be assigned to the CF3 groups located at the positions para to the boron atom (p-CF3). However, the CF3 groups at the positions ortho to the boron atom (o-CF3) of 1 and 2 exhibit one very broad singlet at ca. −53.5 ppm, while the analogous CF3 groups in 3 are characterised by two broad singlets at −51.0 and −56.4 ppm with a 1:1 ratio. The broadening and decoalescence of the signals is ascribed to a dynamic exchange process, in particular, slow interconversion between the two enantiomers of the racemic mixture through rotations of the aryl rings about the boron centre (Fig. 1), according to previous studies of the stereoisomerisation of three-coordinate boron compounds.21 Variable-temperature 19F{1H} NMR measurements of 2 and 3 were carried out to study the stereodynamic process in detail (Fig. 2). At high temperature (353 K for 2, and 373 K for 3), both compounds show a single sharper signal, indicating that the four o-CF3 groups are equivalent on the NMR timescale. Upon cooling, the signal for the o-CF3 groups broadens and decoalesces into two very broad singlets (1:1 integral ratio) at 273 K for 2 and at 323 K for 3; when the temperature is decreased yet further, these two signals sharpen. Interestingly, for 3, the two singlets each split into a quartet upon cooling to 263 K, while no additional splitting could be observed for 2, even at the lowest temperature available to us of 223 K. The sharp resonance signals at low temperature suggest a very slow exchange, which makes the two o-CF3 groups positioned closer to the substituted Ph group (labelled (a) in Fig. 1) inequivalent to the remaining two o-CF3 groups (labelled (b) in Fig. 1) located farther away. A similar dynamic process related to the restricted rotation of an FMes group has been observed in, for example, organometallic compounds containing (FMes)2Ni or (FMes)2Pd moieties.22 Activation energies for the isomerisation process in our system were calculated to be ca. 12.2 and 14.4 kcal mol−1 for 2 and 3, respectively. The two quartets (J = 12 Hz, 223 K) for the o-CF3 groups of 3 observed at low temperature, suggest a 19F–19F coupling between the fluorine atoms of two inequivalent o-CF3 groups. Through-bond 6JFF or 8JFF coupling can be excluded because of the large number of intervening bonds22c,23 and the absence of 6JFF coupling between the o-CF3 and p-CF3 groups in the same FMes moiety. Through-space coupling between the two inequivalent o-CF3 groups of the same FMes moiety is also not feasible because of their large separation of over 5 Å (X-ray crystallography, vide infra) and, again, the absence of coupling to the equidistant p-CF3 groups. Therefore, through-space coupling between two inequivalent o-CF3 groups on different FMes moieties, namely the two o-CF3 groups at the same side of the BC3 plane (see Fig. 1), is the only remaining plausible explanation. Through-space 19F–19F coupling between the CF3 groups of two FMes moieties has also been observed in, for example, the complex [Pd(FMes)2(κ2S,N-SPPh2Py)].22c Such coupling indicates a relatively small distance between the fluorine atoms at low temperature. The two signals for the o-CF3 groups of compound 2 do not show splitting at 223 K, as the peaks are still slightly broadened due to the faster motion of the FMes groups (compared to those of 3) at this temperature, which is related to its lower energy barrier for the dynamic exchange process.
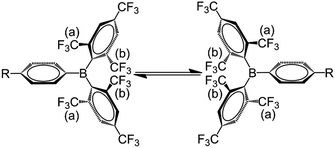 |
| | Fig. 1 Slow exchange between the two enantiomers on the NMR time scale due to restricted rotation of the FMes rings about the boron centre. o-CF3 groups closer to and farther from the substituted Ph group are labelled (a) and (b), respectively. | |
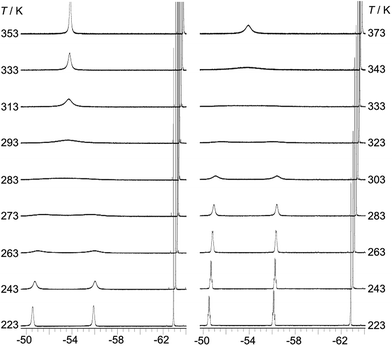 |
| | Fig. 2
19F{1H} NMR spectra (188 MHz) of 2 (left) and 3 (right) in toluene-d8 at various temperatures. | |
Crystal structures
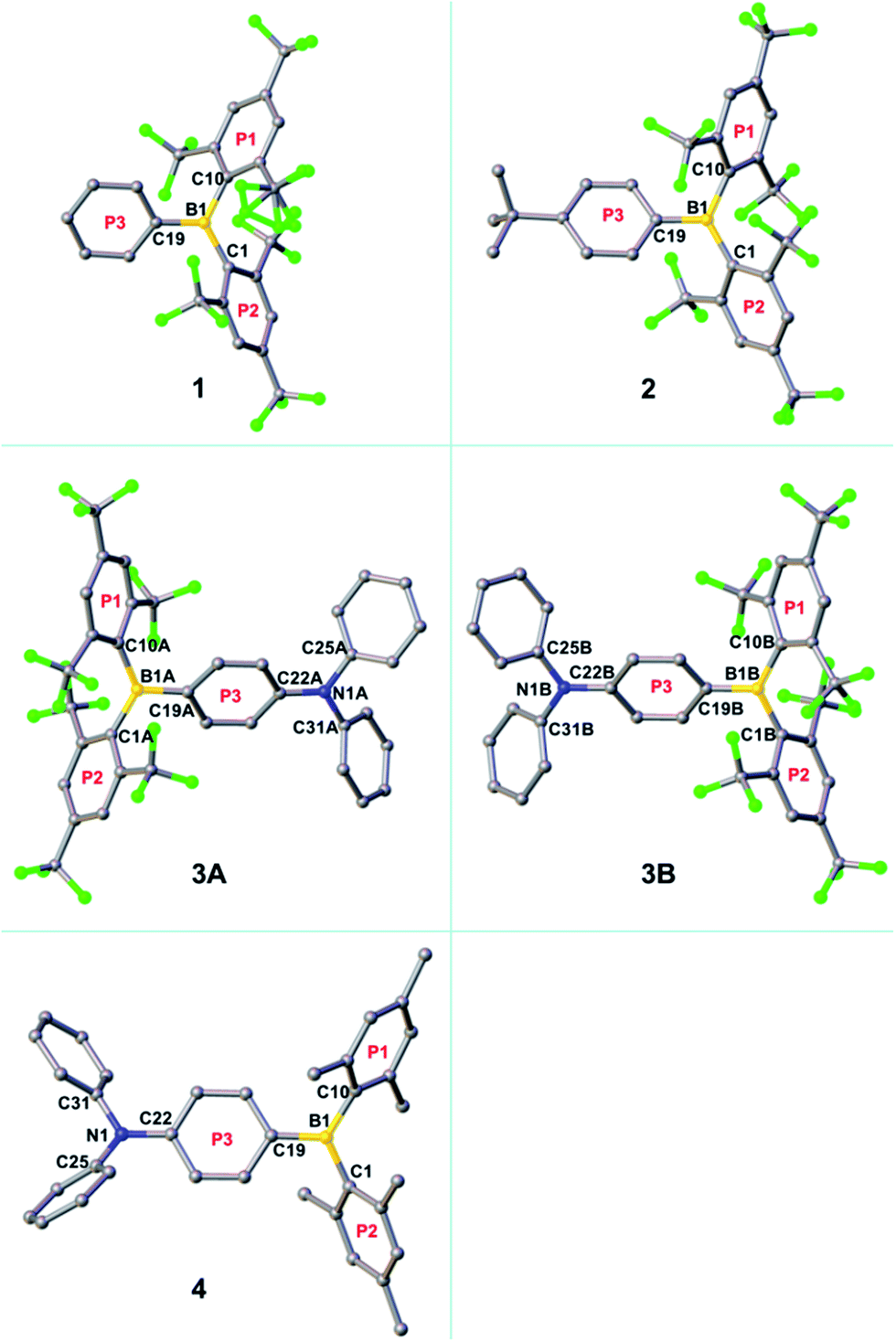
The crystal structure of the starting material [(FMes)Li·Et2O]2 has been re-determined with an improved R1 value and is included as Fig. S1 of the ESI.† The single crystal of 1 suitable for X-ray diffraction was obtained by crystallisation from pentane at −35 °C, while the crystals of 2–4 were obtained by slow evaporation of methanol (for 2 and 4)24 or hexane (for 3) solutions of the respective compounds at room temperature. The molecular structures obtained are shown in Fig. 3 and S2–S5,† and selected bond lengths and dihedral angles are listed in Table 1. The structures of 1, 2 and 4 contain one molecule in the asymmetric unit (Z′ = 1), while two symmetry-independent molecules (labelled 3A and 3B, and related by approximate mirror molecular symmetry) are present for 3 (Z′ = 2). The boron-centred BC3 moieties in these structures are planar, with the sum of the C–B–C bond angles equal to 360°. The NC3 moieties in 3A and 3B are also planar, while that in 4 is slightly pyramidalised, with a sum of 357.3° for the C–N–C bond angles and a distance of 0.137(2) Å between the nitrogen atom and the C22–C25–C31 plane. In 1, 2, 3A and 3B, the BC3 planes and the 2,4,6-trisubstituted rings (P1 and P2) form dihedral angles ranging from 47–66°. These values are similar to those observed for 4 (Table 1) and related p-R–Ph–B(Mes)2 (e.g., R = Me2N, MeO, MeS, Br, I) compounds.3e,3f,5h,25 The third boron-bonded phenyl ring (P3) is twisted out of the BC3 plane by only 25–29°. The dihedral angles between the P3 and NC3 planes are 15.2(2) and 12.5(2)° for 3A and 3B, respectively. It is notable that for 1, 2, 3A, and 3B, the B1–C1 and B1–C10 bond lengths (1.595(7)–1.612(7) Å) are significantly longer than the B1–C19 bond lengths (1.534(7)–1.551(4) Å). This contrasts with 4, which has much closer B1–C1, B1–C10 and B1–C19 bond lengths of 1.579(2), 1.583(2) and 1.561(2) Å, respectively. The larger bond-length difference in the (FMes)2B-containing compounds is likely a result of the electron-withdrawing nature of FMes, which, compared to Mes, leads to a reduced electron delocalisation from these CF3-substituted aryl substituents to the boron centre, and an increased electron delocalisation from the donor moiety to the boron centre via ring P3, lengthening and shortening the respective B–C bonds of the borane. A similar effect of electron-withdrawing C6F5 groups has been observed by Jäkle and coworkers in the compound (C6F5)2B–dithienyl–B(C6F5)2.12a Consistent with the push–pull structures in 2–4, a small ground-state quinoidal distortion of the P3 ring is observed. The quinoidal distortions of the P3 rings, defined as the difference between the averages of the longer and shorter C–C bonds, are approximately 0.02, 0.04, 0.04 and 0.02 Å for 2, 3A, 3B and 4, respectively, which are larger than that observed in Br–Ph–B(Mes)2 (0.003 Å, which is not significantly different to the individual bond-length errors)3f with much weaker push–pull character. The larger quinoidal distortions in 3A and 3B are caused by the combination of the strong amino donor and the strong (FMes)2B acceptor. One might expect that the greater quinoidal distortion of 3 compared to 4 would lead to a more planar structure within the P3–BC3 moiety, that is, the dihedral angle between P3 and the BC3 plane would be smaller; however, this angle is slightly larger for 3 (25–27°) than for 4 (19°), which may be related to the greater steric bulk of the FMes groups. The shortest F–F distances between the two o-CF3 groups on the same side of the BC3 plane are 2.25(1) and 2.528(2) Å in 1, 2.511(2) and 2.539(2) Å in 2, 2.436(5) and 2.667(4) Å in 3A, and 2.629(5) and 2.668(4) Å in 3B. These values are within the distance range suitable for through-space F–F coupling,23 supporting our interpretation of the F–F coupling observed in the low-temperature NMR spectrum of 3. In 1, 2, 3A and 3B, the shortest distances between boron and fluorine atoms of the four o-CF3 groups range from 2.609(6)–2.798(2) Å, which is significantly shorter than the sum of the van der Waals radii of B and F (3.39 Å).26 This observation may suggest the existence of intramolecular C–F⋯B interactions.2p,21a
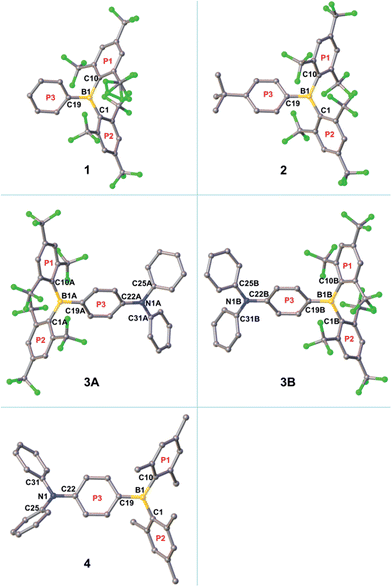 |
| | Fig. 3 Molecular structures of 1–4 from single-crystal X-ray diffraction. Hydrogen atoms are omitted for clarity. With regards to the three aryl rings bonded to boron, the two 2,4,6-substituted aryl rings are labelled P1 and P2, and the remaining ring is labelled P3. | |
Table 1 Selected bond lengths (Å), bond angles (°) and dihedral angles (°) for 1–4 as obtained by single-crystal X-ray diffraction
| |
1
|
2
|
3A
|
3B
|
4
|
| B1–C1 |
1.610(4) |
1.606(3) |
1.596(7) |
1.605(7) |
1.579(2) |
| B1–C10 |
1.604(4) |
1.607(3) |
1.612(7) |
1.595(7) |
1.583(2) |
| B1–C19 |
1.551(4) |
1.546(3) |
1.534(7) |
1.541(7) |
1.561(2) |
| C22–N1 |
— |
— |
1.394(5) |
1.394(6) |
1.408(2) |
| ∠C1–B1–C19 |
119.8(2) |
119.2(2) |
115.8(4) |
115.7(4) |
116.6(1) |
| ∠C10–B1–C19 |
116.6(2) |
115.2(2) |
116.2(4) |
117.2(4) |
119.6(1) |
| ∠C1–B1–C10 |
123.5(2) |
125.7(2) |
127.9(4) |
127.1(4) |
123.8(1) |
| ∠C22–N1–C25 |
— |
— |
122.2(4) |
121.9(4) |
119.6(1) |
| ∠C22–N1–C31 |
— |
— |
121.3(4) |
121.4(4) |
121.0(1) |
| ∠C25–N1–C31 |
— |
— |
116.3(3) |
116.6(4) |
116.7(1) |
| ∠P1–BC3 plane |
63.3(1) |
53.17(7) |
52.3(2) |
50.6(2) |
52.59(5) |
| ∠P2–BC3 plane |
47.1(1) |
50.44(7) |
66.2(2) |
65.2(2) |
56.60(5) |
| ∠P3–BC3 plane |
25.4(1) |
29.19(7) |
26.9(2) |
25.4(2) |
19.47(5) |
| ∠P3–NC3 plane |
— |
— |
15.2(2) |
12.5(2) |
— |
Steric effects on reactivity
The electrophilic borane 1 was combined with one equivalent of tBu3P in dichloromethane (0.06 M) at room temperature, but no interaction was observed by multinuclear NMR spectroscopy, thus establishing that this combination of Lewis acid and base is an FLP. However, exposure of this FLP to H2 (4 atm) resulted in no observable reaction. Moreover, 1 alone or a combination of 1 with DABCO failed to effect the hydrogenation of the prototypical imine PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NtBu under 4 atm of H2, even on heating for 24 hours at 115 °C. This lack of reactivity is in contrast to FLPs generated using (FMes)2BH. Our previous calculations13 disclosed that the LUMO of 1 is similar in energy to that of (C6F5)2BPh,9o a borane that has been used in FLP chemistry, thus excluding an electronic explanation for the low reactivity of FLPs of 1. Therefore, from (FMes)2BH/(C6F5)2BPh to 1, the greater steric congestion about the boron centre is likely responsible for the lack of FLP reactivity. In 1, the o-CF3 groups shield access to the vacant 2pz-orbital on the boron atom, hindering reactivity with even the smallest potential substrate, H2. It is interesting to note that while steric bulk is required to preclude formation of a classical Lewis acid–base adduct and generate an FLP, the present observations also establish that excessive steric congestion suppresses FLP reactivity.
NtBu under 4 atm of H2, even on heating for 24 hours at 115 °C. This lack of reactivity is in contrast to FLPs generated using (FMes)2BH. Our previous calculations13 disclosed that the LUMO of 1 is similar in energy to that of (C6F5)2BPh,9o a borane that has been used in FLP chemistry, thus excluding an electronic explanation for the low reactivity of FLPs of 1. Therefore, from (FMes)2BH/(C6F5)2BPh to 1, the greater steric congestion about the boron centre is likely responsible for the lack of FLP reactivity. In 1, the o-CF3 groups shield access to the vacant 2pz-orbital on the boron atom, hindering reactivity with even the smallest potential substrate, H2. It is interesting to note that while steric bulk is required to preclude formation of a classical Lewis acid–base adduct and generate an FLP, the present observations also establish that excessive steric congestion suppresses FLP reactivity.
Photophysical properties
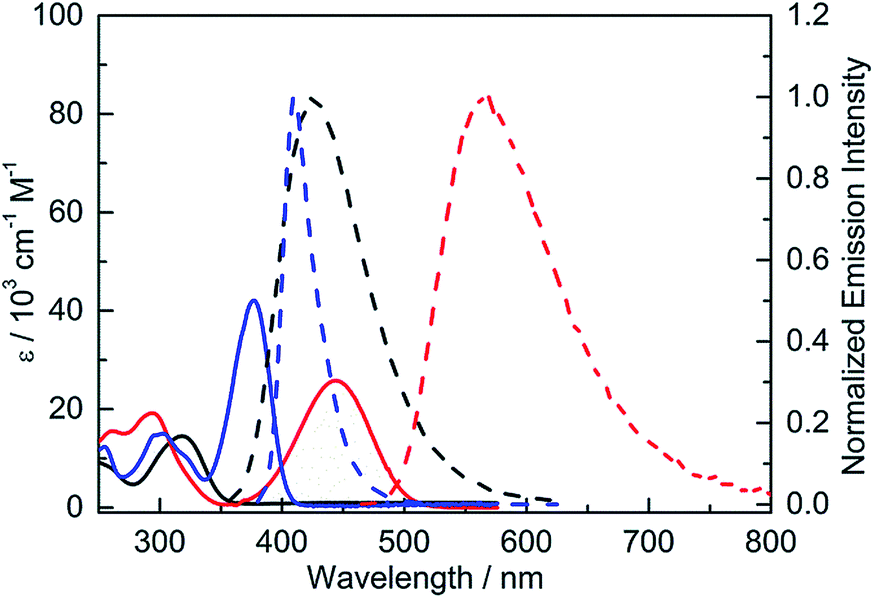
The UV-visible absorption spectra of 2 and 3 have structureless lowest-energy absorption bands with absorption maxima at 318 and 444 nm, respectively, in hexane (Fig. 4 and Table 2). This absorption band is expected to be dominated by an intramolecular charge transfer (ICT) transition between the donor and acceptor moieties. The significantly red-shifted absorption of 3 relative to 2 clearly shows the effect of the stronger donor, NPh2. Compared to 4, which has an absorption maximum at 377 nm, 3 displays a red-shift in the absorption maximum of 67 nm (ca. 4000 cm−1), indicating that incorporation of the stronger acceptor, (FMes)2B, leads to a much lower energy gap. A second band is present at 293 nm for both 3 and 4; the observation that this band is approximately isoenergetic for the two compounds may indicate that this transition involves predominantly the orbitals of the common para-substituted phenyl ring, rather than the Mes or FMes groups.
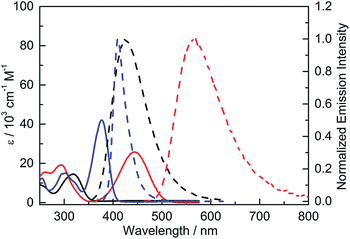 |
| | Fig. 4 UV-visible absorption (solid line) and emission (dashed line) spectra of 2 (black), 3 (red) and 4 (blue) in hexane. | |
Table 2 Photophysical data for 2–4 in solution and in the solid state at room temperature
| |
Medium |
λ
abs
/nm (ε/104 M−1 cm−1) |
λ
em/nm |
Φ
F
|
τ
F/ns |
k
r/107 s−1 |
k
nr/107 s−1 |
Stokes shift/cm−1 |
|
Lowest-energy absorption maximum.
Absolute fluorescence quantum yields measured using an integrating sphere.
Fluorescence lifetimes measured for solid-state samples are estimates.
Not determined due to very weak emission.
|
|
2
|
Hexane |
318 (1.5) |
426 |
0.27 |
9.30 |
2.9 |
7.8 |
8000 |
| Toluene |
321 |
450 |
0.41 |
14.2 |
2.9 |
4.2 |
8900 |
| THF |
317 |
481 |
0.19 |
9.61 |
2.0 |
8.4 |
10800 |
| CH3CN |
315 |
499 |
0.05 |
2.89 |
1.7 |
33 |
11700 |
| Solid |
— |
404 |
0.39 |
4.6c |
8.4 |
13 |
— |
|
3
|
Hexane |
444 (2.6) |
563 |
0.34 |
6.12 |
5.6 |
11 |
4800 |
| Toluene |
448 |
638 |
0.03 |
0.68 |
4.4 |
143 |
6600 |
| THF |
441 |
743 |
—d |
—d |
— |
— |
9200 |
| CH3CN |
434 |
—d |
—d |
—d |
— |
— |
— |
| Solid |
— |
548 |
0.41 |
8.2 (75%), 3.5 (25%)c |
— |
— |
— |
|
4
|
Hexane |
377 (4.2) |
410 |
0.62 |
2.40 |
26 |
16 |
2100 |
| Toluene |
380 |
437 |
0.74 |
3.39 |
22 |
7.7 |
3400 |
| THF |
378 |
462 |
0.70 |
4.96 |
14 |
6.0 |
4800 |
| CH3CN |
375 |
495 |
0.67 |
6.51 |
10 |
5.1 |
6500 |
| Solid |
— |
442 |
0.60 |
4.5 (87%), 1.6 (13%)c |
— |
— |
— |
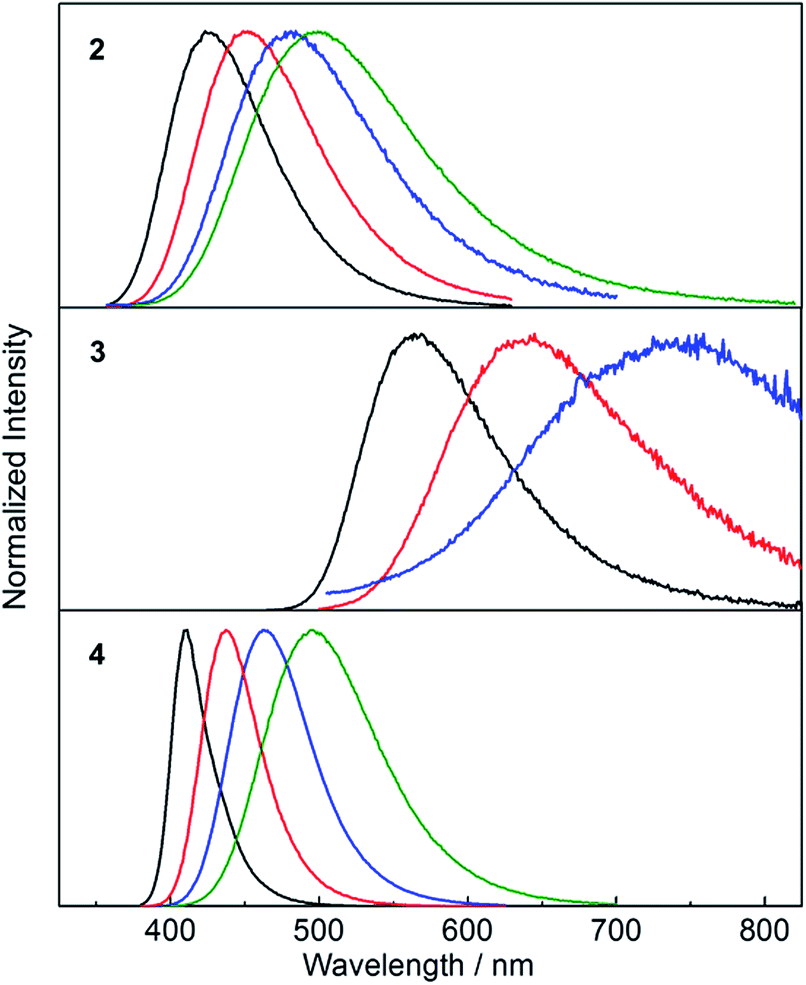
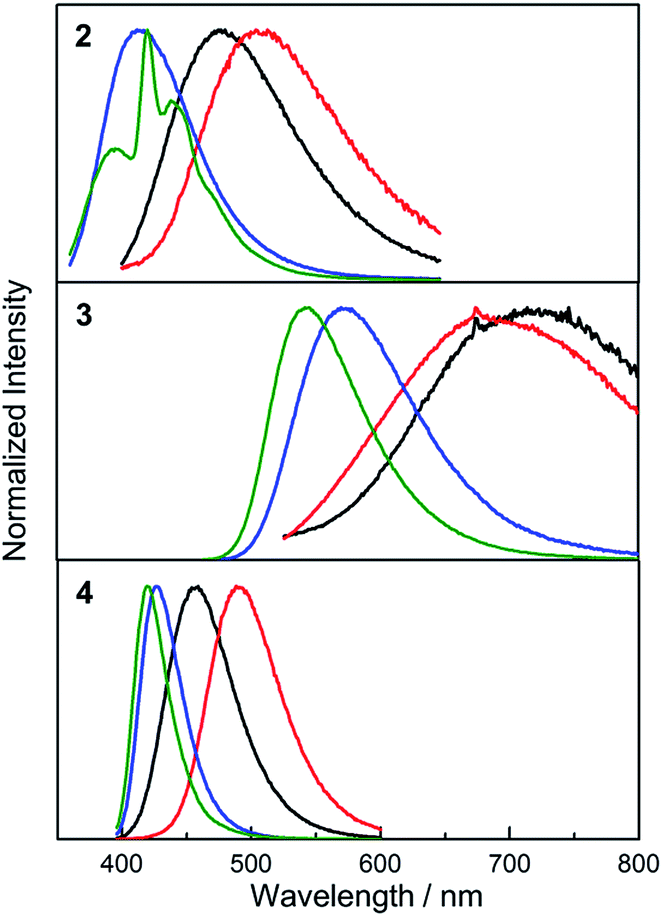
While the absorption spectra of 2, 3 and 4 are only slightly dependent on solvent polarity (negative shifts of up to 720 cm−1 between toluene and CH3CN solutions), the emission spectra of all three compounds exhibit a significant positive solvatochromism (Fig. 5). Upon changing the solvent from hexane to CH3CN, the emission colour of 2 changes from deep blue to blue-green, with the emission maximum shifting from 426 to 499 nm (3400 cm−1 shift). The observation of emission solvatochromism and the lack of strong absorption solvatochromism indicate that 2 has a more polarised first excited state than ground state, consistent with an ICT or TICT transition. The emission of the fluorinated derivative 3 is dramatically red shifted relative to that of 2 by 137 nm (5700 cm−1) in hexane and it shows a stronger sensitivity to changes in solvent polarity. In hexane and toluene, 3 displays yellow (λem = 563 nm) and red (λem = 638 nm) emission, respectively, while in THF, the emission maximum is red-shifted further to 743 nm (a solvatochromic shift totalling 4300 cm−1), but the emission becomes very weak. The stronger solvatochromism of 3 than 2 suggests a larger excited-state dipole in the former, as a result of inclusion of the stronger donor group. It is notable that, in contrast to 3, compound 4 only shows blue emission in hexane (λem = 410 nm) and blue-green emission in CH3CN (λem = 497 nm) (Fig. 5), indicating that the stronger acceptor moiety in 3 is responsible for the large emission red-shift, e.g., 153 nm (6600 cm−1) in hexane and its stronger solvatochromism.
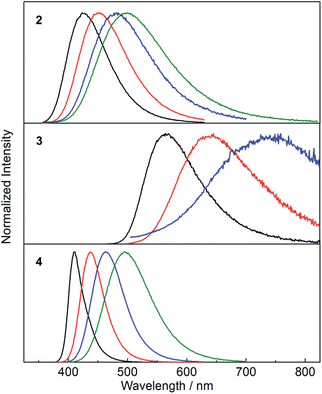 |
| | Fig. 5 Emission spectra of 2–4 in hexane (black), toluene (red), THF (blue) and CH3CN (green) at room temperature. The excitation wavelength used in each case is equal to the maximum of the respective lowest energy absorption band (Table 2). | |
Compound 2 has moderate fluorescence quantum yields in low polarity solvents: 0.27 in hexane and 0.41 in toluene, while at the same time, it maintains a quite large Stokes shift of over 8000 cm−1. With increasing solvent polarity, however, the emission of 2 is effectively quenched (ΦF = 0.05 in CH3CN). Compound 3, possessing stronger push–pull character, displays bright emission only in hexane (ΦF = 0.34). Compared with 2 and 3, compound 4 has much higher ΦF values of 0.62–0.74 in all four solvents used. To understand the factors influencing the quantum yield, fluorescence lifetimes (τF) were measured for these compounds (Table 2). From the ΦF and τF values, the radiative (kr) and nonradiative (knr) decay rate constants were calculated. Compound 2 has very small kr values and relatively larger knr values which, in general, decrease and increase, respectively, with increased solvent polarity, leading to a low ΦF value in CH3CN. Compound 3, has similarly small kr values in hexane and toluene, but a significantly increased value of knr in toluene, leading to very weak emission in this solvent. Compound 4 has a higher kr value in hexane than either 2 or 3, but the value decreases significantly in solvents of increased polarity. Interestingly, from hexane to CH3CN, the knr values of 4 also decrease gradually, which compensates the decrease of kr to give a high ΦF for this compound in both nonpolar and polar solvents. The small kr values of 2 and 3,27 together with their large Stokes shifts16 even in hexane (8000 and 4800 cm−1 for 2 and 3, respectively), suggest a polar and potentially twisted excited state for these two compounds. These potentially twisted excited states might be expected to be related to a large dihedral angle within the Ph–BC3 (in 2) or NC3–Ph–BC3 (in 3 and 4) moieties, such that radiative decay of the excited state is restricted. The increased knr values of 2 and 3 with increased solvent polarity, accompanied by the lower-energy emission, can be understood by the energy-gap law in which nonradiative decay becomes more favourable at smaller energy separations.27a,28 The decreased knr values of 4 in polar solvents indicate an inverse energy-gap law behaviour, which has also been observed by Lambert and coworkers in a related series of organoboron compounds with carbazolyl and diphenylamino donors.29 However, the factors leading to such behaviour for our system are not clear at this time.
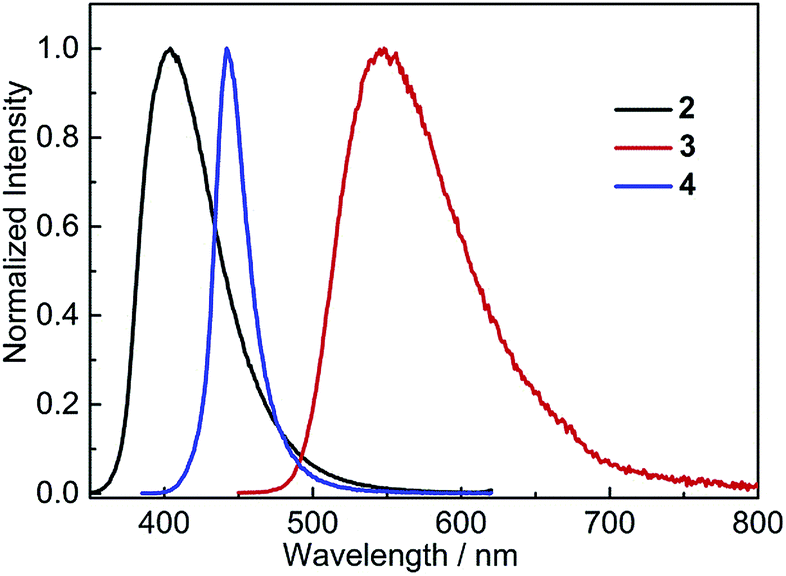
In the solid state, 2 shows bright, deep-blue emission (λem = 404 nm), while 3 displays yellow emission (λem = 548 nm), indicating that the solid-state emission can also be tuned effectively by modifying the donor group (Fig. 6). Comparison of the solid-state emission spectra of 3 and 4 reveals that replacing (Mes)2B (in 4) with (FMes)2B (in 3) results in a spectral red-shift of over 100 nm (4300 cm−1). It is notable that the emission maxima of 2 and 3 in the solid state are blue shifted compared to the corresponding λem in the nonpolar solvent hexane (by 1300 and 500 cm−1, respectively), which is uncommon since the aggregated state often shows red-shifted emission. As disclosed by the crystal structures and theoretical calculations (vide infra), compounds 2 and 3 are relatively planar within the Ph–BC3 (in 2) or NC3–Ph–BC3 (in 3) moieties in the ground state, i.e. they have small dihedral angles about the indicated B–C and C–C bonds, and similar conformations to these are expected to dominate in the powder samples. The blue-shifted solid-state emission is, therefore, considered to be related to the restricted relaxation of the Franck–Condon (FC) state in the rigid medium.30 When either 2 or 3 is excited in hexane solution, the FC state relaxes to a highly twisted conformation, as suggested above; however, in the solid state, the relaxation process through conformational twisting is likely to be restricted. Therefore, in the solid state, excited states with higher energy and higher planarity within the Ph–BC3 (in 2) or NC3–Ph–BC3 (in 3) moieties will be formed, as compared to solution. Moreover, radiative deactivation of the excited state in the solid, via a vertical transition, will reach a FC ground state (S0FC) with a lower energy compared to the S0FC in hexane, because the S0FC in the solid has a more planar conformation that is close in geometry to the ground-state minimum. The larger energy gap, caused by the higher excited-state energy and lower S0FC energy in the solid state, is responsible for the blue-shifted emission of the solid sample. A similar effect was observed by Irle, Yamaguchi and coworkers for carbazole-B(Mes)2 and -B(FMes)2, which show blue shifts of 35 and 30 nm (2000 and 870 cm−1), respectively, in the solid state compared to cyclohexane solutions. It is notable that compounds 2 and 3 retain good ΦF values in the solid state, being 0.39 for 2 and 0.41 for 3, although, again, they are lower than that of 4 (ΦF = 0.60).
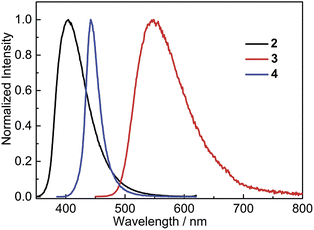 |
| | Fig. 6 Emission spectra of solid (powder) samples of 2–4 at room temperature. The excitation wavelength used in each case is equal to the maximum of the respective lowest energy absorption band in hexane (Table 2). | |
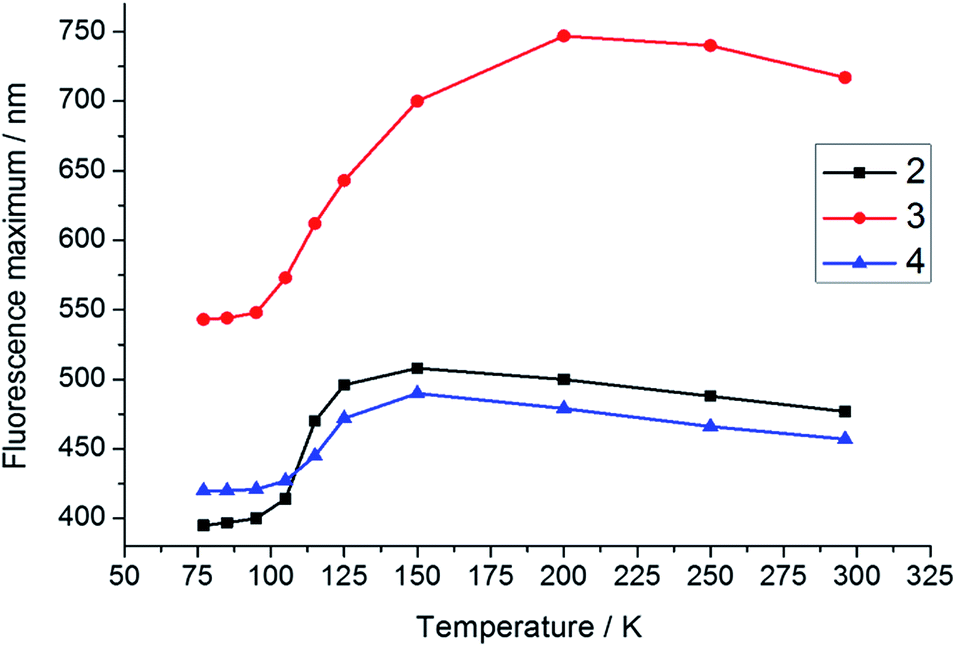
To support our interpretation of the influence of a rigid environment on the emission behaviour, variable-temperature emission spectra (77–296 K) of 2–4 were recorded in toluene and 2-methyltetrahydrofuran (2-MeTHF), as summarised in Table 3 and Fig. S6–S14.† Upon cooling a toluene solution of each compound from 296 to 183 K, a small and continuous red shift of ca. 15–30 nm was observed due to an increase in solvent polarity. However, upon further cooling, passing the melting point of toluene at 178 K, a sudden blue shift occurred, accompanied by an increase in emission intensity at 77 K, most dramatically for 3 (Fig. S6–S8†), in line with expectation for the formation of a restricted excited-state geometry. In addition, inhibition of solvent relaxation around the increased dipole moment in the excited state is also expected to contribute to the blue shift of all three compounds in frozen solutions. Due to the lower temperature at which 2-MeTHF becomes rigid and its higher polarity, the variable temperature data recorded in this solvent were subtly different (Table 3 and Fig. 7 and 8 and S9–S11†). Following an initial red-shift of similar magnitude as that recorded in toluene, a gradual blue shift occurred below 150 K (2 and 4) or 200 K (3) that can be rationalised by a continuous increase in solvent viscosity. At temperatures below 95 K, approximately the glass transition temperature of 2-MeTHF, the emission spectra of 3 and 4 did not vary in a pronounced fashion, while 2 displays additional phosphorescence that will be discussed separately below. It is notable that the blue shift of the fluorescence band from room temperature to 77 K is significantly smaller for 4 (2000 cm−1) as compared with 2 (4300 cm−1) and 3 (4500 cm−1), which reflects a smaller conformational variation between the FC and the relaxed excited states for compound 4. Furthermore, the red shift of the excitation spectra by 6–15 nm in 2-MeTHF upon cooling from 296 to 77 K is relatively small (Fig. S12–S14†), strongly supporting a predominantly excited-state phenomenon. The fluorescence peak of the solid, the 77 K frozen toluene solution and the 77 K 2-MeTHF glass of both 2 and 3 are close in energy, thus implying a related restriction of the geometric relaxation of the FC state in each of these media. The emission spectrum of 4 in the solid state is red-shifted by about 22 nm compared with that in the frozen solutions, which may be due to aggregation in the solid.
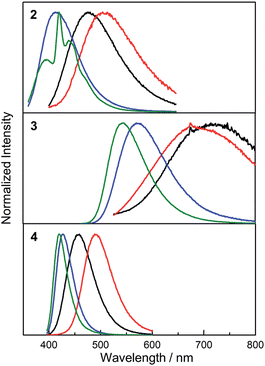 |
| | Fig. 7 Variable-temperature emission spectra of 2–4 in 2-MeTHF recorded at 296 K (black), 150 K (red), 105 K (blue) and 77 K (green). The excitation wavelengths for 2–4 are 330, 443, and 378 nm, respectively. | |
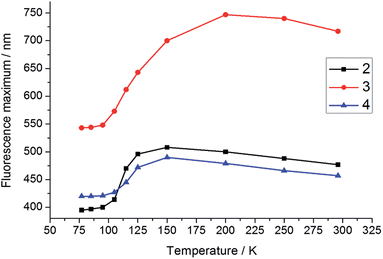 |
| | Fig. 8 Temperature dependence of the fluorescence maxima of 2–4 in 2-MeTHF. The excitation wavelengths for 2–4 are 330, 443, and 378 nm, respectively. | |
Table 3 Photophysical data for 2–4 at 77 K
| |
Medium |
λ
ex/nm |
λ
em/nm |
τ
|
Stokes shift/cm−1 |
|
Percentage values refer to the fluorescence components only.
|
|
2
|
Toluene |
331 |
400 (Sh.), 422, 440 |
— |
5200 |
| 2-MeTHF |
324 |
395, 420, 439 |
3.23 ns (57%),a 6.76 ns (43%),a 2.47 s |
5500 |
|
3
|
Toluene |
465 |
553 |
— |
3400 |
| 2-MeTHF |
450 |
543 |
7.25 ns |
3800 |
|
4
|
Toluene |
393 |
420 |
— |
1600 |
| 2-MeTHF |
392 |
420 |
2.59 ns |
1700 |
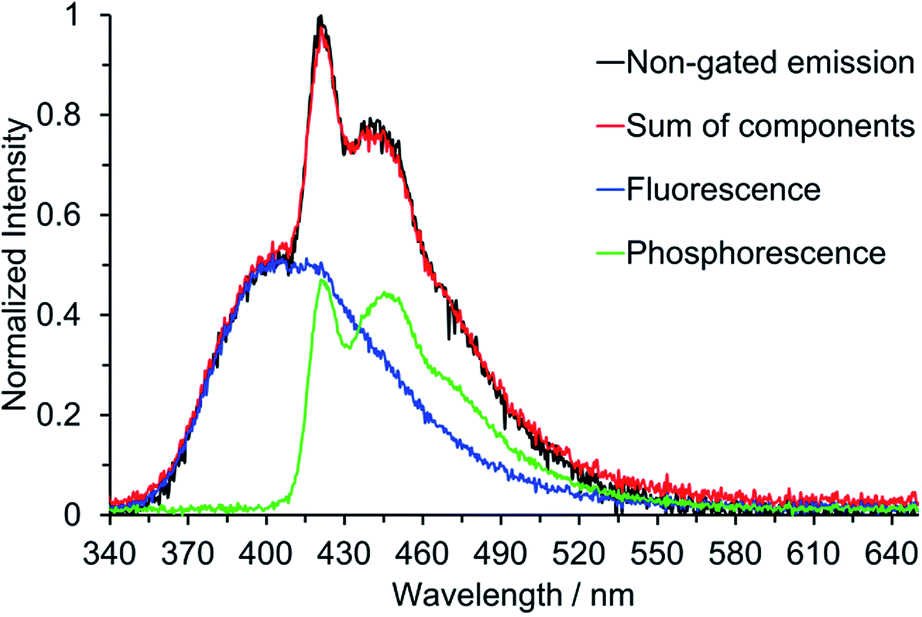
Below the approximate glass transition temperature of 2-MeTHF (95 K), and upon cooling the frozen toluene solution below 133 K, the emission spectrum of 2 splits into two bands; the lower energy band showed an extremely long-lived emission with a lifetime of 2.47 s measured at 420 nm in 2-MeTHF glass at 77 K, which is assumed to be phosphorescence. Phosphorescence lifetimes of this order of magnitude, although not all that common, have been observed for other organic chromophores.27d,31 Furthermore, phosphorescence from various three-coordinate boron compounds with a wide range of lifetimes, spanning nearly six orders of magnitude, has been observed at 77 K in glass matrices or frozen solutions: Wagner and coworkers described the luminescence of 9-hydro-10-mesityl-9,10-diboraanthracence that could be observed for up to 15 s after switching off the excitation source,2q Yamaguchi and coworkers observed phosphorescence with a lifetime of 5.30 ms from a planarised (Ar)3B compound,10d and Wang and coworkers have observed phosphorescence with solvent-dependent lifetimes of 9–11 μs from two trigonal tridurylborane (duryl = 2,3,5,6-tetramethylphenyl) derivatives.2o Compounds 3 and 4, by contrast, have only very minor long-lived components to their emission at 77 K, estimated to account for less than 1% of the total emission that makes no readily perceptible difference to the emission band shape. The phosphorescence lifetimes of 3 and 4 are much shorter than that of 2, but unfortunately they could not be determined accurately. We used time-gated spectroscopy to separate the overlapping fluorescence and phosphorescence spectra of 2 (Fig. 9), and we estimate, based on analysis of these data, that the phosphorescence accounts for ca. 40% of the total steady-state emission spectrum at 77 K. It is interesting to note that the band shapes of the two components of 2 are distinctly different: the fluorescence is broad, indicative of charge transfer, while the phosphorescence is structured with an average vibrational spacing of ca. 1300 cm−1, typical of aromatic ring modes.
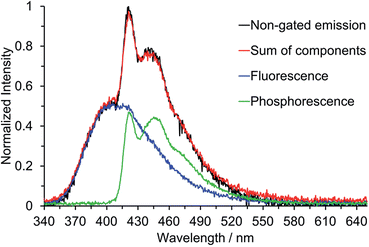 |
| | Fig. 9 Time-gated emission spectroscopy of 2 at 77 K in 2-MeTHF (λex = 330 nm). The normalisation of the fluorescence and phosphorescence spectra have been weighted (ca. 60:40) to reflect their relative contributions to the total emission. The sum of the two individual components is shown in relation to the total emission in the absence of time-gating. | |
Electrochemical properties
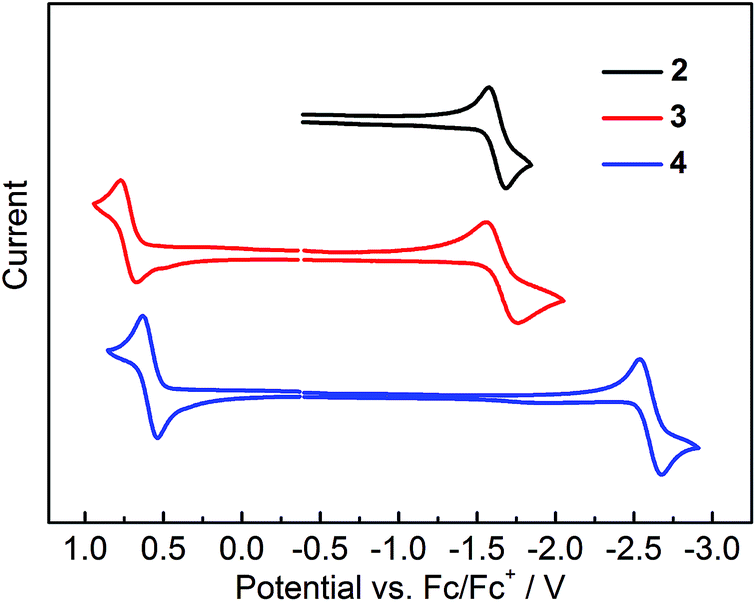
The electrochemical properties of 2–4 were studied by cyclic voltammetry. Compounds 2 and 3 show two reduction waves (Fig. S15†). The first reduction process related to the acceptor moiety is quasi-reversible, occurring at potentials (Ered1/2) of −1.63 and −1.66 V for 2 and 3, respectively (Fig. 10 and Table 4); however, the second reduction is irreversible, showing a broad reduction peak close to the solvent limit (Fig. S15†). For 4, only one quasi-reversible reduction could be observed at Ered1/2 of −2.60 V. The first reduction potential of 3 shows a significant positive shift of ca. 0.94 V compared to that of 4, confirming the much stronger acceptor strength of (FMes)2B in relation to (Mes)2B.13 In addition, the reduction potentials of 2 and 3 are close to that of (Mes)B(C6F5)2 (Ered1/2 = −1.72 V vs. FeCp2+/0),32 further confirming that the acceptor strengths of (FMes)2B and (C6F5)2B are similar, consistent with our previous computational results.13 Moreover, comparison between the reduction potentials of 2, 3, (4-CN-duryl)2B-Ar, (Ered = −1.82 to −1.91 V vs. FeCp2+/0)12f and [4-(Me3N+)-2,6-dimethylphenyl]2B-(Mes) (Ered1/2 = −2.09 V vs. FeCp2+/0)7d indicates that (FMes)2B is a stronger acceptor than (4-CN-duryl)2B or even the cationic [4-(Me3N+)-2,6-dimethylphenyl]2B group. Indeed, the reduction potentials of air-stable 2 and 3 are in the range of those measured for a series of boroles containing nominally antiaromatic BC4R5 cores.33 For the oxidation process, 3 and 4 each show a reversible wave related to the electron-rich aromatic amine moiety, while no oxidation wave could be observed for 2. The oxidation potentials (Eox1/2) of 3 and 4 are +0.72 and +0.59 V, respectively. The positively shifted oxidation potential of 3 indicates that the (FMes)2B group makes the oxidation somewhat more difficult, presumably because the stronger acceptor group leads to a lower electron density at the aromatic amine in the ground state.
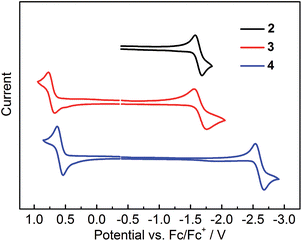 |
| | Fig. 10 Cyclic voltammograms of 2–4. Oxidation and reduction processes were measured in CH2Cl2 and THF, respectively. | |
Table 4 Cyclic voltammetric data,a and related experimental and DFT-calculated (B3LYP/6-31G(d)) HOMO and LUMO energies
| |
E
ox
1/2
/V |
E
red
1/2
/V |
Electrochemicald |
DFT |
| HOMO/eV |
LUMO/eV |
HOMO/eV |
LUMO/eV |
|
Potentials are given vs. ferrocene/ferrocenium (Fc/Fc+).
Measured in CH2Cl2.
The first reduction potentials measured in THF are shown.
Estimated assuming that the HOMO of Fc lies 4.8 eV below the vacuum level.34,35
Calculated from the optical band gap in hexane and the LUMO energy.
|
|
2
|
— |
−1.63 |
−6.70e |
−3.17 |
−6.87 |
−2.68 |
|
3
|
+0.72 |
−1.66 |
−5.52 |
−3.14 |
−5.41 |
−2.48 |
|
4
|
+0.59 |
−2.60 |
−5.39 |
−2.20 |
−5.08 |
−1.50 |
Theoretical calculations
To understand the electronic structures of these compounds further, and to examine the orbitals involved in the electronic transitions, we carried out DFT calculations. The ground-state structures of 2–4 were optimised at the B3LYP/6-31G(d) level of theory using the molecular structures of 2, isomer 3A, and 4 obtained from X-ray diffraction measurements as starting geometries. In the optimised structures of 2 and 3, the B–C bonds to the 2,4,6-trisubstituted aryl rings are about 0.08 Å longer than the B–C bond to the donor substituted aryl rings, while for 4, only a ca. 0.02 Å bond-length difference was found for these two types of B–C bonds, reasonably consistent with the crystallographic data. The ground-state quinoidal distortions of the boron-bonded phenyl rings are also reproduced by the DFT calculations, with values of 0.020, 0.033 and 0.020 Å for 2, 3, and 4, respectively. In addition, similar to the X-ray structures, the N–C (1.400 Å) and B–C (1.527 Å) bonds to the boron-bonded phenyl ring in 3 are shorter than corresponding bonds in 4 (N–C: 1.417 Å; B–C: 1.559 Å), which further support the presence of enhanced quinoidal character for 3, as a direct consequence of the incorporation of the stronger (FMes)2B acceptor. Within the Ph–BC3 moiety in 2 and the NC3–Ph–BC3 moiety in 3, small dihedral angles, less than 28.4°, between the Ph and BC3 or NC3 moieties were observed.
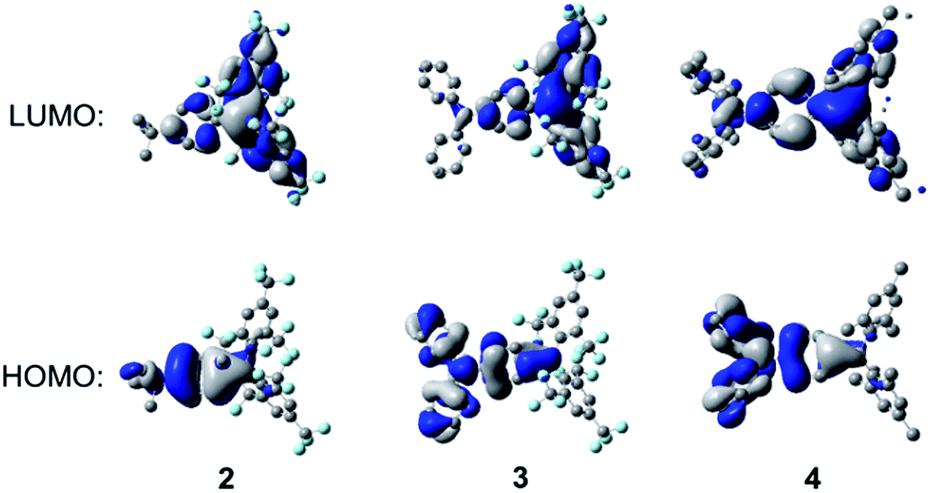
As can be seen from Fig. 11, for all three compounds, the HOMO is localised mainly on the respective 4-tert-butylphenyl or triphenylamine groups, with a small contribution from the nominally empty p-orbital on the boron atom. The LUMO is localised primarily on the boryl group, with some contribution from the boron-bonded phenyl ring, although this contribution is qualitatively smaller in 2 and 3 than in 4. The calculated HOMO and LUMO energy levels and energy gaps show a trend consistent with that obtained from the experimental data (Table 4) but, in all cases, the calculated energy gaps are overestimated by ca. 0.39–0.66 eV. The TD-DFT calculations (CAM-B3LYP/6-31G(d)) show that the S1 ← S0 transitions of these compounds have large oscillator strengths (f = 0.35–0.73), and the excitation wavelengths to the S1 state are calculated to be 293, 369, and 330 nm for 2, 3, and 4, respectively (Table 5). These calculated values overestimate the experimental lowest-energy absorption maxima by some 0.34–0.57 eV (see also Fig. S16–S18†); this phenomenon has been observed previously for 4 and some other (Mes)2B modified triarylamines.5a Excitation to the S1 state in each case is described predominantly by a LUMO ← HOMO transition, which is a transition of ICT character, as can be observed from the orbital distributions. This is consistent with the experimentally observed emission solvatochromism of the compounds, insofar as the enhanced push–pull character of 3 is expected to lead to larger excited-state dipole moments, greater stabilisation of its excited states through solvent reorganisation prior to emission2r and, thus, to increased solvatochromism.
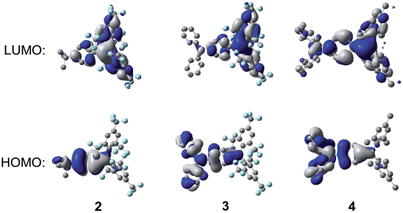 |
| | Fig. 11 DFT calculated frontier orbitals for 2–4 at the B3LYP/6-31G(d) level. Hydrogen atoms have been omitted for clarity. Surface isovalue: ±0.02 [e a0−3]1/2. | |
Table 5 TD-DFT calculated photophysical data for 2–4 at the CAM-B3LYP/6-31G(d) level
| |
Transition (f) |
E
/eV |
λ
/nm |
Dominant componentsb (%) |
|
Values in parentheses are experimental longest-wavelength absorption or emission maxima in hexane.
Components with greater than 10% contribution shown. Percentage contribution approximated by 2 × (ci)2 × 100%, where ci is the coefficient for the particular ‘orbital rotation’.
Taken as the reverse of excitation to S1 from S0 at the optimised S1 geometry.
|
|
Absorption
|
|
2
|
S1 ← S0 (0.347) |
4.24 (3.90) |
293 (318) |
LUMO ← HOMO (93) |
|
3
|
S1 ← S0 (0.606) |
3.36 (2.79) |
369 (444) |
LUMO ← HOMO (87) |
|
4
|
S1 ← S0 (0.726) |
3.76 (3.29) |
330 (377) |
LUMO ← HOMO (85) |
|
|
Emission
|
|
2
|
S1 → S0 (0.018) |
2.76 (2.91) |
449 (426) |
H-SOMO → L-SOMO (94) |
|
3
|
S1 → S0 (0.041) |
2.14 (2.20) |
579 (563) |
H-SOMO → L-SOMO (87) |
|
4
|
S1 → S0 (0.589) |
3.43 (3.02) |
361 (419) |
H-SOMO → L-SOMO (89) |
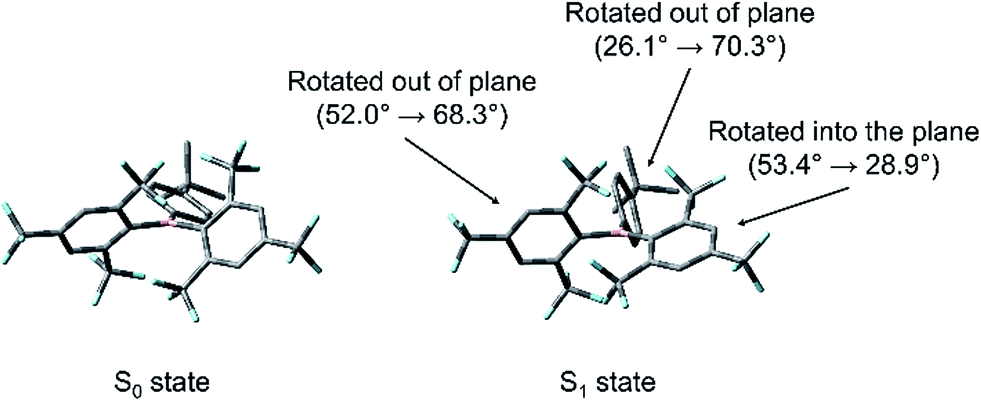
TD-DFT optimisations of the first excited singlet states of 2–4 were carried out to elucidate the nature of the structural relaxation from the FC geometry. The DFT B3LYP/6-31G(d) calculated ground-state optimised geometries were used as the input, and the CAM-B3LYP/6-31G(d) level of theory was employed. These calculations reveal that, in the case of 2 and 3, a distortion of the (FMes)2B group occurs, in which one FMes group becomes more conjugated with the 2pz orbital of the boron atom through a significant reduction of the dihedral angle from 53.4 to 28.9° (2) and from 49.5 to 24.3° (3) with respect to the BC3 plane and shortening of the related B–C bonds by ca. 0.09 Å. Concerted with this, the 4-substituted phenyl ring twists out of conjugation from 26.1 to 70.3° (2) and from 24.2 to 68.9° (3), forming a TICT state, with an elongation of the respective B–C bond of up to 0.05 Å. The second FMes group twists further out of plane than in the ground state, forming dihedral angles of 68.3 and 69.0° for 2 and 3, respectively (Fig. 12 for a representative example). To support these results, we performed further calculations using a range of functionals and starting geometries, but the distortion of the (FMes)2B group was not substantially affected (see ESI† for details).
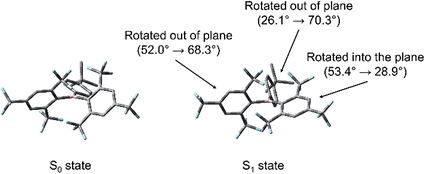 |
| | Fig. 12 Comparison between the geometries of the DFT-optimised S0 state and TD-DFT-optimised S1 state of 2 at the CAM-B3LYP/6-31G(d) level of theory. Changes in the dihedral angles between each of the three aromatic rings and the BC3 plane upon excitation are indicated. Atom colour code: carbon (grey), boron (pink), fluorine (cyan). Hydrogen atoms have been omitted for clarity. | |
No deviation from planarity of either the BC3 or the NC3 plane was observable in the excited state. The calculated planar BC3 moiety is in contrast to the suggestion of Kitamura and coworkers that pyramidalisation at the boron centre occurs in the excited state of a related, but more sterically congested, tridurylborane.36 We note that the (Mes)3B radical anion is planar (X-ray structure)37 and, thus, in general, distortion of a BC3 moiety is not expected upon formal one-electron reduction of the boron atom in either the ground or excited states, except in extremely constrained cases.10d,38
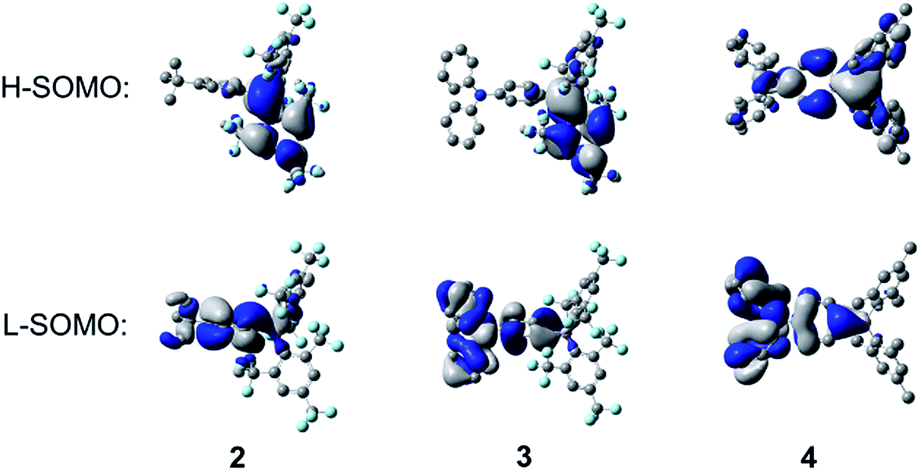
The implication of breaking the C2 symmetry in the excited state can be seen explicitly in the frontier molecular orbitals (Fig. 13), in which the highest singly occupied molecular orbital (H-SOMO) is delocalised over the boron centre and the more planarised FMes moiety, which contrasts with the ground state, where the LUMO is distributed over the whole (FMes)2B group and the substituted phenyl ring (Fig. 11). Furthermore, the lowest singly occupied molecular orbital (L-SOMO) is electronically decoupled from the H-SOMO, i.e. they have little spatial overlap, which accounts for the lower values of kr of 2 and 3. For compound 4, however, there are only small changes in the dihedral angles of the aromatic rings around the boron atom, each varying by less than 8°. The largest change in dihedral angle instead occurs around the C–N bond, increasing from 33.0 to 47.4°. We describe this state in the gas phase as being ICT, in line with previous descriptions. The calculated greater Ph-BC3 planarity of 4 in the excited state leads to a larger change in quinoidal distortion upon excitation (+0.037 Å) than for the twisted structures of 2 and 3 (−0.002 and +0.020 Å, respectively), where conjugation from the phenyl group to the B atom is minimised in the TICT state.
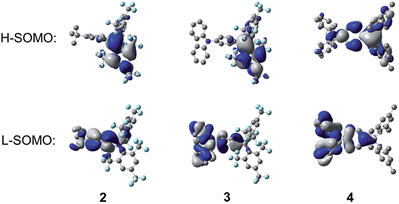 |
| | Fig. 13 Calculated frontier orbitals of the TD-DFT optimised S1 states of 2–4 at the CAM-B3LYP/6-31G(d) level of theory. Hydrogen atoms have been omitted for clarity. Surface isovalue: ±0.02 [e a0−3]1/2. | |
TD-DFT calculated emission energies (Table 5) from the CAM-B3LYP/6-31G(d) optimised geometries correlate well with the experimental values recorded in hexane, particularly for 2 and 3, for which the differences between theory and experiment are 0.15 and 0.09 eV, respectively; for 4 this difference is slightly larger (0.41 eV), but is still within the acceptable range. Furthermore, the significantly larger oscillator strength for 4 than for either 2 or 3 reflects the experimental trend in kr values: 4 has the largest value of kr in hexane by up to an order of magnitude.
We note that, in contrast to the optimised structures of our compounds, no reduction in C2 symmetry was observed in the optimised geometries of carbazole–B(FMes)2 and Ph2N–B(FMes)2 reported by Irle, Yamaguchi and coworkers.17 Furthermore, Song and coworkers have recently performed detailed studies on a series of tridurylboranes in which locally excited, ICT and TICT excited states were identified, with their formation dependent on solvent polarity, viscosity and temperature; however, the nature of the geometrical change on forming the TICT state of these systems was not fully elucidated.12e,12f
Calculated dipole moments at the optimised S0 and S1 geometries and at the Franck–Condon geometries for emission and absorption on these respective surfaces are collected in Table 6. The largest change in dipole moment in the excited state is found for 3, while the values for 2 and 4 are similar, which correlates with the enhanced solvatochromism of 3.
Table 6 TD-DFT calculated dipole moments (debye) for 2–4.a Calculated at the CAM-B3LYP/6-31G(d) level
| |
S0 |
S1FC |
S1 |
S0FC |
S1–S0 |
|
Calculated at the following points on the ground and lowest singlet excited-state surfaces: S0, the optimised geometry of the ground state; S1, the optimised geometry of the first singlet excited state; S1FC, the S1 state at the FC geometry following excitation; and S1FC, the S0 state at the FC geometry following emission.
|
|
2
|
2.2 |
10.4 |
12.3 |
1.4 |
10.1 |
|
3
|
4.8 |
18.8 |
24.6 |
4.2 |
19.8 |
|
4
|
1.8 |
9.6 |
10.4 |
1.0 |
8.6 |
Conclusions
In conclusion, three compounds with an aryl ring directly connected to a (FMes)2B group through B–C bonds have been prepared. The dynamic processes, and related energy barriers, of compounds 2 and 3 have been studied by variable-temperature 19F NMR spectroscopy, and interesting through-space F–F coupling has been observed for compound 3 at low temperature. Compound 1 was found to form FLPs with DABCO and tBu3P. A preliminary test of the catalytic reactivity of 1 alone and the FLP 1·DABCO for the hydrogenation of an imine indicates that, although steric bulk is required to preclude formation of a classical Lewis acid–base adduct and generate an FLP, the large steric congestion in compound 1 suppresses the FLP reactivity, even when using small substrates such as H2. This result provides an upper bound to the degree of steric bulk that can be included in a borane FLP partner before reactivity is inhibited and, thus, should help guide the future design of FLPs based on three-coordinate boron.
Comparison between (Mes)2B- and (FMes)2B-containing donor–acceptor compounds confirms that (FMes)2B is a much stronger acceptor, which leads to: a larger quinoidal distortion, as determined by X-ray crystallography; significantly red-shifted emission in solution and in the solid state; stronger emission solvatochromism; and significantly lower reduction potentials. The photophysical properties of this type of compound can be tuned further and effectively by modification of the donor group. Both compounds 2 and 3 show bright emission in a nonpolar solvent (hexane) and in the solid-state, and they have very strong electron-accepting character, as well as good air-stability. We have also shown by time-gated spectroscopy that the emission spectrum of 2 at 77 K contains a ca. 40% contribution of extremely long-lived (τ = 2.47 s) phosphorescence.
Through a combination of photophysical measurements and theory, we have demonstrated that the excited states of 2 and 3, i.e. those containing a Ph–B(FMes)2 group, relax from the Franck–Condon geometry to a TICT excited state even in the gas phase. TD-DFT optimisation of the excited state has shown that delocalisation of the electron occupying the higher lying SOMO across the boron atom and a single FMes group occurs, facilitated by a reduction in torsion angle between this FMes group and the BC3 plane. Furthermore, concerted twisting of the substituted phenyl group further out of the BC3 plane minimises orbital overlap to the lower lying SOMO, leading to efficient charge separation and a large excited-state dipole moment for 3 in particular. This correlates well with the observed blue shift in the emission in both the solid state and at 77 K in 2-MeTHF glass, wherein geometric relaxation is inhibited, as well as the low kr values in solution. Compound 4, however, is described as undergoing an ICT transition in the gas phase, in which there is much smaller structural reorganisation prior to emission, at least in the nonpolar solvents.
The combined optical and electronic properties of (FMes)2B make it a very promising acceptor moiety for the design of highly efficient, air-stable compounds for, for example, organic light emitting diodes, organic photovoltaics, and two-photon absorbing and two-photon excited fluorescence materials. Investigations into these applications are currently in progress in our laboratory.
Acknowledgements
We are grateful for generous financial support by the Bavarian State Ministry of Science, Research, and the Arts for the Collaborative Research Network “Solar Technologies go Hybrid”. Z. Z. and R. M. E. thank the Alexander von Humboldt Foundation for Postdoctoral Research Fellowships. D. W. S. thanks NSERC of Canada for the award of a Canada Research Chair. L. E. L. is grateful for an NSERC CGS-D and a Walter C. Sumner fellowship. The authors thank Dr Stefan Wagner for mass spectrometry measurements, Dr Rüdiger Bertermann for NMR measurements, and Dr Ivo Krummenacher for the help with electrochemical measurements. We thank Prof. Dr Frank Würthner and Dr Matthias Stolte for the loan of a CCD spectrograph and Prof. Dr Bernd Engels for helpful discussions.
Notes and references
- For reviews, see:
(a) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS;
(b) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS;
(c) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS;
(d) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed;
(e) Z. M. Hudson and S. Wang, Dalton Trans., 2011, 7805–7816 RSC;
(f) F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef PubMed;
(g) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed;
(h) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed;
(i) T. W. Hudnall, C.-W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed;
(j) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef;
(k) M. Elbing and G. C. Bazan, Angew. Chem., Int. Ed., 2008, 47, 834–838 CrossRef CAS PubMed;
(l) N. Matsumi and Y. Chujo, Polym. J., 2008, 40, 77–89 CrossRef CAS;
(m) Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC;
(n) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed.
-
(a) L. Weber, V. Werner, M. A. Fox, T. B. Marder, S. Schwedler, A. Brockhinke, H.-G. Stammler and B. Neumann, Dalton Trans., 2009, 2823–2831 RSC;
(b) H. Braunschweig, T. Herbst, D. Rais, S. Ghosh, T. Kupfer, K. Radacki, A. G. Crawford, R. M. Ward, T. B. Marder, I. Fernández and G. Frenking, J. Am. Chem. Soc., 2009, 131, 8989–8999 CrossRef CAS PubMed;
(c) L. Weber, D. Eickhoff, T. B. Marder, M. A. Fox, P. J. Low, A. D. Dwyer, D. J. Tozer, S. Schwedler, A. Brockhinke, H. G. Stammler and B. Neumann, Chem.–Eur. J., 2012, 18, 1369–1382 CrossRef CAS PubMed;
(d) C.-H. Zhao, A. Wakamiya and S. Yamaguchi, Macromolecules, 2007, 40, 3898–3900 CrossRef CAS;
(e) A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273–4276 CrossRef CAS PubMed;
(f) C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS PubMed;
(g) A. Wakamiya, T. Ide and S. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 14859–14866 CrossRef CAS PubMed;
(h) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2000, 122, 6335–6336 CrossRef CAS;
(i) P. Chen, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 8802–8805 CrossRef CAS PubMed;
(j) Y. Qin, G. Cheng, O. Achara, K. Parab and F. Jäkle, Macromolecules, 2004, 37, 7123–7131 CrossRef CAS;
(k) A. Lorbach, M. Bolte, H. Li, H.-W. Lerner, M. C. Holthausen, F. Jäkle and M. Wagner, Angew. Chem., Int. Ed., 2009, 48, 4584–4588 CrossRef CAS PubMed;
(l) C. Reus, S. Weidlich, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2013, 135, 12892–12907 CrossRef CAS PubMed;
(m) E. Januszewski, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2012, 31, 8420–8425 CrossRef CAS;
(n) N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 5112–5113 CrossRef CAS;
(o) W.-L. Jia, D. Song and S. Wang, J. Org. Chem., 2003, 68, 701–705 CrossRef CAS PubMed;
(p) X. Yin, J. Chen, R. A. Lalancette, T. B. Marder and F. Jäkle, Angew. Chem., Int. Ed., 2014, 53, 9761–9765 CrossRef CAS PubMed;
(q) E. Januszewski, A. Lorbach, R. Grewal, M. Bolte, J. W. Bats, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2011, 17, 12696–12705 CrossRef CAS PubMed;
(r) U. Megerle, F. Selmaier, C. Lambert, E. Riedle and S. Lochbrunner, Phys. Chem. Chem. Phys., 2008, 10, 6245–6251 RSC;
(s) M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, Angew. Chem., Int. Ed., 2007, 46, 4940–4943 CrossRef CAS PubMed;
(t) B. Neue, J. F. Araneda, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2013, 52, 9966–9969 CrossRef CAS PubMed.
-
(a) Z. Yuan, N. J. Taylor, T. B. Marder, I. D. Williams, S. K. Kurtz and L.-T. Cheng, J. Chem. Soc., Chem. Commun., 1990, 1489–1492 RSC;
(b)
Z. Yuan, N. J. Taylor, T. B. Marder, I. D. Williams, S. K. Kurtz and L.-T. Cheng, in Organic Materials for Non-linear Optics II, ed. R. A. Hann and D. Bloor, Royal Society of Chemistry, Cambridge, 1991, pp. 190–196 Search PubMed;
(c) Z. Yuan, N. J. Taylor, Y. Sun, T. B. Marder, I. D. Williams and L.-T. Cheng, J. Organomet. Chem., 1993, 449, 27–37 CrossRef CAS;
(d) Z. Yuan, N. J. Taylor, R. Ramachandran and T. B. Marder, Appl. Organomet. Chem., 1996, 10, 305–316 CrossRef CAS;
(e) Z. Yuan, J. C. Collings, N. J. Taylor, T. B. Marder, C. Jardin and J.-F. Halet, J. Solid State Chem., 2000, 154, 5–12 CrossRef CAS;
(f) Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jové, A. S. Batsanov, J. A. K. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758–2771 CrossRef CAS PubMed.
-
(a) M. Charlot, L. Porrès, C. D. Entwistle, A. Beeby, T. B. Marder and M. Blanchard-Desce, Phys. Chem. Chem. Phys., 2005, 7, 600–606 RSC;
(b) L. Porrès, M. Charlot, C. D. Entwistle, A. Beeby, T. B. Marder and M. Blanchard-Desce, Proc. SPIE, 2005, 92, 5934–5936 Search PubMed;
(c) J. C. Collings, S.-Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L.-O. Pålsson, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W.-Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem.–Eur. J., 2009, 15, 198–208 CrossRef CAS PubMed;
(d) C. D. Entwistle, J. C. Collings, A. Steffen, L.-O. Pålsson, A. Beeby, D. Albesa-Jove, J. M. Burke, A. S. Batsanov, J. A. K. Howard, J. A. Mosely, S.-Y. Poon, W.-Y. Wong, F. Ibersiene, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, J. Mater. Chem., 2009, 19, 7532–7544 RSC;
(e) L. Ji, R. M. Edkins, L. J. Sewell, A. Beeby, A. S. Batsanov, K. Fucke, M. Drafz, J. A. K. Howard, O. Moutounet, F. Ibersiene, A. Boucekkine, E. Furet, Z. Liu, J.-F. Halet, C. Katan and T. B. Marder, Chem.–Eur. J., 2014, 20, 13618–13635 CrossRef CAS PubMed.
-
(a) N. S. Makarov, S. Mukhopadhyay, K. Yesudas, J.-L. Brédas, J. W. Perry, A. Pron, M. Kivala and K. Müllen, J. Phys. Chem. A, 2012, 116, 3781–3793 CrossRef CAS PubMed;
(b) Z. Liu, Q. Fang, D. Wang, G. Xue, W. Yu, Z. Shao and M. Jiang, Chem. Commun., 2002, 2900–2901 RSC;
(c) D. Cao, Z. Liu, Q. Fang, G. Xu, G. Liu and W. Yu, J. Organomet. Chem., 2004, 689, 2201–2206 CrossRef CAS PubMed;
(d) D. Cao, Z. Liu, G. Zhang and G. Li, Dyes Pigm., 2009, 81, 193–196 CrossRef CAS PubMed;
(e) D. Cao, Z. Liu, G. Li, G. Liu and G. Zhang, J. Mol. Struct., 2008, 874, 46–50 CrossRef CAS PubMed;
(f) Z. Liu, Q. Fang, D. Cao, D. Wang and G. Xu, Org. Lett., 2004, 6, 2933–2936 CrossRef CAS PubMed;
(g) Z.-Q. Liu, M. Shi, F.-Y. Li, Q. Fang, Z.-H. Chen, T. Yi and C.-H. Huang, Org. Lett., 2005, 7, 5481–5484 CrossRef CAS PubMed;
(h) Z. Liu, Q. Fang, D. Wang, D. Cao, G. Xue, W. Yu and H. Lei, Chem.–Eur. J., 2003, 9, 5074–5084 CrossRef CAS PubMed;
(i) L. Ji, Q. Fang, M.-S. Yuan, Z.-Q. Liu, Y.-X. Shen and H.-F. Chen, Org. Lett., 2010, 12, 5192–5195 CrossRef CAS PubMed;
(j) M. Lequan, R. M. Lequan and K. C. Ching, J. Mater. Chem., 1991, 1, 997–999 RSC;
(k) M. Lequan, R. M. Lequan, K. C. Ching, M. Barzoukas, A. Fort, H. Lahoucine, B. Bravic, D. Chasseau and J. Gaultier, J. Mater. Chem., 1992, 2, 719–725 RSC;
(l) C. Branger, M. Lequan, R. M. Lequan, M. Barzoukas and A. Fort, J. Mater. Chem., 1996, 6, 555–558 RSC;
(m) M. Lequan, R. M. Lequan, K. Chane-Ching, A.-C. Callier, M. Barzoukas and A. Fort, Adv. Mater. Opt. Electron., 1992, 1, 243–247 CrossRef CAS.
-
(a) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372–11375 CrossRef CAS PubMed;
(b) S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS PubMed;
(c) Y. Kubo, M. Yamamoto, M. Ikeda, M. Takeuchi, S. Shinkai, S. Yamaguchi and K. Tamao, Angew. Chem., Int. Ed., 2003, 42, 2036–2040 CrossRef CAS PubMed;
(d) C.-H. Zhao, E. Sakuda, A. Wakamiya and S. Yamaguchi, Chem.–Eur. J., 2009, 15, 10603–10612 CrossRef CAS PubMed;
(e) M. Varlan, B. A. Blight and S. Wang, Chem. Commun., 2012, 48, 12059–12061 RSC;
(f) Y. Sun, N. Ross, S.-B. Zhao, K. Huszarik, W.-L. Jia, R.-Y. Wang, D. Macartney and S. Wang, J. Am. Chem. Soc., 2007, 129, 7510–7511 CrossRef CAS PubMed;
(g) X. Y. Liu, D. R. Bai and S. Wang, Angew. Chem., Int. Ed., 2006, 45, 5475–5478 CrossRef CAS PubMed;
(h) F. Cheng, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2013, 135, 17286–17289 CrossRef CAS PubMed;
(i) F. Pammer and F. Jäkle, Chem. Sci., 2012, 3, 2598–2606 RSC;
(j) P. Chen and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 20142–20145 CrossRef CAS PubMed;
(k) H. Li, R. A. Lalancette and F. Jäkle, Chem. Commun., 2011, 47, 9378–9380 RSC;
(l) P. Chen, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2012, 51, 7994–7998 CrossRef CAS PubMed.
-
(a) H. Zhao, J. H. Reibenspies and F. P. Gabbaï, Dalton Trans., 2013, 42, 608–610 RSC;
(b) T. W. Hudnall and F. P. Gabbaï, J. Am. Chem. Soc., 2007, 129, 11978–11986 CrossRef CAS PubMed;
(c) C.-W. Chiu and F. P. Gabbaï, Dalton Trans., 2008, 814–817 RSC;
(d) C.-W. Chiu, Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 60–61 CrossRef CAS PubMed;
(e) Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 3363–3369 CrossRef CAS PubMed;
(f) H. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef CAS PubMed;
(g) H. Zhao and F. P. Gabbaï, Organometallics, 2012, 31, 2327–2335 CrossRef CAS.
-
(a) Y. Shirota, M. Kinoshita, T. Noda, K. Okumoto and T. Ohara, J. Am. Chem. Soc., 2000, 122, 11021–11022 CrossRef CAS;
(b) T. Noda, H. Ogawa and Y. Shirota, Adv. Mater., 1999, 11, 283–285 CrossRef CAS;
(c) G. J. Zhou, Q. Wang, X. Z. Wang, C.-L. Ho, W.-Y. Wong, D. G. Ma, L. X. Wang and Z. Y. Lin, J. Mater. Chem., 2010, 20, 7472–7484 RSC;
(d) Z. M. Hudson, C. Sun, M. G. Helander, H. Amarne, Z.-H. Lu and S. Wang, Adv. Funct. Mater., 2010, 20, 3426–3439 CrossRef CAS;
(e) Z. M. Hudson, M. G. Helander, Z.-H. Lu and S. Wang, Chem. Commun., 2011, 47, 755–757 RSC;
(f) Z. Wang, M. G. Helander, Z. M. Hudson, J. Qiu, S. Wang and Z.-H. Lu, Appl. Phys. Lett., 2011, 98, 213301–213303 CrossRef PubMed;
(g) Y. Rao, D. Schoenmakers, Y.-L. Chang, J. Lu, Z.-H. Lu, Y. Kang and S. Wang, Chem.–Eur. J., 2012, 18, 11306–11316 CrossRef CAS PubMed;
(h) C. Sun, Z. M. Hudson, M. G. Helander, Z.-H. Lu and S. Wang, Organometallics, 2011, 30, 5552–5555 CrossRef CAS;
(i) F. Li, W. Jia, S. Wang, Y. Zhao and Z.-H. Lu, J. Appl. Phys., 2008, 103, 034509/1–034509/6 CAS;
(j) W. L. Jia, M. J. Moran, Y. Y. Yuan, Z. H. Lu and S. Wang, J. Mater. Chem., 2005, 15, 3326–3333 RSC;
(k) Z. M. Hudson, C. Sun, M. G. Helander, Y.-L. Chang, Z.-H. Lu and S. Wang, J. Am. Chem. Soc., 2012, 134, 13930–13933 CrossRef CAS PubMed;
(l) S.-B. Ko, J.-S. Lu, Y. Kang and S. Wang, Organometallics, 2013, 32, 599–607 CrossRef CAS;
(m) A. Shuto, T. Kushida, T. Fukushima, H. Kaji and S. Yamaguchi, Org. Lett., 2013, 15, 6234–6237 CrossRef CAS PubMed;
(n) G. Zhou, C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499–511 CrossRef CAS.
-
(a) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed;
(b) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 RSC;
(c) D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC;
(d) D. W. Stephan, Chem. Commun., 2010, 46, 8526–8533 RSC;
(e) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed;
(f) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed;
(g) J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef CAS PubMed;
(h) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396–8397 CrossRef CAS PubMed;
(i) R. L. Melen, M. M. Hansmann, A. J. Lough, A. S. K. Hashmi and D. W. Stephan, Chem.–Eur. J., 2013, 19, 11928–11938 CrossRef CAS PubMed;
(j) M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6422–6432 CrossRef CAS;
(k) R. L. Melen, Chem. Commun., 2014, 50, 1161–1174 RSC;
(l) C. Chen, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2010, 46, 3580–3582 RSC;
(m) R. Liedtke, R. Fröhlich, G. Kehr and G. Erker, Organometallics, 2011, 30, 5222–5232 CrossRef CAS;
(n) D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC;
(o) R. C. Neu, E. Y. Ouyang, S. J. Geier, X. Zhao, A. Ramos and D. W. Stephan, Dalton Trans., 2010, 39, 4285–4294 RSC;
(p) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed.
-
(a) Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed;
(b) S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed;
(c) C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS PubMed;
(d) T. Kushida, C. Camacho, A. Shuto, S. Irle, M. Muramatsu, T. Katayama, S. Ito, Y. Nagasawa, H. Miyasaka, E. Sakuda, N. Kitamura, Z. Zhou, A. Wakamiya and S. Yamaguchi, Chem. Sci., 2014, 5, 1296–1304 RSC;
(e) A. Shuto, T. Kushida, T. Fukushima, H. Kaji and S. Yamaguchi, Org. Lett., 2013, 15, 6234–6237 CrossRef CAS PubMed.
-
(a) Z. Liu, T. Chen, B. Liu, Z.-L. Huang, T. Huang, S. Li, Y. Xu and J. Qin, J. Mater. Chem., 2007, 17, 4685–4689 RSC;
(b) A. Karotki, M. Drobizhev, M. Kruk, C. Spangler, E. Nickel, N. Mamardashvili and A. Rebane, J. Opt. Soc. Am. B, 2003, 20, 321–332 CrossRef CAS.
-
(a) A. Sundararaman, K. Venkatasubbaiah, M. Victor, L. N. Zakharov, A. L. Rheingold and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 16554–16565 CrossRef CAS PubMed;
(b) A. Sundararaman, R. Varughese, H. Li, L. N. Zakharov, A. L. Rheingold and F. Jäkle, Organometallics, 2007, 26, 6126–6131 CrossRef CAS;
(c) H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, C. Hörl, I. Krummenacher, T. Kupfer, L. Mailänder and K. Radacki, J. Am. Chem. Soc., 2012, 134, 20169–20177 CrossRef CAS PubMed;
(d) A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed;
(e) M.-G. Ren and Q.-H. Song, Chem. Commun., 2012, 48, 2970–2972 RSC;
(f) M. Mao, M.-G. Ren and Q.-H. Song, Chem.–Eur. J., 2012, 18, 15512–15522 CrossRef CAS PubMed;
(g) D. J. Parks, R. E. V. H. Spence and W. E. Piers, Angew. Chem., Int. Ed., 1995, 34, 809–811 CrossRef CAS.
- L. Weber, V. Werner, M. Fox, T. B. Marder, S. Schwedler, A. Brockhinke, H.-G. Stammler and B. Neumann, Dalton Trans., 2009, 1339–1351 RSC.
- F. Leroux, ChemBioChem, 2004, 5, 644–649 CrossRef CAS PubMed.
- S. M. Cornet, K. B. Dillon, C. D. Entwistle, M. A. Fox, A. E. Goeta, H. P. Goodwin, T. B. Marder and A. L. Thompson, Dalton Trans., 2003, 4395–4405 RSC.
- T. Taniguchi, J. Wang, S. Irle and S. Yamaguchi, Dalton Trans., 2013, 42, 620–624 RSC.
- J. Wang, Y. Wang, T. Taniguchi, S. Yamaguchi and S. Irle, J. Phys. Chem. A, 2012, 116, 1151–1158 CrossRef CAS PubMed.
-
(a) Z. Lu, Z. Cheng, Z. Chen, L. Weng, Z. H. Li and H. Wang, Angew. Chem., Int. Ed., 2011, 50, 12227–12231 CrossRef CAS PubMed;
(b) Z. Lu, Y. Wang, J. Liu, Y.-J. Lin, Z. H. Li and H. Wang, Organometallics, 2013, 32, 6753–6758 CrossRef CAS;
(c) H. Ye, Z. Lu, D. You, Z. Chen, Z. H. Li and H. Wang, Angew. Chem., Int. Ed., 2012, 51, 12047–12050 CrossRef CAS PubMed.
-
(a) J. C. Doty, B. Babb, P. J. Grisdale, M. Glogowski and J. L. R. Williams, J. Organomet. Chem., 1972, 38, 229–236 CrossRef CAS;
(b) A. Pron, M. Baumgarten and K. Müllen, Org. Lett., 2010, 12, 4236–4239 CrossRef CAS PubMed.
- D. Stalke and K. H. Whitmire, J. Chem. Soc., Chem. Commun., 1990, 833–834 RSC.
-
(a) S. Toyota, M. Asakura, M. Oki and F. Toda, Bull. Chem. Soc. Jpn., 2000, 73, 2357–2362 CrossRef CAS;
(b) J. F. Blount, P. Finocchiaro, D. Gust and K. Mislow, J. Am. Chem. Soc., 1973, 95, 7019–7029 CrossRef CAS.
-
(a) J.-C. Hierso, Chem. Rev., 2014, 114, 4838–4867 CrossRef CAS PubMed;
(b) G. M. Benedikt, B. L. Goodall, S. Iyer, L. H. McIntosh III, R. Mimna, L. F. Rhodes, C. S. Day and V. W. Day, Organometallics, 2001, 20, 2565–2569 CrossRef CAS;
(c) C. Bartolomé, P. Espinet, J. M. Martín-Álvarez and F. Villafañ, Eur. J. Inorg. Chem., 2004, 2326–2337 CrossRef.
- I. E. Kareev, G. S. Quiñones, I. V. Kuvychko, P. A. Khavrel, I. N. Ioffe, I. V. Goldt, S. F. Lebedkin, K. Seppelt, S. H. Strauss and O. V. Boltalina, J. Am. Chem. Soc., 2005, 127, 11497–11504 CrossRef CAS PubMed.
- Although the single crystal of 2 was obtained from methanol, the compound selectively decomposes after a period of several days in this solvent to give the borinic ester (FMes)B(OMe)(C6H4-4-tBu), as observed by GC/MS (m/z = 456 [M]+). We note here also that in wet THF, the analogous borinic acid (GC/MS m/z = 442 [M]+) forms on a similar timescale, while 2 is stable in standard grade hexane, toluene and CH2Cl2 for extended periods of time and is stable in the solid state for a period of longer than six months without any detectable decomposition by NMR, even when stored under air.
-
(a) W. L. Jia, D. R. Bai, T. McCormick, Q. D. Liu, M. Motala, R. Y. Wang, C. Seward, Y. Tao and S. Wang, Chem.–Eur. J., 2004, 10, 994–1006 CrossRef CAS PubMed;
(b) N. Wang, Z. M. Hudson and S. Wang, Organometallics, 2010, 29, 4007–4011 CrossRef CAS;
(c) A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem.–Eur. J., 2012, 18, 5022–5035 CrossRef CAS PubMed;
(d) W. Z. Yuan, S. Chen, J. W. Y. Lam, C. Deng, P. Lu, H. H.-Y. Sung, I. D. Williams, H. S. Kwok, Y. Zhang and B. Z. Tang, Chem. Commun., 2011, 47, 11216–11218 RSC.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
-
(a) U. Resch-Genger, Y. Q. Li, J. L. Bricks, V. Kharlanov and W. Rettig, J. Phys. Chem. A, 2006, 110, 10956–10971 CrossRef CAS PubMed;
(b) V. A. Kharlanov, W. Abraham and W. Rettig, J. Photochem. Photobiol., A, 2001, 143, 109–117 CrossRef CAS;
(c) W. Rettig, V. Kharlanov and M. Maus, Chem. Phys. Lett., 2000, 318, 173–180 CrossRef CAS;
(d) M. Maus, W. Rettig, D. Bonafoux and R. Lapouyade, J. Phys. Chem. A, 1999, 103, 3388–3401 CrossRef CAS;
(e) M. Mausand and W. Rettig, Chem. Phys., 1997, 218, 151–162 CrossRef.
- W. Siebrand, J. Chem. Phys., 1967, 46, 440–447 CrossRef CAS PubMed.
- R. Stahl, C. Lambert, C. Kaiser, R. Wortmann and R. Jakober, Chem.–Eur. J., 2006, 12, 2358–2370 CrossRef CAS PubMed.
- Q. Wu, T. Zhang, Q. Peng, D. Wang and Z. Shuai, Phys. Chem. Chem. Phys., 2014, 16, 5545–5552 RSC.
-
(a) S. Menning, M. Krämer, B. A. Coombs, F. Rominger, A. Beeby, A. Dreuw and U. H. F. Bunz, J. Am. Chem. Soc., 2013, 135, 2160–2163 CrossRef CAS PubMed;
(b) M. Levitus, G. Zepeda, H. Dang, C. Godinesz, T.-A. V. Khuong, K. Schemieder and M. A. Garcis-Garibay, J. Org. Chem., 2001, 66, 3188–3195 CrossRef CAS PubMed;
(c) S. Menning, M. Krämer, A. Duckworth, F. Rominger, A. Beeby, A. Dreuw and U. H. F. Bunz, J. Org. Chem., 2014, 79, 6571–6578 CrossRef CAS PubMed.
- S. A. Cummings, M. Iimura, C. J. Harlan, R. J. Kwaan, I. V. Trieu, J. R. Norton, B. M. Bridgewater, F. Jäkle, A. Sundararaman and M. Tilset, Organometallics, 2006, 25, 1565–1568 CrossRef CAS.
-
(a) H. Braunschweig, V. Dyakonov, J. O. C. Jiminez-Halla, K. Kraft, I. Krummenacher, K. Radacki, A. Sperlich and J. Wahler, Angew. Chem., Int. Ed., 2012, 51, 2977–2980 CrossRef CAS PubMed;
(b) H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Chem.–Eur. J., 2012, 18, 8430–8436 CrossRef CAS PubMed.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- We note that the HOMO energy of Fc estimated by electrochemical methods in CH2Cl2 (−5.11 eV) differs from the value of −4.8 eV used here, see: D. Reitzenstein, T. Quast, F. Kanal, M. Kullmann, S. Ruetzel, M. S. Hammer, C. Deibel, V. Dyakonov, T. Brixner and C. Lambert, Chem. Mater., 2010, 22, 6641–6655 CrossRef CAS . However, −4.8 eV was adopted because it is widely used in the literature and will, therefore, facilitate comparison of the energy levels of the present compounds with previously reported compounds.
- E. Sakuda, Y. Ando, A. Ito and N. Kitamura, J. Phys. Chem. A, 2010, 114, 9144–9150 CrossRef CAS PubMed.
- M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1986, 108, 4235–4236 CrossRef CAS.
- T. Kushida and S. Yamaguchi, Organometallics, 2013, 32, 6654–6657 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. Additional views of the molecular structures from X-ray diffraction. Cyclic voltammograms of 2 and 3. Plots of calculated lowest energy transitions versus UV-visible absorption spectra. Variable-temperature emission and excitation spectra. Copies of all NMR spectra. Cartesian coordinates of all optimised geometries. CCDC 1010408–1010412. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02410a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence