
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-dimensional copper based colloidal nanocrystals: synthesis and applications
Nilotpal
Kapuria
 ,
Niraj Nitish
Patil
,
Kevin M.
Ryan
and
Shalini
Singh
*
,
Niraj Nitish
Patil
,
Kevin M.
Ryan
and
Shalini
Singh
*
Department of Chemical Sciences and Bernal Institute, University of Limerick, Limerick, Ireland. E-mail: Shalini.Singh@ul.ie
First published on 27th January 2022
Abstract
Two-dimensional (2D) semiconductor nanocrystals display unconventional physical and opto-electronic properties due to their ultrathin and unique electronic structures. Since the success of Cd-based photoemissive nanocrystals, the development of sustainable and low-cost nanocrystals with enhanced electronic and physical properties has become a central research theme. In this context, copper-based semiconductor 2D nanocrystals, the cost-effective and eco-friendly alternative, exhibit unique plasmonic resonance, transport properties, and high ionic conductivity beneficial for sensing, energy storage, conversion, and catalytic applications. This review summarizes recent progress in the colloidal synthesis, growth mechanisms, properties, and applications of 2D copper-based nanostructures with tunable compositions, dimensions, and crystal phases. We highlight the growth mechanisms concerning their shape evolution in two dimensions. We analyse the effectiveness of cation exchange as a tool to synthesize multinary nanocrystals. Based on the preparation of Cu-based chalcogenide and non-chalcogenide compositions, we discuss synthesis control achieved via colloidal approaches to allow dimension tunability, phase engineering, and plasmonic and thermoelectric property optimization. Furthermore, their potential in various applications of catalysis, energy storage, and sensing is reviewed. Finally, we address the current challenges associated with 2D Cu-based nanocrystal development and provide an outlook pertaining to unexplored research areas.
 Shalini Singh | Dr Shalini Singh is a Lecturer at the Department of Chemical Sciences and the Bernal Institute at the University of Limerick, Ireland. She received her PhD in chemistry from the University of Limerick in 2016 and was an FWO (Research Foundation, Flanders, Belgium) Postdoctoral Fellow at Ghent University, Belgium until 2019. Since 2020, she has been leading the Functional Nanomaterial Research Group at the University of Limerick. Her research interest is focused on the development of novel colloidal semiconductor and metallic nanocrystals for energy conversion and storage applications. |
1. Introduction
The advances in colloidal synthesis approaches over the past decade have resulted in unprecedented synthesis control over several solution-processed two-dimensional (2D) nanocrystals (NCs), with layered and non-layered structures, that display extraordinary physical and optoelectronic properties.1–8 In the layered structures, each monolayer with a covalently bonded atomic arrangement is vertically stacked together via the weak van der Waals (vdW) force. Using colloidal approaches, a range of layered transition metal chalcogenides of ME2 (M = Mo, W, Zr, Hf), SnS, GeS, InS, and GaS have been successfully synthesised.1,3,9–12 Controlling the 2D thickness of these nanostructures resulted in high mechanical stability, optical transparency, the transition of indirect to direct bandgap, and stabilisation of metastable phases. The non-layered structures possess covalent bonds in three dimensions. For non-layered NCs, the 2D nature is defined by strong electron–hole confinement in one dimension or by growth limitation in one of the directions. Quantum confinement resulting from controlling the thickness below the Bohr radius leads to several exciting optoelectronic properties such as sharp emissions, large laser threshold, enhanced oscillator strength, fast exciton dynamics, etc. Colloidal nanosheets, nanobelts, or rectangular nanoplatelets of CdE, ZnE, PbS, PbSe, HgSe, and HgTe are among the most explored systems in this category.6,13–20The sustainability and toxicity issues associated with heavy elements constituting a significant portion of nanomaterials (e.g. Cd and Pb) have shifted the current research focus towards cost-effective and eco-friendly NCs with a 2D morphology such as copper chalcogenides, copper phosphide (Cu3−xP), and metallic alloys.21–26 Owing to their high electron conductivity and thickness controlled optoelectronic properties, they have found applications in thermoelectrics, alkali-ion battery, sensing, etc.27–31 These NCs with a 2D morphology also exhibit a high surface area for efficient catalysis and further allow the manipulation of plasmonic properties by doping, defect engineering, or heterostructure formation.32 For the semiconductor and metallic NCs that do not display quantum confinement, the 2D shape is largely defined by the morphological appearance in the form of discs, plates, or sheets (Scheme 1a). The nanocrystals with high symmetry, such as hexagonal, are more inclined to form hexagonal and triangular plates, or discs, and NCs with cubic or orthorhombic crystal structures tend to form platelets. In terms of quantum confinement, nanosheets display strong out of plane confinement and zero in-plane confinement as the length and width of nanosheets are at least 20 times larger than the thickness,6,21 whereas nanoplatelets display weak electron confinement along the width and length.7 Likewise, nanodiscs and nanoplates exhibit weak in-plane electron confinement. For nanoplates and nanodiscs confinement along the vertical direction can range from weak to strong depending on the thickness. The least passivated narrow edges of these 2D NCs are often critical for active species binding during electrochemical reactions.
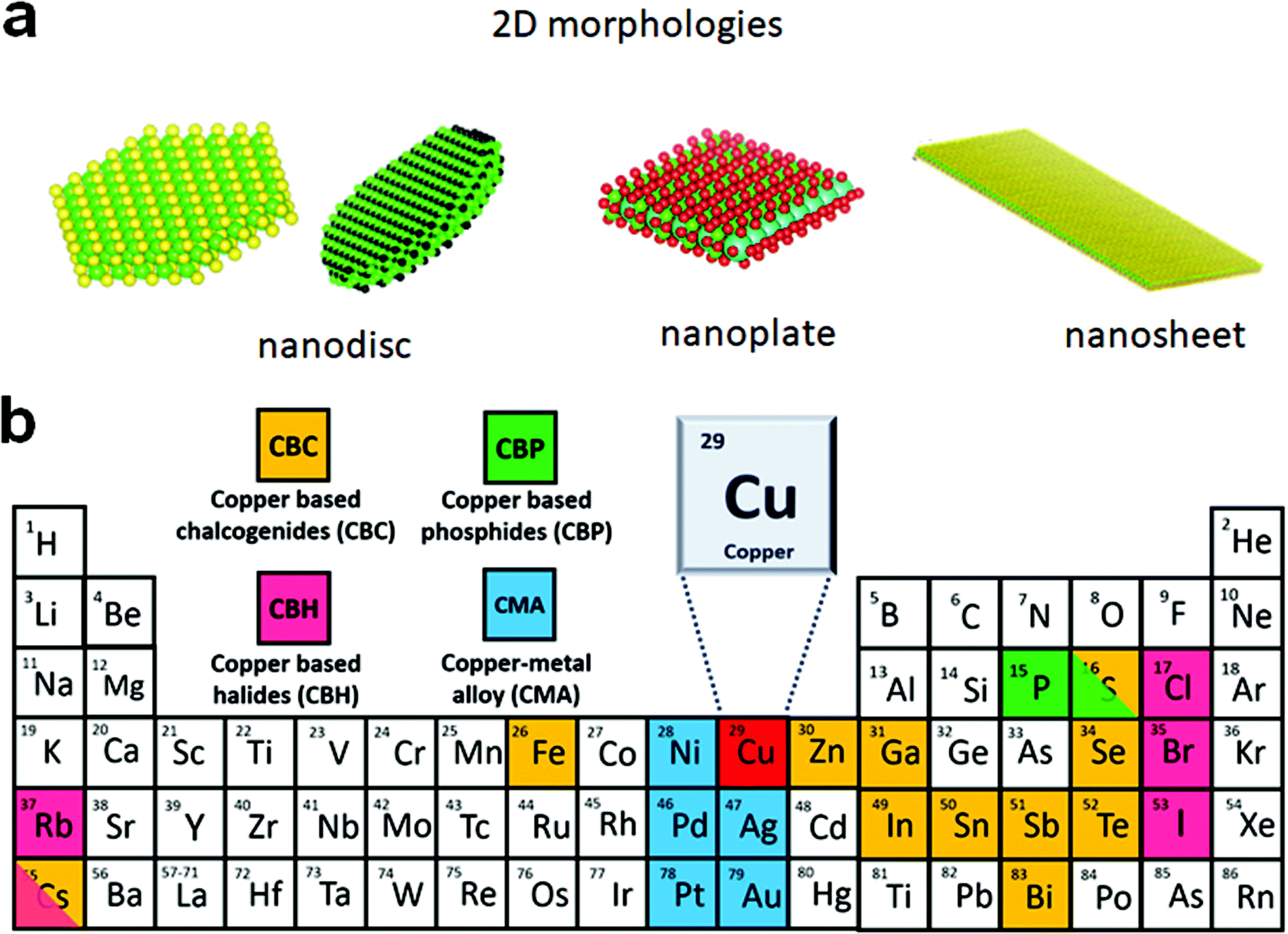 | ||
| Scheme 1 (a) Possible morphologies of colloidal 2D NCs where charge carriers are confined within a plane but can move along the plane; (b) composition of Cu-based 2D NCs. | ||
The emerging ternary alkali metal (Cs, Rb) copper halides that display strong electron–phonon coupling and structural distortion upon photoexcitation result in deeply self-trapped emission.33,34 Given their photoluminescence properties, they are applied in blue LED fabrication. Moreover, they are also predicted as phonon glass electron crystals with a thermoelectric figure of merit ∼ 2.6.35 In addition, the high ionic mobility of Cu+ in a Cu-based NC lattice benefits foreign cation substitution forming multinary NCs, which is not feasible for most other 2D NCs.36,37 These Cu-based NCs can possess both layered and non-layered structures. For example, in orthorhombic CuSbS2 each layer of interconnected CuS3 and SbS2 chains weakly interacts via the vdW force.27 Interestingly, a layered copper sulphide that exhibits temperature-dependent photoluminescence exists constituting monolayers of two (Cu–S)n chains interconnected by Cu–Cu bonds, and where each layer is interconnected by alkanethiol.38 Given the significant advancement of synthesis control and applicability of the colloidal Cu-based 2D NCs that have been achieved to date, it is timely to review the progress made, challenges remaining, and future outlook.
The present review aims at providing an overview of the multifaceted advancements that have emerged in the growth mechanism, synthesis, properties, and application of colloidal 2D Cu based NCs. First, we briefly discuss the various compositions of 2D Cu based NCs achieved using colloidal approaches. We discuss the growth mechanism of these NCs, highlighting the effect of ligand passivation, the effect of alkanethiol and halide anions in copper chalcogenide formation and cation exchange for multi-elemental NC formation. This is followed by the discussion on synthesis and growth parameters, with a specific focus on copper-based chalcogenides, copper phosphides, copper-based halide NCs, and metal NCs. We describe their plasmonic and thermoelectric properties and applications in photocatalysis, electrocatalysis, energy storage, and sensors. We conclude the review with an outlook of the different synthetic approaches and compositions for 2D copper based NCs in view of their applications.
2. Colloidal 2D copper based NCs
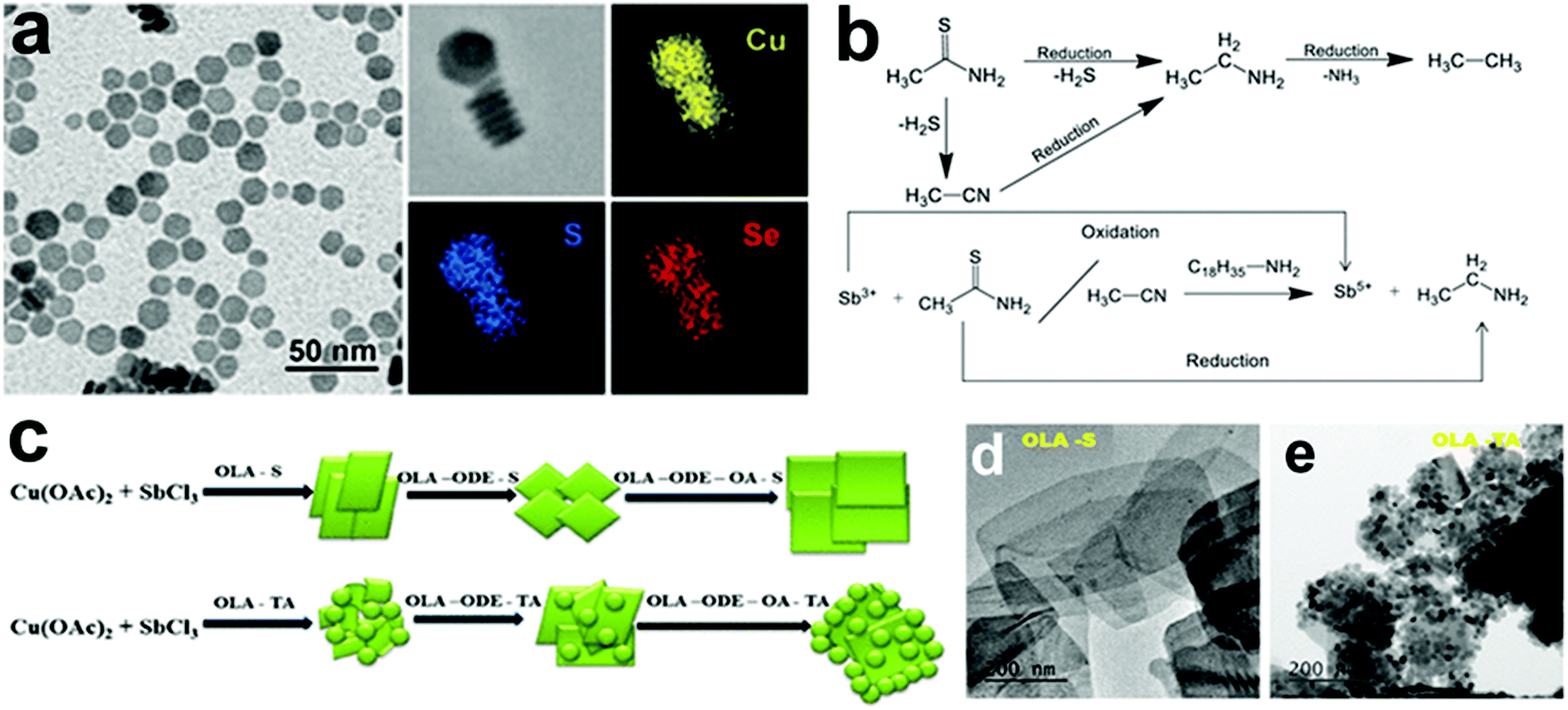
In recent years, bottom-up approaches have gained prominence in synthetic chemistry, particularly with the advancement of synthetic techniques for producing nanomaterials with well-defined size, morphology, phase, and characteristics.39,40 So far, bottom-up methods, such as the physical deposition method and wet chemical synthesis, have been comprehensively investigated to develop 2D nanostructures.8,41 In particular, colloidal synthesis approaches have shown significant utility in synthesising solution processable 2D NCs with the morphology ranging from nanosheets, nanodiscs to nanoplates.40,42,43 In a typical colloidal synthesis, the organic anions and metal salts or organometallic precursors as metal sources are combined with the organic solvent with boiling point up to 360 °C under inert conditions. The heat-up technique and the hot injection approach are the two ways often used in colloidal systems. The heat-up technique includes gradually heating the precursors in the presence of ligands and organic solvents.44 In contrast, the hot injection method induces supersaturation by creating a temperature gradient between anionic and metal precursors. The rapid injection of an anionic source into a hot metal precursor solution in the presence of ligands and organic solvents, or vice versa, starts the nucleation.45,46 Heat-up synthesis is beneficial for the gram-scale synthesis of NCs due to its simplicity. However, in the heat-up process, the nucleation and growth step are not separated well due to the continuous supply of the monomer with an increment of thermal energy. The extended monomer saturation stage induces broad size distribution, especially for multinary systems. In contrast, hot-injection induces a supersaturation that accounts for single burst nucleation providing more control over the shape, size, and composition.To date, using colloidal approaches, a number of 2D NCs of binary and multinary Cu chalcogenides (Cu2−xE, E = S, Se, Te), binary Cu phosphides (Cu3−xP) and Cu–metal alloys (Scheme 1b) have been synthesised. The binary Cu2−xS, Cu2−xSe, Cu2−xTe, and Cu3−xP are the most explored compositions for 2D morphologies.47–50 The introduction of group III metals such as In and Ga in the binary Cu chalcogenides resulted in the second most explored compositions of ternary CuInS2 and CuGaS2 and their selenide alternatives in 2D mrophology.51–53 Here the partial replacement of In for Ga and S for Se has resulted in quaternary compositions of CuIn1−xGaxS2, CuInS2xSe2−2x and a more complex composition of CuIn1−xGaxS2xSe2−2x.54 Introduction of transition metals, group IV and group V elements into Cu-chalcogenide 2D NCs resulted in the following compositions of CuSbS2, Cu3SbS3, Cu3BiS3, Cu2SnS3, Cu2SnSe3, Cu2GeS3, CuFeS2, Cu2SnZnSe4, Cu2GeZnSe4, etc.22,37,55–61 More recently, metal halides of Cu with compositions of Cs3Cu2I5, Cs3Cu2Br5 and Rb2CuX3 (X = Cl, Br) have also been synthesised.35,62 Among the metallic Cu 2D NCs seeded growth materialised NCs with PdCu and PdPtCu compositions.63 Apart from these, many other unconventional compositions such as Cu3P1−xSx and CsCu5S3 have also been achieved using colloidal methods.64,65 All the compositional outcomes that are achieved using the colloidal approach are highlighted in Scheme 1b.
2D NCs of Cu2−xS, Cu2−xSe, Cu3−xP, and Cu2−xTe have been synthesised using heat-up and hot-injection approaches in different phases.23,47,48,66–69 They are mainly investigated for their plasmonic and electronic properties. Though initial approaches for synthesising binary Cu2−xS were solely based on traditional hot-injection and heat-up techniques, later soft templates of Cu-thiolates and other amphiphilic ligands were improvised to achieve NCs with ultrathin thickness. For binary Cu3−xP, heat up techniques are devised based on the decomposition of organophosphorous compounds such as trioctylphosphine, tris(diethylamino)phosphine and triphenyl phosphite. High temperatures above 300 °C are used for trioctylphosphine but other precursors could be decomposed below 300 °C. While the hot-injection synthesis of Cu3−xP requires the injection of highly reactive precursors such as tris(trimethylsilyl)phosphine. Among the other semiconductor NCs are multinary CuInS2 nanodiscs, nanosheets, CuSbS2 nanosheets, and Cu3BiS3 nanoplatelets using heat-up and hot-injection approaches.21,52,55,56,70 Initial approaches for synthesising CuInS2 were based on the heat-up of the metal precursor and S source. But extension of the elemental composition to quaternary by the introduction of Ga and Se required a more controlled hot-injection approach where cation and anion introduction windows can be separated based on the injection temperature. For group V based NCs, heat up techniques are based on the decomposition of diethyldithiocarbamate precursors of metals.55,56 Using the cation exchange of Cu2−xS and Cu2−xSe 2D templates several heterostructures and multinary nanoplates with more complex compositions involving Au, Ag, Cd, Zn, Hg, Pb, Pd, In, Ga, Fe, Sn, and Ge have also been achieved in recent times.61 The recent addition to the ion exchange series is the anion exchange of the Cu3−xP template for the materialisation of Cu3P1−xSx nanorings.64 Alloyed Cu-based multimetallic NCs such as PtPdCu nanorings have also been synthesised via Cu2+ introduction of the PtPd template.63 The milestones achieved in terms of synthesis in this field are highlighted in Scheme 2.
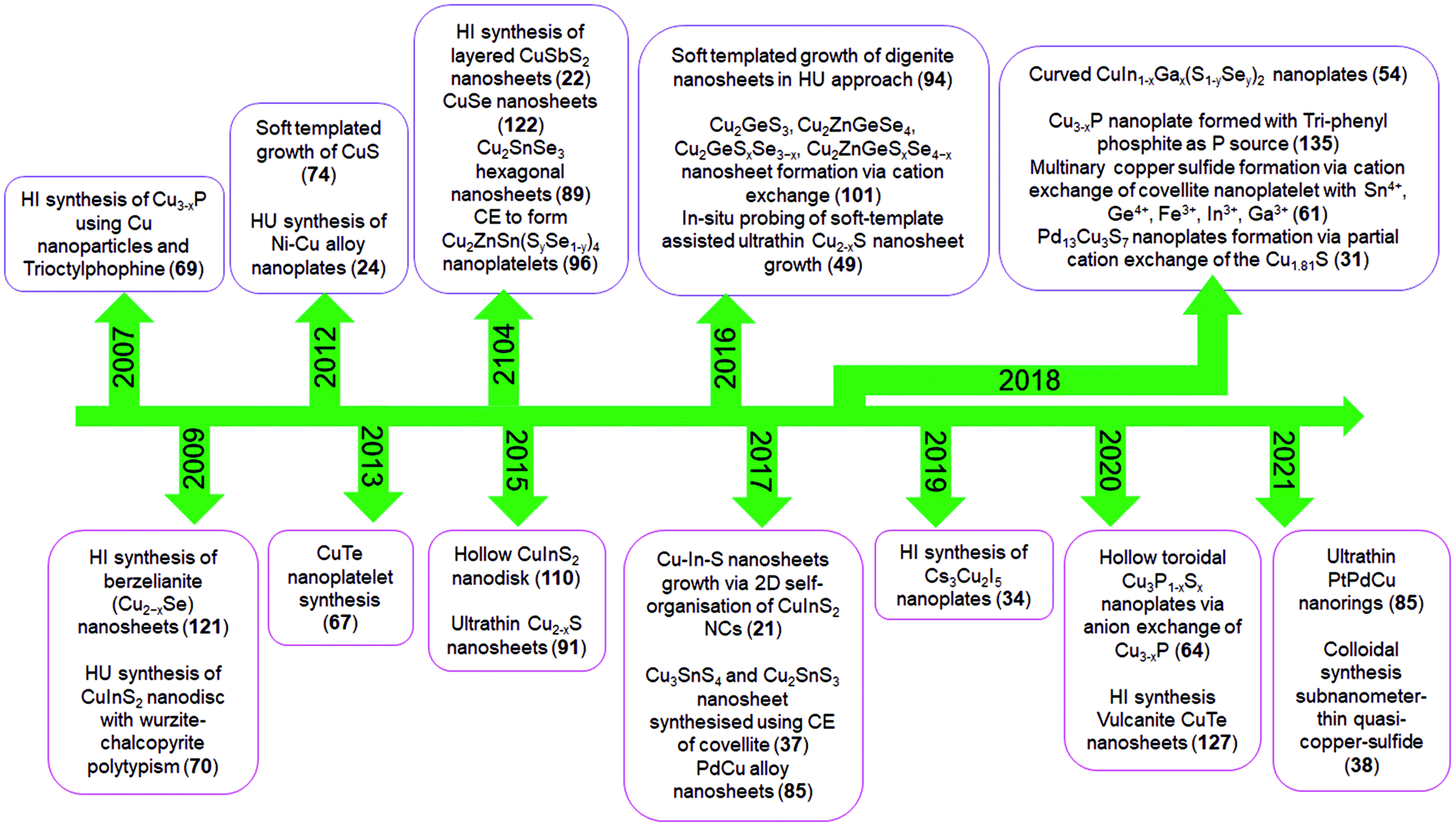 | ||
| Scheme 2 Literature reports on colloidal 2D Cu-based nanocrystals. Here HI, HU and CE denote hot-injection, heat-up and cation exchange, respectively. | ||
3. Growth mechanism
Controlling the morphology in two dimensions in the form of colloidal nanoplates, nanodiscs, or nanosheets is generally achieved through oriented attachments, soft template-assisted growth, cation exchange (CE) of 2D templates or due to the ligand passivation of certain facets and restriction from crystal symmetry.6,21 In oriented attachment/self-organisation, the energetically under-passivated crystal facets of NCs attach to form a superstructure assembly which further goes through recrystallisation to form larger nanocrystals.71–73 However, the 2D morphology will form when the preferential binding of ligands to the facet perpendicular to the growth facet constricts the assembly in a two-dimensional template. In the oriented attachment driven growth of 2D nanocrystals, Schliehe et al. describe an egg tray-like assembly of PbS quantum dots via attachment of (110) facets where the oleic acid bound to the (100) facets formed a bilayer stacking resulting in PbS nanosheets.73 The surface energy is a controlling parameter to decide the shape of NCs in solution. The Gibbs free energy for an individual NC formation is described as ΔG = ΔGbulk + γδA, where ΔGbulk = ΔHbulk − TΔSbulk, γ is the surface energy per unit area of NCs and A is the total area of NC. The surface energy (γ) contributes significantly to the total Gibbs free energy for NC formation. The surface energy of individual NCs depends on the passivation of certain facets, and the ligands play a critical role in determining which facets will grow. Therefore, ligand passivation is regarded as a significant driving factor for forming anisotropic NCs.7 In the case of soft templated growth, ligands such as OLA and 1-DDT, or an amphiphilic surfactant such as cetrimonium bromide that dissolve the cationic precursors can form lamellar templates sandwiching the metal cations between the top and bottom layer alkyl chain coordinated via their functional groups.74 The stability of such a lamellar structure facilitates metal chalcogenide formation in the 2D confined framework upon the introduction of anionic precursors.74–76 Another method, the cation exchange, which requires topotactic cationic replacement of 2D template NCs to form a new composition, has also been used to synthesize several multinary 2D NCs from 2D binary metal chalcogenide templates.77 The formation of anisotropic metal nanocrystals proceeds via a slow reduction process, providing a better chance of overcoming the more thermodynamically favored reactions toward forming truncated nanocubes due to their intrinsically higher energy compared to other isotropic shapes.78,79 Additionally, the anisotropic growth of multimetallic metal NCs is vastly influenced by the nature and shape of the initial seeds.80,81 In contrast to semiconductors, the seeded growth of multi-metallic NCs initiates via the deposition of foreign cations on the seed and their subsequent interatomic diffusion forms the alloy. The following sub-sections will discuss the crucial mechanisms that drive the growth and 2D morphology evolution in the colloidal synthesis of Cu based NCs.3.1. Effect of ligand passivation
In colloidal synthesis the choice of the ligand is a crucial factor in directing the shape of NCs during growth. Depending on the metal–anion framework of the terminating facets, the polarity of each facet varies; thus, the surface energy is also different for each facet.82,83 The strong binding of the ligand to certain facets inhibits monomer deposition on those facets compared to other facets and influences anisotropic growth. The ligands can be small molecules generated upon precursor decomposition such as halides, carbonyls, solvents such as oleylamine (OLA), trioctylphosphine oxide (TOPO), or metal–ligand complexes.51,84 During the growth of metal NCs with the cubic crystal phase, CO generated upon metal carbonyl precursor decomposition binds explicitly with the (111) facet to inhibit further monomer deposition from forming 2D NCs enclosed with {111} planes. For example, Yang et al. used Mo(CO)6 for generating CO in situ, which influenced the nanosheet morphology during CuPd NC growth.85 As another class of small molecules, halide ions act as surface-active species that govern the shape of NCs. The halide ions can influence the anisotropic development of metal nanocrystals in numerous ways, including modification of the redox potentials of the metal ions, serving as face-specific capping agents, and/or influencing the degree of metal underpotential deposition at the nanocrystal surface. For example, the presence of halide ions was shown to control the 2D morphology in hexagonal CuInS2 and CuGaS2 NCs.52,86,87 Liu et al. showed the formation of 2D CuGaS2 NCs by using GaCl3 as the Ga3+ source whereas an identical synthesis involving Ga(acetylacetonate) [Ga(acac)3] formed nanorods.52 The favourable adsorption of Cl− on basal {001} facets of wurtzite CuGaS2 influenced the 2D growth (Fig. 5e and f). Tang et al. demonstrated the influence of halide ions on the morphology-controlled synthesis of Cu nanocrystals in the aqueous phase.88 The type of halide ion (Cl−, Br−, I−) solely controlled the morphology of nanocrystals (2D plates, 1D wires, and 3D polyhedral particle). DFT study showed that the co-adsorption strength of ammonium and halide ions on Cu(100) compared to Cu(111) was a critical factor in regulating the shape of the Cu NCs. As a result, the selective adsorption of halide ions on certain facets of Cu NCs would contribute to morphological differences.Coordinating ligands can affect the NC morphology immensely by adsorbing onto partially charged facets. OLA was shown to passivate the polar (001) facet of wurtzite CuGaS2 NCs inhibiting the growth along the 〈001〉 direction promoting 2D growth. For example, Adhikari et al. demonstrated a non-coordinating solvent in the reaction system formed wurtzite CuGaS2 nano-tadpoles.51 In contrast, when a coordinating solvent OLA was used, flat 2D nano tadpoles were formed. Similarly, our group observed that using coordinating solvents such as OLA and TOPO in the reaction system for wurtzite CuIn1−xGax(S1−ySey)2 synthesis formed nanoplates.54 However, NCs with intrinsically asymmetric crystal structures such as klockmannite and covellite crystal phases seldom get affected by ligand interaction.89 In klockmannite, covellite, one CuE3 unit sandwiched between two CuE4 units, comprises a trilayer interconnected to the another trilayer up and down via di-chalcogenide bonds along the vertical axis. For such structures, new metal–chalcogenide bond formation can only take place along the linear direction forming 2D NCs.
3.2. Effect of alkanethiol and halide anions on copper chalcogenide synthesis
According to Pearson's hard–soft acid–base theory, alkanethiols as soft Lewis bases display a strong affinity towards soft Lewis acidic CuI cations. This favourable interaction is responsible for forming columnar or lamellar Cu-thiolate complexes.48,49,91–93 In the lamellar framework, CuI stays confined between hydrophobic layers of alkanethiols.49 The alkane chain length decides the integrity of the lamellar structure. In the columnar structure, four CuI interact with four alkanethiol forming discs that stack with each other.48 These polymeric complexes can break down above a certain temperature e.g., columnar framework of CuI-1-DDT breaks down beyond 190 °C. The complexes subsequently undergo an isotropic phase transition to form monomers upon thermolysis of the C–S bond resulting in the isotropic nucleation of pseudospherical NCs. The presence of halide anions can prolong the tenacity of such CuI-alkanethiolate complexes up to the onset of nucleation.49,91,94 van der Stam et al. observed a CuI-DDT lamellar structure, which generally melts beyond 150 °C, but remained intact in the presence of chlorides up to 230 °C.49As a result, the nucleation in the 2D lamellar template formed thin nanosheets of Cu2−xS (Fig. 1a and b). It was proposed that the presence of Cl− as a poly coordinated bridging ligand strengthens the lamellar structure. In another instance, Zhai et al. observed the effect of chloride ion on the morphology by varying the ratios between Cu(NO3)2 and CuCl2 precursors (Fig. 1c).90 Keeping the total copper precursor concentration constant, a lower concentration of CuCl2 with respect to Cu(NO3)2 (CuCl2/Cu(NO3)2 = 0.53) materialized 1D nanostructures whereas a higher concentration of CuCl2 (CuCl2/Cu(NO3)2 = 9) solely formed 2D nanoplates. Similarly, Wu et al. varied HCl in a reaction system containing Cu(OAc)2, TOPO, and ODE to see a transition from spherical NPs for zero or low HCl to triangular nanoplates at 2 mmol HCl.95 Apart from the Cu-halides other metal halides (e.g. SnBr4, SnCl4) have also been explored to induce a 2D morphology where their presence instigated 2D templated growth by stabilizing the lamellar complex.91 Depending on the halide species, the shape of the NCs can be modulated. In the presence of Br−, mostly hexagonal plates or sheets are formed whereas Cl− predominantly forms triangular shapes (Fig. 1d).91,95 In contrast, I− species which is a soft base does not form any regular shapes. Such disparity can be attributed to the strong interaction between Cu+ and I−, which hampers the formation of Cu-alkanethiolate. However, it is unclear if halide anions can affect the 2D growth of multinary Cu-chalcogenides by forming a soft template as nuclei.
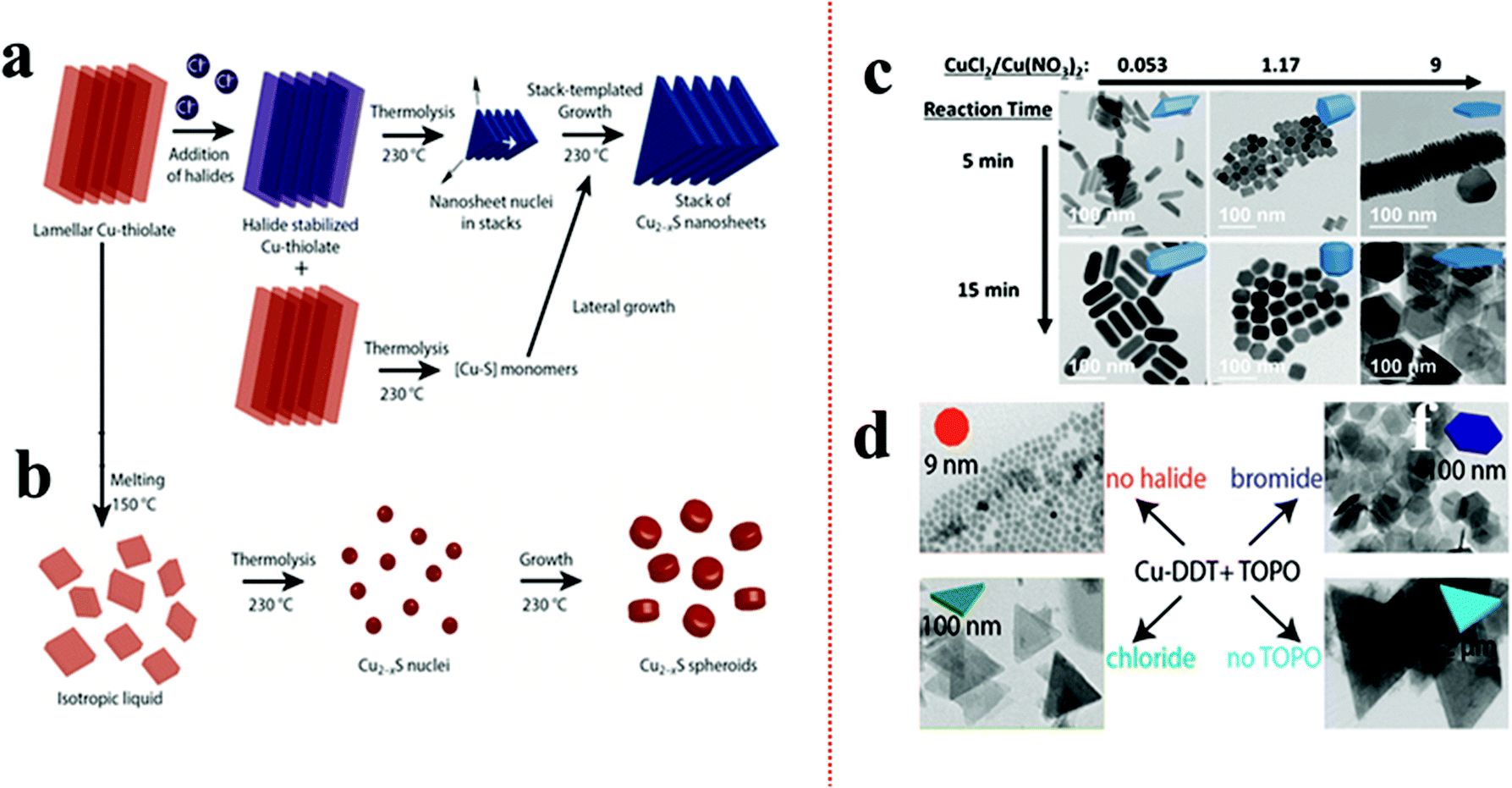 | ||
| Fig. 1 (a) Schematic of controlling the 2D shape of the Cu2−xS using Cl− source. The Cl− stabilized the lamellar Cu-alkanethiolate structure close to the nucleation stage influencing a 2D templated nucleation; (b) in the absence Cl− the lamellar structure melts and provides monomers for Cu2−xS nuclei formation resulting in spheroids. Reproduced from ref. 49 with permission from ACS, copyright 2016; (c) effect of ratio variation between CuCl2 and Cu(NO3)2 on morphology control of Cu2−xS. Reproduced from ref. 90 with permission from ACS, copyright 2017; (d) effect of SnX4 (X = Cl, Br) additives on nanoplate formation of Cu2−xS. The Br− source formed hexagonal plates whereas the Cl− source produced triangular plates. Reproduced from ref. 91 with permission from ACS, copyright 2016. | ||
3.3. Cation exchange (CE)
The topotactic cationic replacement in 2D Cu2−xS or Se nanoplates are usually used to materialize multi-elemental NCs of different compositions in a two-dimensional morphology.61,96,98 The high mobility of cations on a rigid anionic lattice is mostly responsible for creating channels for cation replacement.99 Sequential cationic incorporation in the anionic lattice avoids the unnecessary formation of secondary impurities while keeping the morphology intact. The compositional outcome mostly depends on the following:(1) Incoming foreign cation valency and its ability to adapt the coordination environment of the template lattice.
(2) The vacancy of Cu cations in the template to facilitate the movement of cations on the rigid anionic lattice.
(3) The presence of mildly reducing soft Lewis base (e.g. alkanethiols, tri-alkyl phosphine) to scavenge Cu+ using favourable soft acid–soft base interaction.
Cu2−xS or Se possesses a rich and diverse phase diagram having monosulphide (Cu2−xS, x = 0–0.4) and disulphide anions with tetrahedral and trigonal sites. A ternary combination involving a divalent cation (Cd2+, Zn2+, Hg2+etc.) and a monovalent cation cannot satisfy the charge balance in tetrahedral coordination, thus the divalent cations partially or completely replace the Cu cation to form heterostructures or a new binary phase.36,77 Liu et al. outlined the formation of heterostructure nanoplates of Cu2S–CdS on a covellite CuS template by using Cd2+ as the foreign cation with 1-DDT. In this study, 1-DDT acted as the Cu+ extractor and reducing agent for CuS to Cu2S (Fig. 2a).61 In contrast, trivalent cations such as In3+, Ga3+ can satisfy the charge balance and alloy to form ternary NCs. Mu et al. demonstrated the formation of thin CuInS2 nanoplates using CE of the djurleite Cu2−xS template where Cu+ and In3+ share the tetrahedral coordination site.100 Moreover, with the tetravalent cations (Sn4+, Ge4+) alloying has also been possible with templates having trigonal sites along with the tetrahedral site. One such example is the cation exchange of CuS nanoplates with Sn4+ in the presence of a mild reducing agent, 1-DDT (Fig. 2b).37 In CuS, two tetrahedral units of the CuS4 sandwich a trigonal CuS3 unit to form a single layer where each layer is connected to the other layer via a disulphide bond. Here 1-DDT is crucial to reduce the disulphide bond to expose holes for cation incorporation. Additionally, 1-DDT being a soft base scavenged Cu+ cations to create cation vacancies in the lattice. Upon disulphide bond breakage, Sn4+ replaces Cu+ from the trigonal site and keeps the tetrahedral sites intact to form Cu2SnS3 nanoplates. In contrast, using Sn2+ with 1-DDT in the same process generated SnS. However, in the absence of 1-DDT, Sn2+ acts as a reducing agent and reduced the disulphide bond in a two-electron reduction process converting all the trigonal sites into tetrahedral co-ordination. Upon the two electron-reduction process of the Cu6S6 covellite unit, two Sn2+ entities convert themselves into Sn4+ and incorporate in the tetrahedral sites to form Kuramite Cu3SnS4 nanoplates. Using 1-DDT as the reducing agent and CuS nanoplates as a template, Liu et al. synthesized Cu2GeS3, CuInS2, CuGaS2, and CuFeS2 nanoplates (Fig. 2c).61 Once the coordination restriction relaxes upon the introduction of a second cation, divalent cation incorporation is facilitated to form quaternary or multinary nanoplatelets. Lesnyak et al. demonstrated the formation of Cu2ZnSnSeyS1−yvia a stepwise cation exchange of Cu2−xSeyS1−y template using TOP as a soft Lewis base (Fig. 2d).96 Similarly, the compositions of Cu2ZnSnS4, Cu2ZnGeSe4, Cu2GeSeyS3−y, and Cu2ZnGeSeyS4−y were also explored via CE.101,102 Besides providing uniform elemental compositions, CE preserves the anionic sublattice and metal coordination environment in most cases.103–105 Consequentially, the kinetically trapped phases which are not accessible via conventional synthesis approaches could also be materialized. Generally, a template with hexagonal packing of anions will generate the hcp arrangement of anions in the resulting product. This leads to the preferred formation of a wurtzite, monoclinic or rhombohedral crystal phase. Whereas a cubic (ccp, fcc, bcc) packing will result in zinc blende, orthorhombic, and tetragonal crystal phases. For example, using the djurleite Cu1.94S template which possesses a hexagonal S packing, CE with In3+ formed wurtzite CuInS2.57 Similarly, using a digenite Cu2−xS template which possesses cubic close packing (ccp) of S, CE with In3+ formed zinc blende CuInS2.104 Despite having control over the composition and crystal structure of di-, tri-, and tetra-valent cations, the outcome of CE with monovalent cations is less clear.106–108 Depending on the crystal structure of the template used, the outcome will vary. For example, Swihart and co-workers observed at a lower Ag+ concentration that Ag2S domains were formed at the corners of the covellite CuS plate when used as a template (Fig. 2e).97 The high surface energy at the corner lowers the nucleation barrier, due to which most of the CE starts at the corner and gradually reaches the core. The disulphide bond in CuS allowed for the redox reaction and reorganisation that lead to Ag2S domains to grow further to take a pseudospherical morphology. In contrast, the djurleite Cu1.94S template facilitated the formation of alloyed Ag3CuS2 plates. In another instance, Kim et al. explored the Cu29S16 template to materialise segregated Cu31S16–Ag2S plates which evolve into an Ag2S shell on Cu31S16 plates and later again to segregated Cu31S16–Ag2S plates upon Pt shell formation on the basal plane.109 They suggested the transition of Cu29S16 into Cu31S16 and strain in Ag2S facilitated Ag+ migration from the core to shell and again to the core. It is worth mentioning that once the crystal phase reaches a stable structure, exchange of cation ceases. However, high temperature, high lattice strain, and vast difference between the anionic lattice of the template and the desired product can promote reorganisation of the lattice structure and change the shape of the NCs with metastable structures.
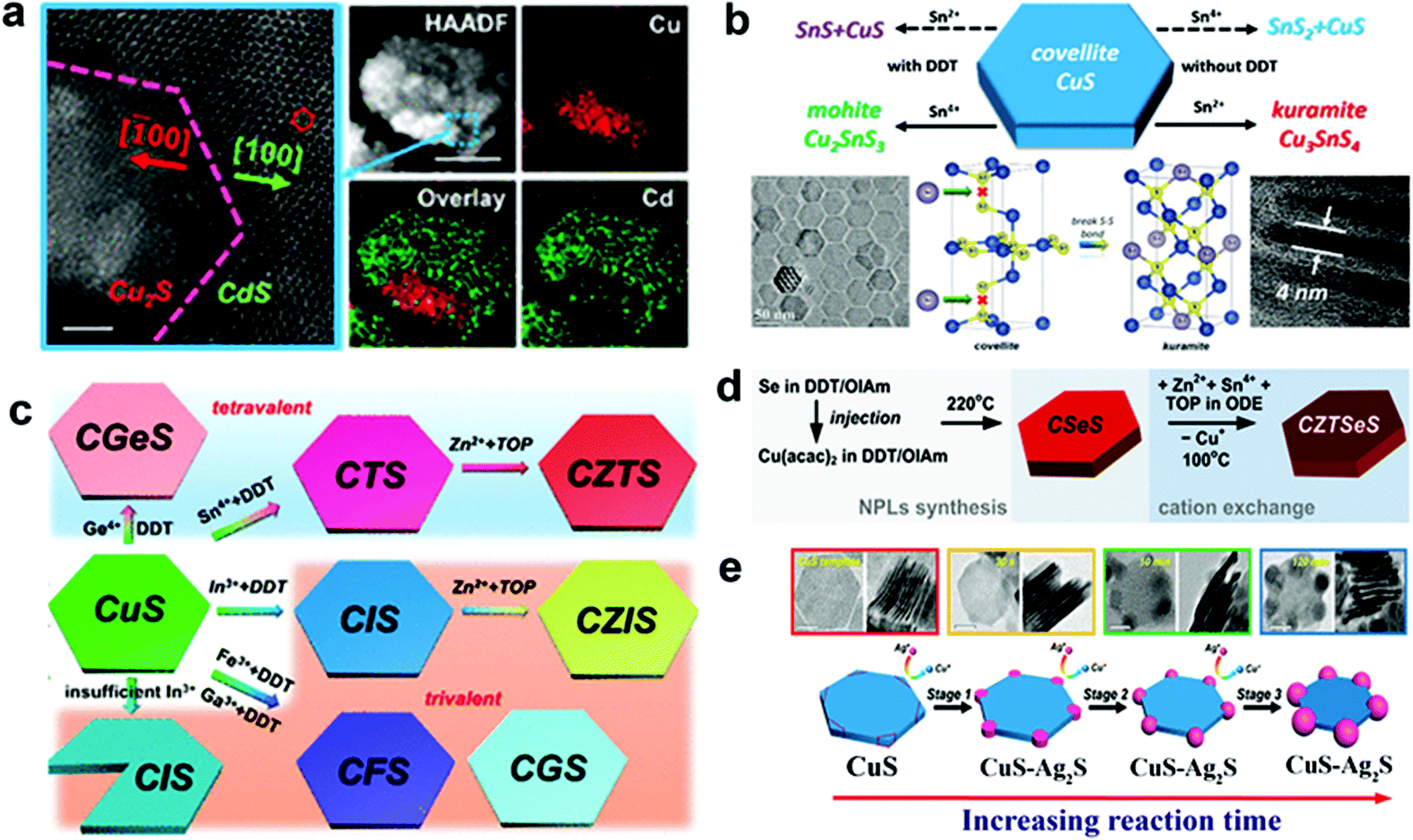 | ||
| Fig. 2 (a) CE of the covellite CuS template with Cd2+ forming a heterostructure of Cu2S–CdS; reproduced from ref. 61 with permission from ACS, copyright 2018; (b) illustration of outcomes from CE of the covellite CuS template with Sn2+ and Sn4+ cations in the presence and absence of dodecanethiol as the reducing agent; reproduced from ref. 37 with permission from ACS, copyright 2017; (c) schematic of the CE product from CuS using tri and tetravalent cations. Reproduced from ref. 61 with permission from ACS, copyright 2018; (d) sequential CE of the Cu2−xSeyS1−y template forming multi-elemental Cu2ZnSnSeyS1−y nanoplates. Reproduced from ref. 96 with permission from ACS, copyright 2014; (e) CE of Ag+ on the CuS template leading to segregated Ag2S formation at the corners of CuS nanoplates. Reproduced from ref. 97 with permission from ACS, copyright 2018. | ||
In templated cation exchange, variation in the solvation of incoming and outgoing ionic species alters the diffusion rate.111–113 Due to the difference between diffusion rates of slow incoming foreign cations and fast outgoing host cations, the host cations deplete and form hollow structures in a process called the Kirkendall effect.111,114 The nanoscale Kirkendall effect is well explored for metal oxide and chalcogenide NCs. For the nanoplate morphologies, when the cation exchange starts from the edges and the outgoing host cation replacement is faster than the incoming ionic species incorporation rate, voids will form in the 2D templates to generate biconcave or toroidal shapes. It is worth mentioning that the nanoscale Kirkendall effect is discernible for a larger template as CE occurs from the edges in an anisotropic template. Hence, the diffusion rate at the basal plane will be significantly different from edges only when the diameter is very large. In Cu2−xE templates, Cu+ is highly mobile; hence, the mobility of the incoming cation will be crucial to control the hollow shape. Mu et al. utilized a Cu2−xS template to form perforated CuInS2 plates.110 The role of In3+ was crucial to control the Kirkendall effect (Fig. 3a). They suggested the ability to generate In3+ upon nucleophilic attack of S2− varied as In(OAc)3 ∼ In(acac)3 > InCl3 > InI3. Thus, the slow release of In3+ from InI3 creates a significant difference in the diffusion rate of outgoing fast Cu+ and S2− compared to slower In3+ resulting in nanoplates with holes (Fig. 3b). Similarly, Hong et al. explored the roxbyte Cu29S16 template for a hollowing desulphurisation process using tri(ethyldiamino)phosphine.64 A Cu migration process occurs following the desulphurisation forming Cu3P1−xSx toroidal rings (Fig. 3c). The utilisation of this concept in 2D materials is fairly new, thus opening a window to explore new compositions with hollow 2D morphologies.
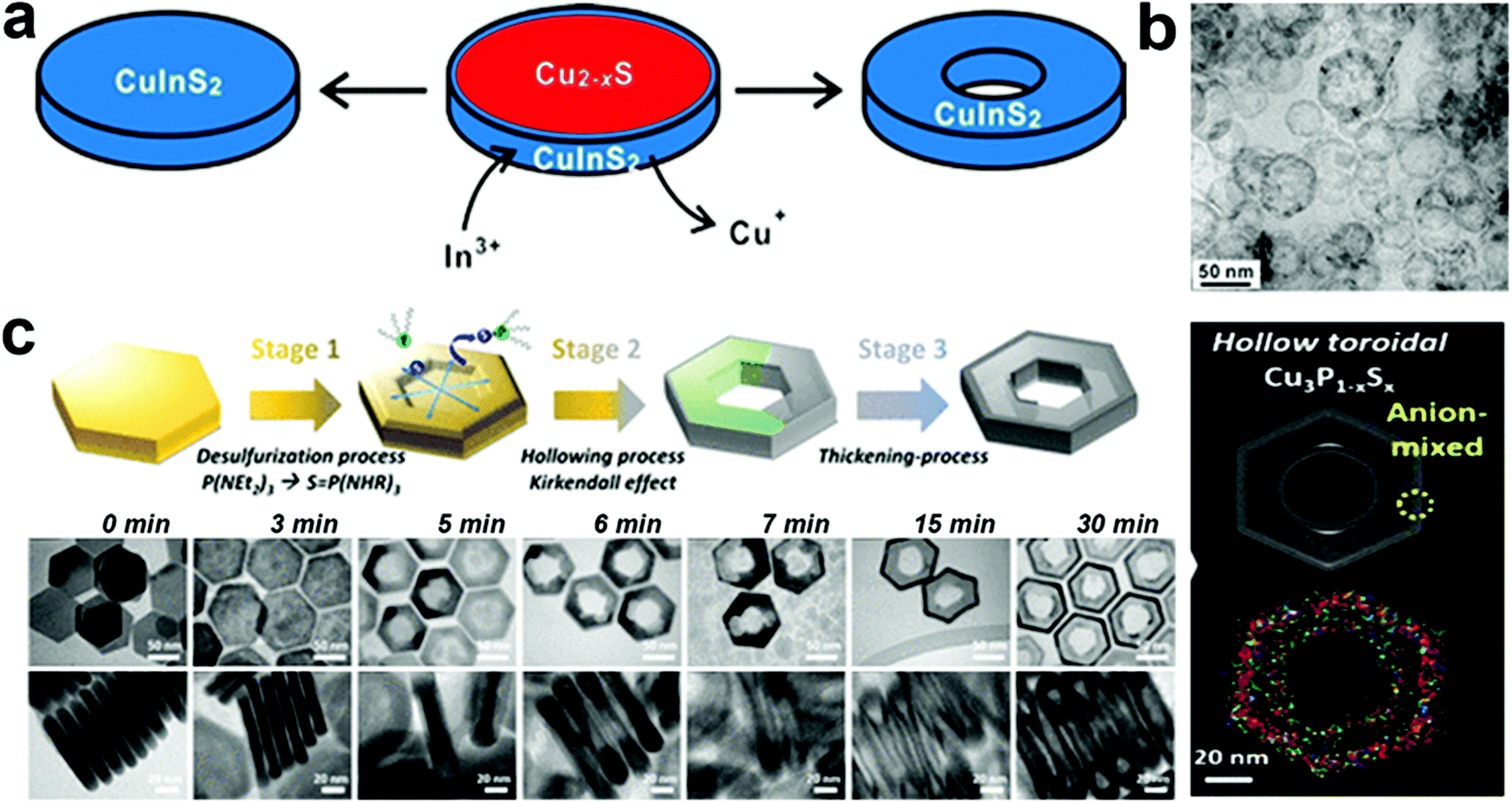 | ||
| Fig. 3 (a) Illustration of the nanoscale Kirkendall mechanism on the Cu2−xS template for forming (b) hollow CuInS2 nanoplates; reproduced from ref. 110 with permission from ACS, copyright 2015; (c) schematics and low resolution TEM of Cu3P1−xSx toroidal ring formation mechanism. The STEM-EDS elemental mapping displays the homogeneous signal of Cu (red), P (green) and S (blue) on the toroidal ring. Reproduced from ref. 64 with permission from ACS, copyright 2020. | ||
4. Colloidal synthesis and key growth parameters for 2D morphology
In colloidal synthesis, NC formation proceeds in three stages, (1) decomposition of the precursors and formation of metal anion complexes. (2) Nucleation of monomers; depending on the techniques, the nucleation stage can be either continuous (heat-up) or discrete (hot-injection). For hot-injection, rapid precursor introduction and a temperature gradient between precursor mixtures present in the flask and precursor in the injection induces a supersaturation which leads to a discrete nucleation stage. (3) Growth from monomer deposition on nucleated NCs occurs in two stages where the initial stage is rapid, and the second stage of growth is slow. In the heat-up approach, the first stage of growth and nucleation overlap. In hot injection, the rapid deposition of monomers is essential to terminate nucleation. Here the choice of precursors and ligands is a crucial factor to avoid the formation of secondary phases or to induce the formation of beneficial soft templates. Precursor chemistry can be regulated majorly based on the hard–soft-acid–base theory initially developed by Pearson. Based on the tendency to accept (metal source) or donate electron (ligand), charge on the species, size, and the polarisability of the electron cloud, the cations and anions can be categorised as hard, soft and borderline. For example, metal ions with high charge, small size, and low polarisability tend to be hard acids. The anions and cations with a similar nature i.e. hard–hard or soft–soft tend to show superior interactions than other combinations, whereas the multifarious role of ligands has been discussed in the previous sections. Another crucial factor is temperature. Modulation of the reaction temperature is a gateway to provide enough energy to control cationic diffusion and crystal phase transition. The following section presents the progress made to synthesise 2D copper-based NCs using colloidal synthesis. We examine their growth process and how certain factors play a vital role in regulating morphologies. The focus is on how various reaction factors such as precursor ratio, ligands classes and their concentration, reaction kinetics, and reaction temperature will alter the 2D morphologies (Table 1). The section is split into Cu chalcogenide and non-chalcogenide based 2D NCs. Copper chalcogenide NCs are further categorized into two sub-sections: binary copper chalcogenides and multinary copper chalcogenides (ternary and quaternary). Furthermore, the section of non-chalcogenide based NCs covers copper phosphides, copper-based alkali metal halides and metal–metal alloys.| Products | Method | Crystal phase | Reactants, ligands, and solvents | Temperature and duration | Ref. |
|---|---|---|---|---|---|
| 2D-copper-based binary chalcogenides | |||||
| Copper sulfide | |||||
| CuxS nanoprisms and nanosheets | Hot-injection approach | Hexagonal (covellite) | CuI/CuOAc, S, OLA, S | 120–150 °C/30 min | 115 |
| CuS nanosheets | Hot-injection approach | Hexagonal (covellite) | CuCl, S, OLA, octylamine | 95 °C/18 h | 74 |
| Sn doped Cu31S16 nanosheets | Heat up approach | Monoclinic djurleite | Cu(acac)2, n-dodecanethiol, SnCl4·5H2O | 200 °C/48 min | 93 |
| Cu2S nanodiscs | Hot-injection approach | Hexagonal | CuOAc, 1-dodecanethiol (1-DDT), tri-n-octylphosphine oxide (TOPO), ODE | 200–220 °C/5–540 min | 116 |
| CuS nanodiscs | Hot-injection approach | Hexagonal (covellite) | CuCl, S, OLA, OA, ODE | 180 °C/10 min | 117 |
| Cu2−xS nanodiscs & nanoplates | Hot-injection approach | Monoclinic (roxbyite) | CuCl2·2H2O, di-tert-butyl disulfide, OLA | 180–220 °C/10–60 min | 118 |
| CuS nanoplates | Hot-injection approach | Hexagonal (covellite) | CuCl2·2H2O, ammonium sulfide, OLA | 200 °C/10 s to 160 min | 119 |
| Cu2−xS nanoplates | Heat up approach | Hexagonal (covellite) | CuCl, S, TOPO, OLA | 85 °C/1 h | 120 |
| Copper selenide | |||||
| Cu2−xSe nanosheets & nanoplates | Hot-injection approach | Cubic (berzelianite) | CuCl, Se powder, 2-ethylhexanoic acid, OLA, paraffin liquid | 250 °C/30 min | 121 |
| Cu2−xSe & CuSe nanosheets | Hot-injection approach | Cubic/Cu2−xSe hexagonal/CuSe | CuCl2·2H2O, Se powder, hydroxylamine hydrochloride, 1-DDT, OLA | 80 °C/3 h | 122 |
| CuSe nanoplates | Hot-injection approach | Hexagonal | CuCl, Se, OLA, OA, ODE | 300–330 °C/15 min | 68 |
| Cu2−xSe nanoplates | Heat up approach | Hexagonal (weissite like) | Cu(acac)2, Ph2Se2, OLA | 220 °C | 123 |
| Cu2−xSe nanodiscs | Hot-injection approach | Mix phase | CuCl, selenourea, OLA | 200–240 °C/10–90 min | 124 |
| Cu2−xSe nanodiscs | Hot-injection approach | Cubic | CuCl2, dimethylimidazoline-2-selenone, OLA, methylene chloride | 175 °C/10 min | 125 |
| Copper telluride | |||||
| CuTe nanoplates | Hot-injection approach | — | CuCl, TOP–Te, LiN(SiMe3)2, TOPO, OLA | 190 °C/15 min | 67 |
| Cu2Te nanodiscs | Hot-injection approach | — | Cu(acac)2, TOP–Te, C12SH, OA | 180 °C/2 min | 126 |
| CuTe nanosheets | Heat up approach | — | Cu(acac)2, didodecyl ditelluride, dioctyl ether | 155 °C/1 h | 127 |
| 2D-copper-based multinary chalcogenides | |||||
| Cu2−xSySe1−y nanodiscs | Hot-injection approach | Monoclinic | CuCl, Se powder, DDT, OLA, ODE | 190 °C/1 h | 128 |
| CuSySe1−y nanoplates | Heat up approach | Hexagonal | Cu(NO3)2, S powder, Se powder, ethanol, NaOH | 80–95 °C/10 h | 129 |
| CuSbS2 rectangular nanosheets | Heat up approach | Orthorhombic | Cu(diethyldithiocarbamate)2, Sb(diethyldithiocarbamate)3, OLA, 1-DDT | 220 °C/1 h | 130 |
| Cu3SbS3 rhombic nanosheets | Heat up approach | Monoclinic | Cu(diethyldithiocarbamate)2(phenanthroline), Sb(diethyldithiocarbamate)3, OLA, 1-DDT | 220 °C/1 h | 130 |
| CuSbS2 nanoplates | Hot-injection approach | Orthorhombic | Cu(acac)2, SbCl3·6H2O, S, OLA | 170–280 °C/10–30 min | 22 |
| CuSbS2 nanoplates | Hot-injection approach | Orthorhombic | Cu(OAc)2·H2O, SbCl3, S, OLA | 190 °C/45 min | 55 |
| Cu3BiS3 nanoplatelets | Hot-injection approach | Orthorhombic | Bi(diethyldithiocarbamate)3, Cu(diethyldithiocarbamate)2, OLA, 1DDT | 220 °C/15 min | 56 |
| CuFeS2 nanoplates | Hot-injection approach | Tetragonal | Cu-oleate, Fe-stearate, S, OLA, ODE | 240 °C/1 h | 131 |
| CuInS2 nanodiscs | Heat up approach | Hexagonal | CuCl, InCl3, thiourea, OLA | 240 °C/1 h | 70 |
| CsCu5S3 nanoplatelets | Hot-injection approach | Orthorhombic | Cs2CO3, Cu(acac)2, S, OLA, OA | 260 °C/15 min | 65 |
| Cu2ZnSnS4 nanoplates | Hot-injection approach | Hexagonal wurtzite | CuCl2·2H2O, SnCl4·5H2O, ZnCl2, DDT, OLA, OA | 240 °C/1 h | 132 |
| Cu2ZnSnS4 nanosheets | Heat-up approach | Hexagonal wurtzite | Cu(acac)2, Zn(acac)2, SnCl4·5H2O, 1-DDT | 250 °C/25 min | 60 |
| Cu2ZnSnSe4 nanoplates | Hot-injection approach | Hexagonal wurtzite | Cu(acac)2, Zn(Ac)2, Sn(Ac)2, diphenyl diselenide, OLA | 250 °C/15 min | 133 |
| Cu2InxGa1−x(SySe1−y)2 nanoplates | Hot-injection approach | Hexagonal wurtzite | CuI, In(acac)3, Ga(acac)3, diphenyl diselenide, TOPO, 1-DDT, OLA, ODE | 175 °C/25 min | 54 |
| 2D-copper based non-chalcogenides | |||||
| Copper phosphides | |||||
| Cu3−xP nanoplates | Hot-injection approach | Hexagonal | Cu NCs solution, TOP, TOPO, OLA | 320 °C/1 h | 69 |
| Cu3P nanoplates | Heat-up approach | Hexagonal | CuCl, TOP, TOPO, octylamine, OLA | 350 °C/10 s to 25 min | 23 |
| Cu3P nanoplatelets | Heat up approach | Hexagonal | CuCl, PH3 gas TOP, TOPO, octylamine, OLA | 200–230 °C/15 min | 134 |
| Cu3−xP nanodiscs | Hot-injection approach | Hexagonal | CuCl, (TMS)3P, TOP, OLA, ODE | 300 °C/10 min | 50 |
| Cu3−xP nanodiscs | Heat up approach | Hexagonal | CuCl2, triphenyl phosphite, hexadecylamine, ODE, EtOH | 300 °C | 135 |
| Cu3−xP nanoplates | Heat up approach | Hexagonal | CuCl, P(NEt2)3, OLA, ODE, trioctylamine | T f ≈ 280 °C | 136 |
| Ternary Cu-based halides | |||||
| Cs3Cu2I5 nanodiscs | Hot-injection approach | Orthorhombic | CuI, Cs-oleate, OLA, ODE, OA | 70–80 °C/30 s | 34 |
| Cs3Cu2Br5 nanoplates | Hot-injection approach | Orthorhombic | CuBr, Cs-oleate, OLA, ODE, OA | 70–80 °C/30 s | 34 |
| Cs3Cu2I5 nanoplates | Hot-injection approach | Orthorhombic | CuI, Cs-oleate, OLA, ODE, OA | 110 °C/10 s | 62 |
| Cs3Cu2Br5 nanoplates & nanodiscs | Hot-injection approach | Orthorhombic | CuBr, Cs-carboxylate, ODE, {(hexanoic acid/nonylamine) for nanoplates}, {(nonanoic acid/OLA) for nanodiscs} | 70 °C/25 s | 137 |
| Rb2CuX3 (X = Cl, Br) | Mix-up approach | Orthorhombic | RbX (X = Cl, Br), CuX (X = Cl, Br), DMSO, OA, toluene | Room temperature | 33 |
| Metallic Cu-based NCs | |||||
| Cu nanoplate | Heat up approach | Cubic | CuSO4·5H2O, ascorbic acid, CTAB, NaOH, water | 85 °C/2.5 h | 138 |
| Cu nanosheet | Heat up approach | Cubic | Cu(NO3)2·3H2O, ascorbic acid, CTAB, hexamethylenetetramine, water | 80 °C/3 h | 139 |
| Cu nanoplate | Heat up approach | Cubic | Cu(OAc)2·H2O, poly(vinyl pyrrolidone), DMF, hydrazine, water | 60 °C/3–4 min | 140 |
| Cu nanoplate | Heat up approach | Cubic | CuBr2, ascorbic acid, branched polyethyleneimine water | 90 °C/10 h | 88 |
| Ni–Cu alloy nanoplates | Heat up approach | Defected cubic | CuCl2·2H2O, Ni(acac)2, OLA, TOP, dibenzyl ether | 175–240 °C/1 h | 24 |
| PdCu alloy nanosheets | Heat up approach | Cubic | Cu(acac)2, Pd(acac)2, Mo(CO)6, TOPO, DMF, OA | 60 °C/18 h | 85 |
| Cu–Ag alloy NCs | Heat up approach | Cubic | Cu(acac)2, Ag(Ac), OLA, OA, ODE | 180 °C/30 min | 141 |
4.1. 2D copper based chalcogenides
Cu chalcogenides (S, Se and Te) NCs exhibit a wide range of compositions and diverse crystal structures.40 Therefore, tuning the composition, size and shape is the key to alter their optoelectronic properties for several applications such as thermoelectrics, photovoltaics, catalysis, etc. Over the past decade, significant control over the morphology and crystal phases was achieved using colloidal synthesis, including several metastable phases of Cu chalcogenide NCs.59,133,142,143 This review section will summarize the synthesis advancements in the 2D morphology of binary and multinary i.e., ternary and quaternary NCs.P-Type Cu2−xS is the most studied binary copper chalcogenide due to its inherent Cu vacancies. The binary Cu–S system has a very rich phase diagram,144 with a wide range of equilibrium crystal forms having different 2D morphologies. Copper-rich phases such as chalcocite (Cu2S), djurleite (Cu31S16 or Cu1.94S), digenite (Cu9S5 or Cu1.8S), and anilite (Cu7S4 or Cu1.75S) usually form as nanoplates or nanodiscs, whereas copper-poor phases including covellite (CuS) and villamaninite (CuS2) exhibit nanosheet-like morphologies. The availability of different S sources provides further accessibility for tuning the size and shape of NCs. For instance, Lesyuk et al. employed a hot-injection method to synthesise triangular and hexagonal-like 2D NCs. Altering precursor ratios, reaction temperature, and time resulted in size and shape change (Fig. 4a).115 They investigated the effect of the Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S precursor on the shape of nanostructures. With a high Cu to S precursor ratio, irregular spherical covellite nanodiscs were produced, which progressively transformed into faceted triangles as the Cu:S precursor ratio decreased. The thermodynamic growth mechanism played an important role in the change in morphology. At the start of the reaction with a sufficient supply of sulfur, the most reactive positions (vertex tips) of the triangular NCs were formed, promoting the triangular shape of the initial NCs under kinetic growth. As the reaction proceeds, under equilibrated monomer supply NCs developed with all six facets to minimise the system's surface energy at the expense of smaller triangles, which gradually dissolve to produce hexagonal platelets. Furthermore, the gradual addition of Cu and S precursors into the reaction system formed nanosheets up to 2 microns. In another instance, Du et al. materialised CuS nanosheets by reacting sulfur powder and CuCl in the presence of OLA and octylamine at 95 °C for 18 h.74 The formed nanosheets were found to have an average plane length of 453 ± 6 nm and thickness of 3.2 ± 0.2 nm. The nanosheet formation was attributed to nucleation in a confined lamellar structure due to the reaction of OLA and octylamine with CuCl (Fig. 4b). Gao and co-workers illustrated the formation of Sn-doped ultrathin Cu31S16 nanosheets with lateral dimensions >200 nm by varying the quantity of SnCl4 in the reaction medium.93 The authors proposed that SnCl4 can form stable complexes with dodecanethiol, which acts as surface capping agents, preferentially adhering to the djurleite's (100) facets, resulting in ultrathin 2D nanosheets. Wang et al. reported the synthesis of Cu2S nanodiscs.116,120 These nanodiscs were formed by injecting 1-dodecanethiol (1-DDT) into a heated solution consisting of copper acetate (CuOAc), tri-octyl phosphine oxide (TOPO), and 1-octadecene (ODE). The rise in injection temperature from 160 to 190 °C increased the size of nanoparticles from 13.1 ± 0.9 nm to 16.9 ± 2.3 nm, whereas monodisperse nanodiscs were obtained with a further increase in temperature to 200 and 220 °C. Xie and co-workers fabricated anisotropic CuS nanodiscs with low size distribution by injecting S–OLA solution in a hot colloidal mixture of CuCl, oleic acid, OLA and 1-ODE.117 In another study, Li et al. revealed that an oriented attachment is critical for the precise control of the size and shape of Cu2xS NCs.118 At the initial stage of the reaction, Cu2xS spherical nanoparticles were formed by injecting di-tert-butyl disulphide (TBDS) into a heated CuCl2 solution. These spherical nanoparticles evolved further into circular nanodiscs and hexagonal Cu1.96S nanodiscs with prolonged reaction times (Fig. 4c). Higher temperatures and longer reaction times enhanced the crystallinity and diameter of the nanodiscs, with minor effects on their thickness, crystallographic phase, and composition. The room temperature synthesis of CuS nanoplates by multiple injections of ammonium sulphide into a mixture of CuCl2, OLA, and toluene was reported by Liu et al.119 The authors achieved a wide variety of NC sizes by varying the number of ammonium sulphide injections, ranging from large nanosheets to smaller nanoparticles. A single ammonium sulphide injection resulted in large irregularly shaped plates with high polydispersity. A series of smaller injections with an overall constant volume of ammonium sulphide solution resulted in NCs with decreased average size and low polydispersity. Heating the copper precursor and sulfur powder demonstrated a simple synthesis protocol for the formation of monodisperse Cu2−xS nanoplates at 85 °C in a solution containing OLA and TOPO.120
:S precursor on the shape of nanostructures. With a high Cu to S precursor ratio, irregular spherical covellite nanodiscs were produced, which progressively transformed into faceted triangles as the Cu:S precursor ratio decreased. The thermodynamic growth mechanism played an important role in the change in morphology. At the start of the reaction with a sufficient supply of sulfur, the most reactive positions (vertex tips) of the triangular NCs were formed, promoting the triangular shape of the initial NCs under kinetic growth. As the reaction proceeds, under equilibrated monomer supply NCs developed with all six facets to minimise the system's surface energy at the expense of smaller triangles, which gradually dissolve to produce hexagonal platelets. Furthermore, the gradual addition of Cu and S precursors into the reaction system formed nanosheets up to 2 microns. In another instance, Du et al. materialised CuS nanosheets by reacting sulfur powder and CuCl in the presence of OLA and octylamine at 95 °C for 18 h.74 The formed nanosheets were found to have an average plane length of 453 ± 6 nm and thickness of 3.2 ± 0.2 nm. The nanosheet formation was attributed to nucleation in a confined lamellar structure due to the reaction of OLA and octylamine with CuCl (Fig. 4b). Gao and co-workers illustrated the formation of Sn-doped ultrathin Cu31S16 nanosheets with lateral dimensions >200 nm by varying the quantity of SnCl4 in the reaction medium.93 The authors proposed that SnCl4 can form stable complexes with dodecanethiol, which acts as surface capping agents, preferentially adhering to the djurleite's (100) facets, resulting in ultrathin 2D nanosheets. Wang et al. reported the synthesis of Cu2S nanodiscs.116,120 These nanodiscs were formed by injecting 1-dodecanethiol (1-DDT) into a heated solution consisting of copper acetate (CuOAc), tri-octyl phosphine oxide (TOPO), and 1-octadecene (ODE). The rise in injection temperature from 160 to 190 °C increased the size of nanoparticles from 13.1 ± 0.9 nm to 16.9 ± 2.3 nm, whereas monodisperse nanodiscs were obtained with a further increase in temperature to 200 and 220 °C. Xie and co-workers fabricated anisotropic CuS nanodiscs with low size distribution by injecting S–OLA solution in a hot colloidal mixture of CuCl, oleic acid, OLA and 1-ODE.117 In another study, Li et al. revealed that an oriented attachment is critical for the precise control of the size and shape of Cu2xS NCs.118 At the initial stage of the reaction, Cu2xS spherical nanoparticles were formed by injecting di-tert-butyl disulphide (TBDS) into a heated CuCl2 solution. These spherical nanoparticles evolved further into circular nanodiscs and hexagonal Cu1.96S nanodiscs with prolonged reaction times (Fig. 4c). Higher temperatures and longer reaction times enhanced the crystallinity and diameter of the nanodiscs, with minor effects on their thickness, crystallographic phase, and composition. The room temperature synthesis of CuS nanoplates by multiple injections of ammonium sulphide into a mixture of CuCl2, OLA, and toluene was reported by Liu et al.119 The authors achieved a wide variety of NC sizes by varying the number of ammonium sulphide injections, ranging from large nanosheets to smaller nanoparticles. A single ammonium sulphide injection resulted in large irregularly shaped plates with high polydispersity. A series of smaller injections with an overall constant volume of ammonium sulphide solution resulted in NCs with decreased average size and low polydispersity. Heating the copper precursor and sulfur powder demonstrated a simple synthesis protocol for the formation of monodisperse Cu2−xS nanoplates at 85 °C in a solution containing OLA and TOPO.120
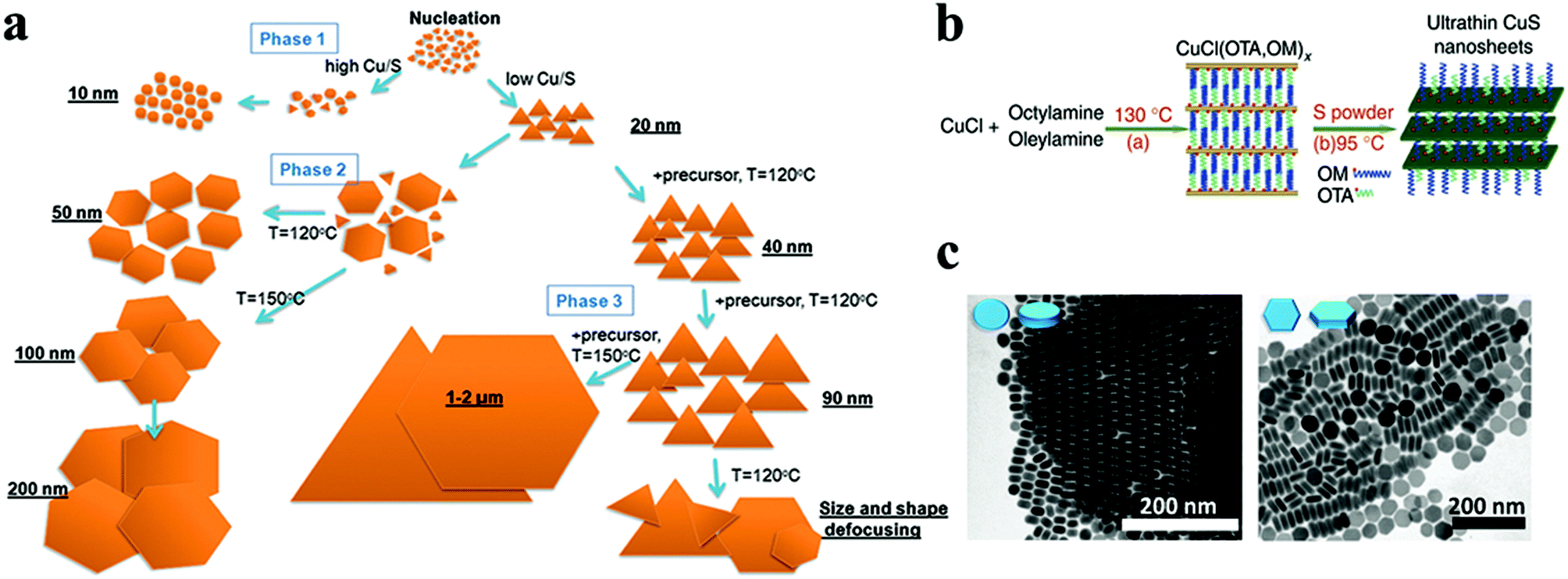 | ||
| Fig. 4 (a) The shape transformations during the OAm–S based hot-injection copper sulphide synthesis are shown in a scheme, along with methods to control the shape. Reproduced from ref. 115 with permission from RSC, copyright 2018; (b) schematic illustration of fabrication of ultrathin CuS nanosheets.74 ©2012 Nature (c) TEM images showing the morphology evolution of Cu2−xS NCs. Reproduced from ref. 118 with permission from RSC, copyright 2011 ©2011 RSC. | ||
Copper selenide is another notable p-type binary semiconductor with potential uses in solar cells, thermoelectric converters, solar cells, photothermal treatment, and photocatalytic activity.41,123 Copper selenide has a range of crystal structures dictated by stoichiometric and non-stoichiometric phases. It exhibits a variety of phases, ranging from Cu-deficient hexagonal klockmannite CuSe to copper-rich bellidoite cubic Cu2Se. There are few reports on 2-D copper selenide among the various 2-D nanostructures of metal chalcogenides. Deng et al. synthesised berzelianite Cu2−xSe nanoplates and nanosheets by injecting Cu(I)-complex precursor solution into a hot reaction mixture of selenium powder and paraffin oil.121 Higher copper concentrations resulted in nanosheets with diameters of 1.0–2.5 μm, whereas lower copper concentrations resulted in nanoplates. In the case of lower copper content, the facets of the NCs are likely to be extensively passivated, resulting in a reduced in-plane diameter. Another study reported the formation of cubic Cu2−xSe nanosheets via simply heating micro-sized CuSe nanosheets in the presence of Cu(I) cations without any morphological modification.122 The authors proposed that the structure of klockmannite CuSe, which has a low Cu cation occupancy, functioned as a suitable template for additional Cu cation insertion, which led to the formation of antifluorite Cu2−xSe (Fig. 5a). Different selenium sources have been reported to synthesise 2D Cu2−xSe NCs with morphologies like disc and plates. For instance, Deka and co-workers reported a hot-injection approach to fabricate stoichiometric CuSe hexagonal nanoplatelets using Se–ODE solution as a Se source.68 Injecting Se–ODE solution into a colloidal solution of CuCl in OLA and ODE formed nanoplatelets with dimensions of over 100 nm. Alternatively, selenourea has been employed as a Se source in a hot injection synthesis derived nanodisc with a diameter of 16 nm.49 Furthermore, a diaryldichalogenide precursor, diphenyl diselenide, directed the synthesis of hexagonal nanoplates of copper selenide.123 In a seminal work, an air-stable selenium source, namely 1,3-dimethylimidazoline-2-selenon has been synthesised and employed for the hot-injection synthesis of Cu2xSe nanodiscs.125 The authors suggested that the N-heterocyclic carbenes formed from imidazoline-2-selenone and oleylamine worked together to form nanodiscs by interacting with copper on a crystal plane (Fig. 5b).
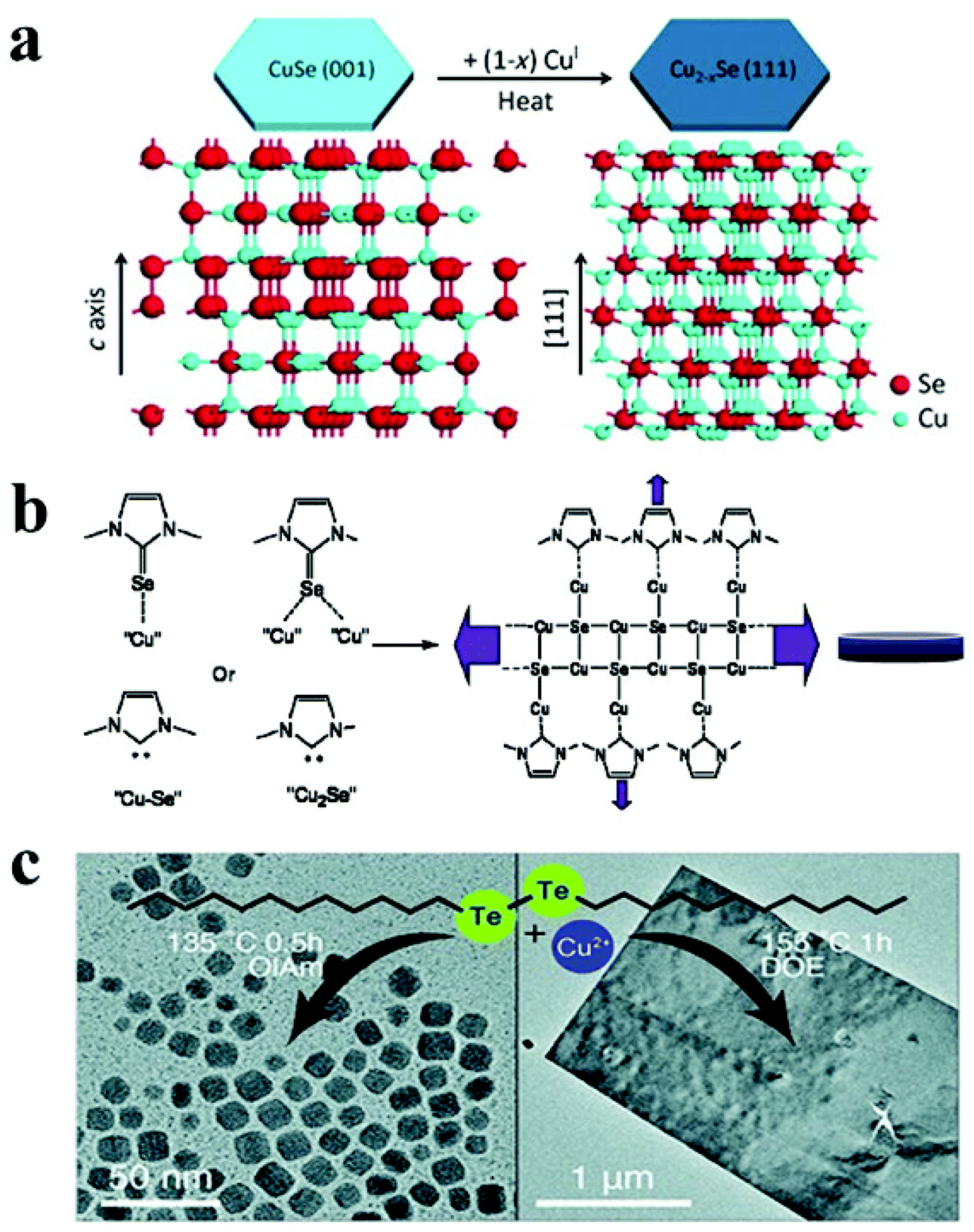 | ||
| Fig. 5 (a) Schematic illustrating transformation from hexagonal CuSe to cubic Cu2−xSe under heat treatment due to insertion of CuI; reproduced from ref. 122 with permission from Wiley, copyright 2014; (b) mechanism of cubic Cu2−xSe nanodisc formation via imidazoline-2-selenone interaction with NC surface; reproduced from ref. 125 with permission from ACS, copyright 2010; (c) formation of Cu2−xTe NCs and vulcanite nanosheets under different reaction condition using di-dodecyl ditelluride as the Te precursor. Reproduced from ref. 127 with permission from RSC, copyright 2020. | ||
Similar to Cu2−xS, Cu2−xTe exhibits Cu-deficiency-dependent plasmonic behaviour, high ionic conductivity, and low thermal conductivity and finds applications in photothermal therapy, surface-enhanced Raman scattering probes, and thermoelectric devices.31,145,146 Cu2−xTe possesses a complex phase diagram with many stoichiometries and crystal structures. For example, Cu2Te (hexagonal), Cu7Te4 (trigonal), Cu3Te2 (tetragonal), and CuTe (orthorhombic) are a few notable members of this family. Controlling the 2D shape of the Cu2−xTe NCs remains challenging due to the limited Te precursor availability. Li et al. utilized tri-octyl phosphine (TOP)–Te as the Te source with lithium bis(trimethylsilyl)amide and CuCl in the presence of OLA and TOPO to materialize rectangular Cu2−xTe nanoplates at 190 °C.67 Lithium bis(trimethylsilyl)amide was suggested to activate the Cu-oleylamido complex responsible for Cu2−xTe formation. In another approach, Li et al. synthesized hexagonal Cu2−xTe nanoplates by injecting TOP–Te solution in a hot colloidal mixture of Cu(acac)2, 1-DDT and OLA.126 Switching to a longer chain length thiol (octadecanethiol) led to the formation of nanodots suggesting the suppression of growth for Cu2−xTe NCs. Robinson et al. used di-dodecyl ditelluride as the Te source to synthesise vulcanite CuTe nanosheets in dioctyl ether (DOE) at 155 °C for 30 min to 1 h.127 A low temperature of 135 °C and co-ordinating solvents such as OLA and OA produced Cu1.5Te NCs. In contrast, using DOE at an elevated temperature of 155 °C formed nanosheets predominantly (Fig. 5c).
 | ||
| Fig. 6 (a) TEM image and EDS elemental mapping of Cu2−xSySe1−y NCs synthesized in the presence of 5 mL of 1-DDT; reproduced from ref. 128 with permission from ACS, copyright 2017; (b) schematic showing reduction thioacetamide and oxidation of Sb3+ to Sb5+, (c) reaction scheme for the formation of (d) CuSbS2 nanoplates and (e) the CuSbS2–Cu3SbS4 composite using S–OLA and thioacetamide respectively. Reproduced from ref. 55 with permission from ACS, copyright 2019. | ||
Bera et al. reported the formation of Cu3BiS3 nanoplatelets by using single source precursors, Bi(diethyldithiocarbamate)3 and Cu(diethyldithiocarbamate)2 decomposition in OLA.56 The shape of the NCs was controlled either by adding both metal sources together in the flask or by adding a small quantity of the Cu source in the flask ahead of the Cu and Bi-source mixture injection(Fig. 7b). The shape of the platelets was determined by the local concentration of the Cu7S4 NCs formed initially around the Cu3BiS3 NCs to go through a ripening process (Fig. 7a and b). Conditions that favoured the high concentration of Cu7S4 NCs near the Cu3BiS3 nucleation sphere formed flat and longer nanoplatelets. For example, a high amount of the Cu-source in the first addition, high temperature, and lower solvent volume resulted in the formation of Cu7S4 NCs in high numbers facilitating the ripening of Cu3BiS3 NCs to form flat nanoplatelets, whereas a low amount of the Cu-source in the first addition, lower temperature, and a high volume of solvent resulted in the formation of Cu7S4 NCs in a low number, hence obtaining tapered nanoplatelets (Fig. 7c).
 | ||
| Fig. 7 (a) Schematic presentation of the number density variation reactions of Cu7S4 and Cu3BiS3 ternary nuclei forming flat platelets for high number density of Cu7S4 nuclei and tapered platelets for low number density of Cu7S4 (b) the active reaction sphere model showing the growth of Cu3BiS3 nanoplatelets in the presence of Bi(III) precursor and Cu7S4 platelets, (c) TEM images for the formation pathways collected at different growth times of flat and tapered Cu3BiS3 nanoplatelets. Reproduced from ref. 56 with permission from ACS, copyright 2020. | ||
Among other tri-valent cations, Fe3+ and In3+ have also been explored to form ternary Cu-chalcogenide based 2D NCs.21,53,70,131 For example, Gabka et al. injected S–OLA solution into a Cu-oleate and Fe-stearate mixture present in ODE to materialize CuFeS2 nanoplates.131 In another work, CuInS2 nanodiscs were synthesized using a heat-up approach where CuCl, InCl3, and thiourea in OLA were heated up to 240 °C for 1 h.70 Due to the low energy difference between the wurtzite and cubic phase of CuInS2 (∼0.1 eV per atom), polytypism was observed along the c-axis of these nanodiscs. Berends et al. prepared In-poor Cu–In–S nanosheets via self-organisation of pre-formed trigonal pyramidal shaped cubic CuInS2 in alkylamine and S–ODE solution (Fig. 8a–d).21 The reactive sulphur species, generated in situ via the reaction of alkylamine and elemental sulphur, extracted In3+ cations to form In-poor CuInS NCs. These In-poor NCs underwent oriented attachment to minimize their surface energy and form hexagonal In-poor CuInS nanosheets with lateral dimensions of 20 nm to 1μm with a thickness of ∼3 nm (Fig. 8a). Among other compositions, Yang et al. reported colloidal hexagonal CsCu5S3 nanoplatelets with a narrow size distribution of 32.3 ± 4.3 nm via a hot injection method.65 To obtain nanoplatelets, S–OLA solution was injected as a chalcogenide source into the mix of Cs-carbonate and Cu-acetate precursors at 260 °C. The synthesised NCs showed an orthorhombic structure with a direct bandgap of 1.40 eV. Quaternary Cu2ZnSnS4 nanoplates were prepared using the injection of metal thiolates in a solution mixture of 1-DDT and oleic acid.132 Using a heat up approach, Zhang et al. prepared Cu2ZnSnS4 nanosheets in the wurtzite phase with lateral dimensions of 350 ± 50 nm and a thickness of ∼5 nm.60 In this work, the metal precursors were mixed with 1-DDT and heated to 250 °C. Upon heating Cu2−xS nuclei formed around 220 °C, which transformed into Cu2ZnSnS4 nanosheets gradually. Ren et al. used a dual injection process in which Sn(OAc)2, diphenyldiselenide, and OLA were used as the first injection mixture, and Zn(OAc)2 and OLA as the second injection mixture to synthesize wurtzite Cu2ZnSnSe4 nanoplates.133 The double injection procedure ensured the segregation of Cu and Se nucleation with the rest of the cations and made room for uninterrupted sequential incorporation of Sn and Zn. Coughlan et al. synthesized curved Cu2InxGa1−x(SySe1−y)2 with contours using TOPO as the co-ordinating solvent (Fig. 8e).54 The use of a coordinating ligand was determined to be necessary to synthesize metastable wurtzite nanoplates. As the prismatic planes of the nanoplates are nonpolar, the specific binding of TOPO to metal rich basal (001) facets influenced a higher growth rate on the prismatic planes compared to the well-passivated basal planes forming the contours. However, replacing TOPO with another co-ordinating solvent i.e., OLA generated nanoplates without contours, and using a combination of both OLA and TOPO generated nanoplates with shallow curves (Fig. 8f). It was hypothesised that the linear branch of OLA passivates flat surfaces, whereas the bulky TOPO preferred binding to curved surfaces (Fig. 8g).
 | ||
| Fig. 8 (a) Schematic representation of the formation of Cu–In–S nanosheets generated via oriented attachment of CuInS2 nanopyramids, TEM image depicting the evolution of the nanosheets collected after (b) 1 min, (c) 2 min (d) 5 min; Reproduced from ref. 21 with permission from ACS, copyright 2017; (e) TEM image of the Cu2InxGa1−x(SySe1−y)2 disc with bend contours synthesised using TOPO as the coordinating solvent, (f) TEM image of the Cu2InxGa1−x(SySe1−y)2 disc with shallow bend contours synthesised using TOPO and OLA as the solvent, (g) TEM image showing lateral view of the nanodisc where the cartoon depicts the prismatic and basal planes. Reproduced from ref. 54 with permission from ACS, copyright 2018. | ||
Nevertheless, there are some challenges regarding the synthesis of colloidal Cu chalcogenide NCs, particularly in the case of selenides and tellurides, for which there are limited anion precursors. The alternative can be the more convenient alkahest solutions of Se and Te and di-orgnyl Se and Te precursors.47,148 Soft-templated growth is a crucial way to achieve ultrathin NCs. However, the multinary Cu chalcogenide NCs with ultrathin thickness are difficult to materialise from the existing soft-template regimes used for 2D nanostructure formation which degrades typically at higher temperatures (>220 °C), restricting the uniform incorporation of elements in the synthesised NCs. A viable approach will be to employ a double injection approach where the initial injection will ensure the formation of a soft template, and second injection just after the Cu–S bond formation will ensure the diffusion of cations into the pre-nucleated template to form multinary compositions. Another approach could be to design heterometallic single-source precursors which can provide a 2D template during metal–sulfide bond formation.149
4.2. Cu based non-chalcogenides
Besides copper chalcogenides, there are few examples of 2D anisotropic copper NCs. Copper phosphide (Cu3−xP), alkali metal copper halide, and copper-based alloys are some NCs that have shown 2D anisotropic shapes in colloidal synthesis. This section describes the key synthesis insights from recent reports on 2D Cu-based non-chalcogenide NCs.:Cu molar ratio of at least 11:1 was used, with no apparent intermediary formation of Cu NCs. Furthermore, increasing the amount of TOP causes particles to become more polydisperse and more prominent in size, primarily due to Ostwald ripening. At the same time, lower TOP/Cu ratios resulted in crystalline Cu NCs.
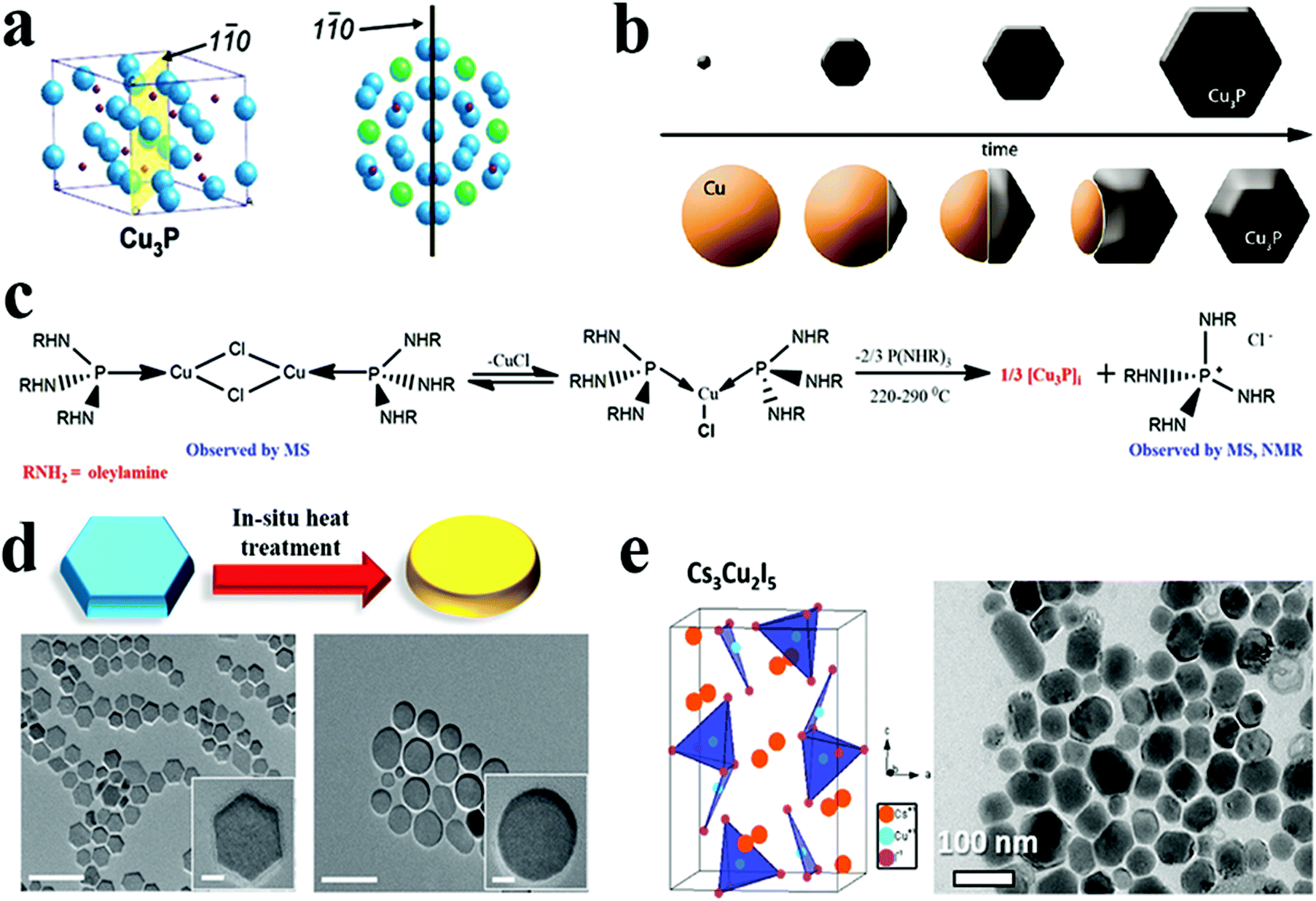 | ||
Fig. 9 (a) Cu3−xP unit cell with the 1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0 plane highlighted, and top view of numerous unit cells illustrating how the Cu3−xP crystal structure is connected to the nanocrystal hexagonal morphology (Cu atoms with hexagonal patterns emphasised in green, whereas other Cu atoms are blue and phosphorus atoms are red). Reproduced from ref. 69 with permission from ACS, copyright 2007; (b) schematic view of two different synthesis approaches to Cu3−xP NCs. Reproduced from ref. 23 with permission from ACS, copyright 2012; (c) reaction scheme showing the synthesis of Cu3−xP via intermediate formation. Reproduced from ref. 136 with permission from ACS, copyright 2021; (d) a schematic representation and TEM images illustrating morphological change that occurred following in situ heat treatment of Cu3−xP NCs (scale bars are 100 nm and scale bars in the insets are 10 nm). Reproduced from ref. 50 with permission from Wiley, copyright 2016; (e) crystal structure and transmission electron micrographs of Cs3Cu2I5 NPs. Reproduced from ref. 62 with permission from ACS, copyright 2019. 0 plane highlighted, and top view of numerous unit cells illustrating how the Cu3−xP crystal structure is connected to the nanocrystal hexagonal morphology (Cu atoms with hexagonal patterns emphasised in green, whereas other Cu atoms are blue and phosphorus atoms are red). Reproduced from ref. 69 with permission from ACS, copyright 2007; (b) schematic view of two different synthesis approaches to Cu3−xP NCs. Reproduced from ref. 23 with permission from ACS, copyright 2012; (c) reaction scheme showing the synthesis of Cu3−xP via intermediate formation. Reproduced from ref. 136 with permission from ACS, copyright 2021; (d) a schematic representation and TEM images illustrating morphological change that occurred following in situ heat treatment of Cu3−xP NCs (scale bars are 100 nm and scale bars in the insets are 10 nm). Reproduced from ref. 50 with permission from Wiley, copyright 2016; (e) crystal structure and transmission electron micrographs of Cs3Cu2I5 NPs. Reproduced from ref. 62 with permission from ACS, copyright 2019. | ||
Manna et al. reported a semiconducting and plasmonic 2D platelet-shaped CuI phosphide using a relatively low-temperature synthesis procedure.134 In this study, ex situ generated phosphine gas was explored as the P source, TOPO as a solvent, and TOP as the nucleation controlling agent. Using more reactive PH3 instead of traditional P sources, such as TOP or TOPO lowered the reaction temperature to 200–230 °C. The tuning of the concentration of TOP resulted in nanoplates of varying sizes from nano to micro. The number of nucleation events varies with the amount of TOP used; a higher amount of TOP reduces the number of nucleation events, resulting in platelets of varying sizes. Furthermore, Liu et al. replaced PH3 with (TMS)3P, which increases the reactivity of phosphorus and, consequently, lowers the reaction temperature to as low as 60 °C.50 They discovered that varying the concentration of TOP affected the average size of hexagonal platelets (Fig. 9d). Subsequently, the colloidal synthesis of Cu3−xP NCs has been reported with the direct nucleation of Cu3−xP using inexpensive, low-toxic, and air-stable triphenyl phosphite as a source of phosphorus by Liu et al.135 This simplified synthesis procedure formed disc-shaped Cu3−xP with a size around 17 nm. Recently, Rachkov et al. reported a colloidal synthesis of Cu3−xP nanoplatelets from copper halide salts and tris(diethylamino)phosphine [P(NEt2)3] in a one-pot approach.136 The mass spectrometry and nuclear magnetic resonance spectroscopy study revealed that aminophosphine is first transaminated with a long-chain primary amine. Then the aminophosphine-coordinated metal is disproportionated to give Cu3−xP NCs along with some byproducts (Fig. 9c). Larger nanocrystal sizes were obtained by raising the P(NEt2)3/Cu, and OLA/phosphorus molar ratios, respectively, but the size of NCs decreased as the reaction temperature decreased. Thus, considerable progress on understanding ex situ mechanistic details of growth has been made for Cu3−xP formation. Additionally, the exclusion of TOP as a P source facilitated synthesis at comparatively lower temperatures (<250 °C).
Cheng and colleagues reported the first investigation on the colloidal synthesis of caesium copper halide NCs using a hot-injection method.34 By treating a Cs oleate precursor with Cu(I) halide in ODE, colloidal NCs were synthesised. During the synthesis, CsCu2I3 nanorods and Cs3Cu2I5 nanodiscs were synthesised at two distinct reaction temperatures. Furthermore, they utilised a similar approach to synthesise Cs3Cu2Br5 nanoplatelets by using CuBr as a precursor, expanding the library of copper-based two-dimensional anisotropic nanomaterials. Also, Vashishtha et al. reported the synthesis of shape- and composition-controlled Cs3Cu2I5 nanoplates via tailoring the reaction temperature and OLA/oleic acid proportions (Fig. 9e).62 The injection of Cs-oleate in a solution of copper iodide with a ligand at a lower temperature around 110 °C formed Cs3Cu2I5 nanoplates. In contrast, injection at 160 °C resulted in CsCu2I3 with a nanorod morphology. Here, the reaction temperature was crucial for precise shape tuning, whereas growth time was found to be ineffective for shape tuning, given the high growth rate of these metal halides. Recently, Le et al. reported the hot injection synthesis of Cs3Cu2Br5 NCs with different chain-length ligands.137 Acid- and amine-based hydrocarbon ligands were used to improve the solubility of the precursor CuBr. The combination of ligands with different carbon chain lengths (C6, C9 and C18) led to different morphologies. A short ligand (C6) resulted in unevenly shaped, polydisperse NCs due to less control over nucleation and growth with inadequate surface passivation. In comparison, rectangular nanoplates and nanodiscs were developed by combining acid and amine-based ligands with lengths C6/C9 and C9/C18, respectively. The first colloidal Rb2CuX3 (X = Cl, Br) NCs were synthesised utilising a room-temperature ligand assisted re-precipitation technique by Vashishtha et al. The addition of toluene and oleic acid ligands to Rb and Cu halide salts dissolved in DMSO resulted in the formation of NCs.33 Both structures exhibited a nanoplate-like morphology, with average sizes of 7.7 nm and 7.5 nm for Rb2CuBr3 and Rb2CuCl3, respectively. Despite having advantages such as being less toxic and stable, these copper-based halides have poor light-emitting characteristics in comparison with halide perovskites. In addition, a thorough understanding of the influence of morphology on the light-emitting characteristics of Cu based halides is currently missing.
For the synthesis of Cu NCs in the 2D morphology, cetyltrimethylammonium bromide (CTAB), an amphiphilic ligand, is commonly employed. It acts both as a stabilizing and shape-directing agent. For instance, Wang et al. achieved the fabrication of single-crystalline Cu nanoplates via reducing Cu+ using ascorbic acid in the presence of CTAB.138 Cu NCs with a triangular plate morphology were obtained by varying the pH of the reaction mixture and the concentration of CTAB. The pH of the environment closely governs the ascorbic acid reduction potential and morphology of the NC. At the same time, CTAB forms micelles in the reaction mixture, which serves as a soft template for forming the triangular plate structure (Fig. 10a). Similar observations were reported using ascorbic acid and CTAB by Luc and co-workers where they fabricated freestanding high-quality Cu nanosheets.139 The Cu nanosheets were found to be triangular in shape with an average edge length of 1.7 ± 0.5 μm. Furthermore, atomic force microscopy revealed the thickness of the Cu nanosheet to be 5 nm. The Cu nanosheets were shown to be stable under ambient conditions, attributed to surface bound ascorbic acid, which inhibited oxidation. In another example, copper(II) acetate was reduced with hydrazine in the presence of N,N-dimethylformamide (DMF) and polyvinylpyrrolidone (PVP) to produce Cu nanoplates with a diameter of approximately 50 nm and an average thickness of around 20 nm (Fig. 10b).140 While DMF has been shown to be vital for shape regulation, its specific function remained anonymous.
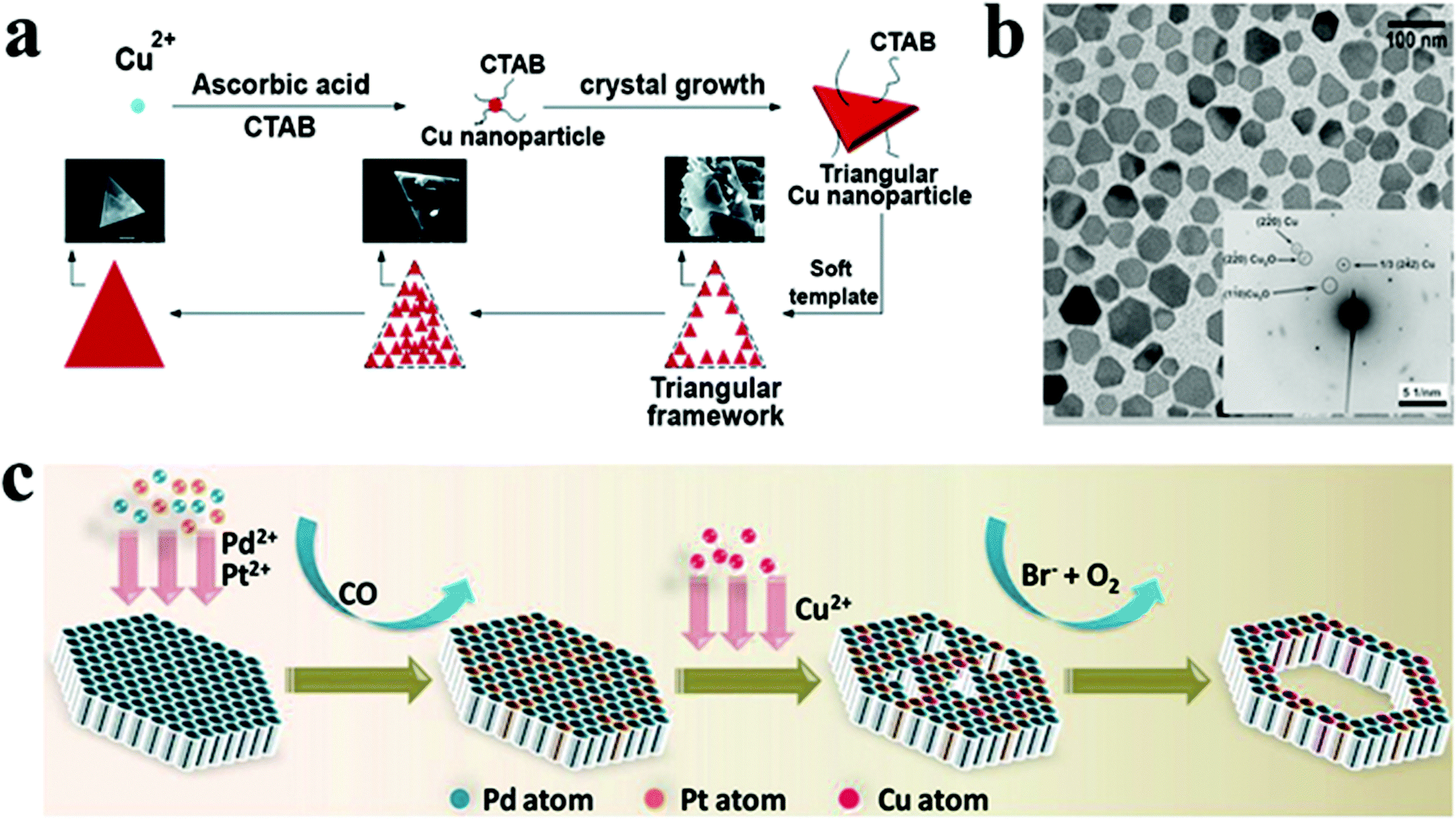 | ||
| Fig. 10 (a) Schematic illustrating growth mechanism of Cu nanoplates. Reproduced from ref. 138 with permission from RSC, copyright 2017; (b) TEM image of Cu nanoplates synthesized by the reduction of the Cu precursor by hydrazine in DMF using PVP as the stabilizer. Reproduced from ref. 140 with permission from Wiley, copyright 2009; (c) schematic model illustrating the formation of PdPtCu nanorings. Reproduced from ref. 63 with permission from Wiley, copyright 2021. | ||
Copper has been found to be an ideal candidate for alloying with other metals such as Pt, Pd, and Ni. Bimetallic and multimetallic alloy NCs outperform monometallic NCs due to the synergistic impact between/among various metals. For example, ternary FePtCu alloy nanorods surpass binary FePt counterparts and commercial Pt catalysts in catalytic activity and durability in the oxygen reduction reaction (ORR).156 Cu alloyed with noble metals such as Au provides good stability, optical tunability, and strong catalytic activity and has been utilised in many catalytic processes, including CO2 reduction, p-nitrophenol reduction, and catalytic oxidation of benzyl alcohol, CO, and propene.157–159 Cu has been associated with numerous high cost and low abundant metals such as Pd, Pt, and Au as cost-effective and highly efficient catalysts. Only a few colloidal copper-based metal–metal alloys have been reported to date that exhibit a two-dimensional morphology. For example, Guo et al. reported the facile one-pot synthesis of hexagonal and triangular Ni–Cu alloy nanoplates.24 Distinct Ni(II) reduction and oxidative etching rates at different reaction temperatures influenced the nanoplatelet shapes from hexagonal to triangular. In the same study TOP, a capping agent, also impacted the kinetics of the reaction and has been shown to control anisotropic shapes. TOP lowered the reaction kinetics, which aided in the formation of the nanoplates, however when TOP was not utilized, the nanosphere shape was formed owing to the rapid reaction kinetics.
Cu has also been alloyed with Pd for heterogeneous catalyst application. For the direct formation of 2D structures,160 various studies have documented the use of carbonyl salts such as W(CO)6 and Mo(CO)6 as reductants to generate CO molecules that passivate the (111) planes of Pd-based nanosheets. For example, Li et al. reported PdPtCu ultrathin nanorings using W(CO)6 as a reductant.63 Initially, Pd-rich ultrathin nanosheets were synthesized, and these nanosheets were alloyed with Pt to form PdPt nanosheets. Furthermore, PdPt nanosheets were employed as seeds for PdPtCu nanoring formation. When Cu salt is introduced to the reaction system, it starts etching the nanosheet's surface (Fig. 10c). The nanosheets were etched more severely as the reaction continued. After 4 hours, the PdPtCu nanoring morphology was observed. In another study, Yang et al. demonstrated the high-yield fabrication of ultrathin 2D PdCu alloy nanosheets with various Cu/Pd atomic ratios.85 Mo(CO)6 was used initially to reduce Pd(II), which in turn accelerated the reduction of Cu(II), resulting in small PdCu clusters. Furthermore, the PdCu nanosheets were eventually formed due to a seed growth process, and more Cu atoms were embedded into the nanosheets as the process progressed. Very recently, Zhang and coworkers synthesized a PdCu nanoflower shape comprised of multiple 2D nanosheets.161
Ag has also been alloyed with Cu among other noble metals to form 2D NCs. For instance, Balkan et al. have investigated a one-pot wet-chemical technique for the composition-controlled fabrication of monodisperse CuAg alloy NPs.141 The fcc type Cu–Ag alloy nanoparticles were formed by co-reducing Cu and Ag metal precursors at 180 °C. The presence of OLA in the reaction facilitated the metal precursor reduction, while oleic acid assisted in narrowing down the size distribution giving monodisperse nanoparticles. Seminal progress has been made towards achieving 2D Cu based metal alloys. However, there are a plethora of compositions such as CuNiPd, PdCuCo, CuNiPt6, CuFeNi, FePtCu, etc., that need to be exploited as 2D NCs to enhance the catalytic efficiency and reduce the cost of production.
5. Properties and applications
Owing to the unique characteristics related to their structural features, copper-based 2D NCs have received a lot of interest in plasmonics, heterogeneous catalysis, thermoelectrics and metal-ion batteries.28,63,141,150,162,163 2D nanostructures outperform 0D and 1D nanomaterials in architectural modification, charge separation, light harvesting and tunability.7,164 Extensive research has been done to develop effective catalysts with 2D nanomaterials, inspired by the unique optical, electronic properties and high surface area.63,162,165 2D nanostructures with a high surface-area-to-volume ratio and a high density of exposed atoms on their surface hold great promise for enhancing plasmonic resonance and catalytic processes. With the variable valence states, high defect densities, and vast compositional ranges, these NCs have become an ideal choice for the domain of catalysis. With excellent optoelectronic characteristics, Cu based NCs have been exploited as photocatalysts to harvest solar energy.28,52 They have also been employed as electrocatalysts in oxygen reduction and evolution reactions. In addition, they were found to be suitable for catalysts in synthetic organic chemistry. They are regarded as efficient thermoelectric materials due to the high ionic mobility of Cu+ in the chalcogenide templates that stems for phonon–liquid electron crystal behaviour resulting in suppressed thermal conductivity.166,167 The 2D shape of these materials stems from complex electronic transport behaviour.168–170 The in-plane direction possesses a smaller effective mass for electronic transport than the out of plane direction, resulting in greater carrier transport. In the following sub-sections, we cover the range of technological applications of colloidal 2D Cu based NCs.5.1. Plasmonics
The collective oscillation of free-charged carriers related to surface plasmon resonance is the source of plasmonic properties in semiconductor and noble metal bulk materials.175 In the case of NCs, surface plasmon resonance is localised (localised surface plasmon resonance, LSPR) and can resonate in-phase with the electromagnetic field of light to produce an intense electromagnetic field.176,177 The enhanced electromagnetic field can convert into heat that functions as heat mediators in photothermal therapy or radiate light that finds application as a concentrator in solar cells, imaging probes, or sensors. Cu based chalcogenides and phosphides represent an exciting class of plasmonic materials where the collective oscillation of holes is the origin of such behaviour.40,178 2D anisotropy in these NCs results in two separate LSPR modes i.e., low energy in plane (longitudinal) and high energy out of plane (transverse). The in-plane LSPR peak shifts to a higher wavelength with increased aspect ratio, which can be separated from the out-plane mode, which shifts to a lower wavelength at high aspect ratios.179 Xie et al. observed that the out-plane and in-plane LSPR peaks remain unresolved at a lower aspect ratio of 2 in CuS nanodiscs.117 However, increasing the aspect ratio two-fold resolved the low intensity out-of plane LSPR peak from the red-shifted in-plane LSPR peak. Recently, Sun et al. devised a heat-up approach for synthesising CuS nanodiscs with variable aspect ratio up to ∼27. Similarly, they observed red shift in longitudinal LSPR peak from NIR to deep mid-IR with increasing aspect ratio of CuS nanodiscs (Fig. 11a).171 For the nanodiscs with an aspect ratio of ∼27 a low intensity transverse LSPR peak was observed around 900 nm. For low symmetry, 2D morphologies such as triangular and hexa-triangular the transverse peak is more prominent. For instance, Castro et al. observed two LSPR peaks at 943 and a longitudinal mode at 1346 nm for triangular CuS nanodiscs. With increasing aspect ratio, a redshift in the longitudinal peak and a blue shift in the transverse LSPR peak were observed (Fig. 11b).172 However, for more symmetric structures and low dimensions, axial LSPR modes are poorly separated, resulting in broad full width at half maximum.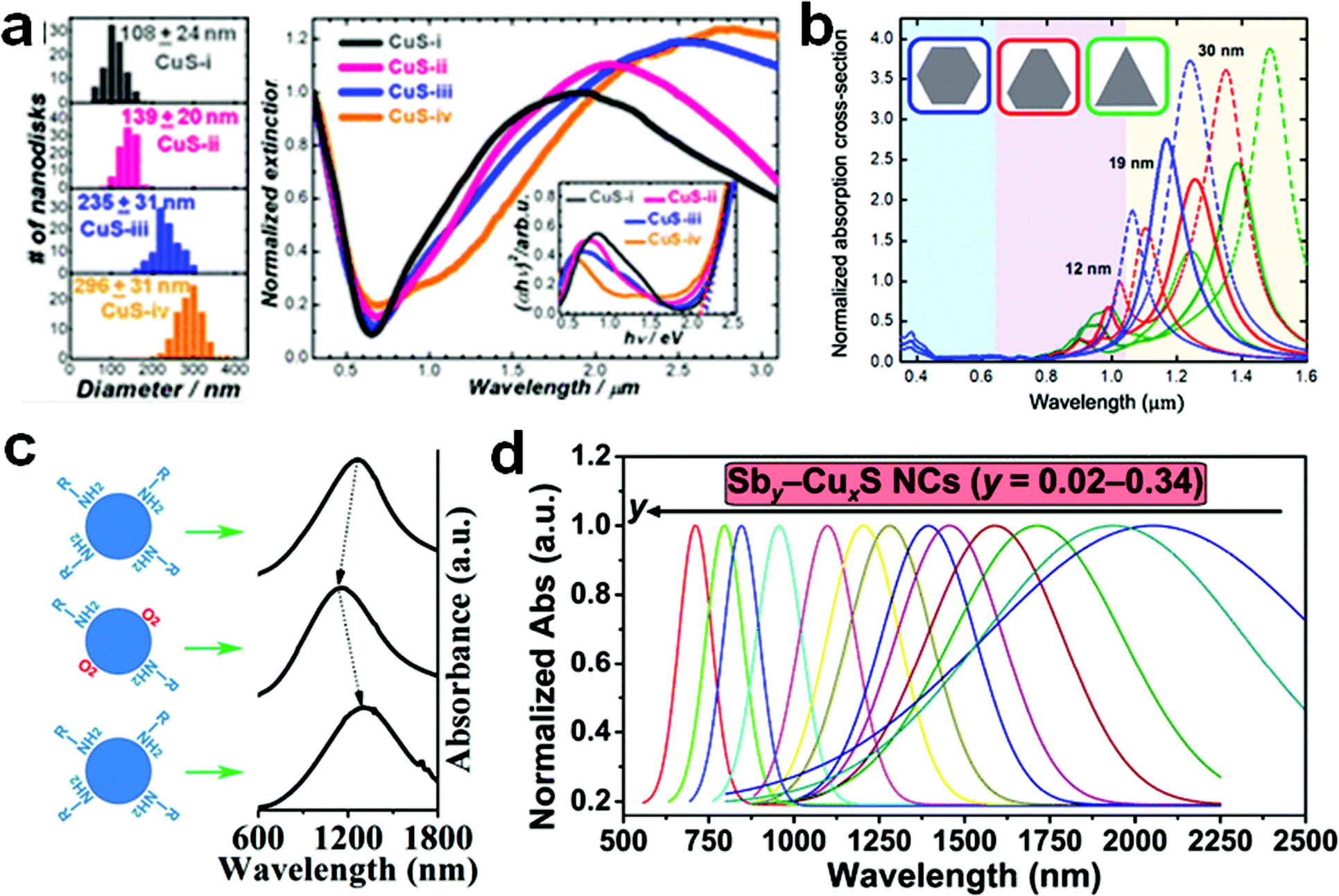 | ||
| Fig. 11 (a) Optical extinction spectra displaying a red-shift in the longitudinal LSPR peak with increment in the aspect ratio (fixed thickness of ∼11 nm). Reproduced from ref. 171 with permission from ACS, copyright 2021; (b) normalized absorption spectra for CuS nanocrystals of hexagonal, triangular, and hexa-triangular shapes for different lateral dimensions: (dashed line) 12 nm, (solid line) 19 nm, and (dash-dotted line) 30 nm displaying red and blue shift in longitudinal and transverse LSPR peak, respectively. Reproduced from ref. 172 with permission from ACS, copyright 2019; (c) tuning of the LSPR peak by the hole density in CuS air exposure and OLA passivation changing. Reproduced from ref. 173 with permission from ACS, copyright 2013; (d) LSPR tuning of CuxS via Sb3+ doping. Reproduced from ref. 174 with permission from ACS, copyright 2021. | ||
Tunability of free charge carrier (∼1016 to 1021 cm−3) concentration by doping can alter the absorption window from visible to near-infrared to (NIR) mid-infrared (MIR) for Cu based chalcogenides which is not possible for noble metals.180 For example, for Cu-deficient Cu2−xS, which possess a diverse phase diagram, the LSPR frequencies can be tuned based on the free hole concentration depending on the Cu/S ratio.181 Xie et al. showcased how LSPR frequency changed depending on hole concentration by adding Cu+ into CuS nanoplatelet templates.171 Each subsequent addition of Cu+ into the CuS template until Cu/S is 2/1 reduced the hole concentration resulting in fading of LSPR intensity and a red shift in the peak position. In contrast, for Cu2S and Cu2Se oxidation induced Cu vacancy increases the hole concentration to modulate LSPR frequency. Wei et al. demonstrated the effect of electron donating (Lewis base) and electron accepting (Lewis acid) ligands on modulating the hole density in CuS nanodiscs.173 Lewis acids such as molecular O2 (from air exposure) and oleic acid, which can act as electron acceptors, increase the hole density by attaching to the NC surface and result in a blue shift of the LSPR peak (Fig. 11c). At the same time, passivation with OLA restores the peak position. Apart from oxidation and reduction-related hole carrier density modulation in self-doped CuxS, foreign metal cation doping has also been used to tune LSPR.182,183 Liu et al. doped Sb3+ in CuxS nanodiscs to modulate the LSPR frequency from NIR to mid visible range (Fig. 11d).174 Sb3+ doping induced high carrier density originating from copper vacancy and aliovalent Sb3+ and resulted in the LSPR peak shift to a higher energy and narrowing of the full width at half maximum. Thus, stoichiometry is a vital parameter to modulate the hole density for tuning the LSPR frequency. Doping of foreign cations can alter the hole density as well. However, a more controlled way of tuning LSPR frequency is achieving greater control in synthesis, which will ensure desirable stoichiometries with the required morphologies especially with higher asymmetry and aspect ratio for more intense low energy and high energy LSPR modes.
5.2. Thermoelectrics
Thermoelectric (TE) materials are promising as a renewable source for converting waste heat to electricity and vice versa.186,187 However, the conversion efficiency denoted by the TE figure of merit (ZT) is yet to reach the benchmark efficiency value of >4 for broader commercial applications.188 The ZT (=σS2T/k) depends on the electrical conductivity (σ), Seebeck coefficient (S), and total thermal conductivity (k), where the total thermal conductivity is defined by the lattice thermal conductivity (kL) and electrical thermal conductivity (ke). A high-power factor (σS2) and low thermal conductivity are required to achieve a high ZT value. However, controlling these transport properties remains challenging due to their interdependent nature. In recent times, the Cu-based chalcogenides having reached ZT values >2 attracted immense attention as viable TE materials.163,167,185 The copper based binary chalcogenides, especially Cu2−xS and Cu2−xSe display reversible phase transition to highly symmetric phases upon heating to a certain temperature.166,189 For example, α-Cu2Se possesses a low symmetry monoclinic crystal structure at room temperature transitions to the cubic β-Cu2Se phase above 400 K (Fig. 12a).166 At temperatures above 373 K, Cu+ become highly disordered on the symmetric chalcogen framework resulting a liquid like behaviour responsible for their intrinsic low thermal conductivity. Additionally, nanostructuring of these materials can further reduce the thermal conductivity to a theoretical low value by scattering low and mid frequency phonons (Fig. 12b). Yang et al. achieved a ZT value of 1.82 for β-Cu2Se nanoplates by reducing the kL 0.11 W m−1 K−1via forming a high density of small angle grain boundaries and dislocation beneficial for phonon scattering.190 Similarly, Gahtori utilized nanostructuring of Cu2Se to reduce the thermal conductivity to 0.34 W m−1 K−1 which is 65% lower than the bulk thermal conductivity (Fig. 12c) and achieved a ZT value of 2.1 at 973 K.185 Doping of foreign cations has also been explored as a mean of reducing thermal conductivity or modulating carrier concentration. Liao et al. reduced the carrier concentration of α-Cu2Se from 4.1 × 1020 to 2.0 × 1020 cm−3 at room temperature by doping Bi as the electron donor that reduced ke and optimized the ZT value to 0.43 at near room temperature of 373 K.191 Similar to the Cu2−xSe, the liquid-like motion of Cu+ in Cu2−xS results in low κL and high ZT values.192,193 However, Cu2−xS faces a major problem of S volatilization upon annealing leading to S loss and formation of phases with low Cu deficiency resulting in a low carrier concentration.194,195 Zhang et al. showcased that 0.3% doping of Pb could stabilize the Cu deficient Cu1.8S phase up to 880 K during Pb:Cu2−xS phase transition enabling a high carrier concentration compared to Cu2−xS (Fig. 12d).163 Thus, stabilization of the Cu1.8S phase during annealing resulted in a high power factor and a high ZT of 2.03 at 880 K (Fig. 12e and f). More complex structures of copper chalcogenides such as tetrahedrite Cu12Sb4S13 have also been explored extensively where a ZT of more than 1 was attained.194 Doping of divalent transition elements by forming Cu12−xMSb4S13 is also a popular way to control their ZT by suppressing the number of holes that eventually reduces the ke resulting in low thermal conductivity. However, most 2D Cu based materials remain unexplored for thermoelectrics. For instance, high entropy multinary NCs and layered NCs which display low thermal conductivities are yet to be utilised.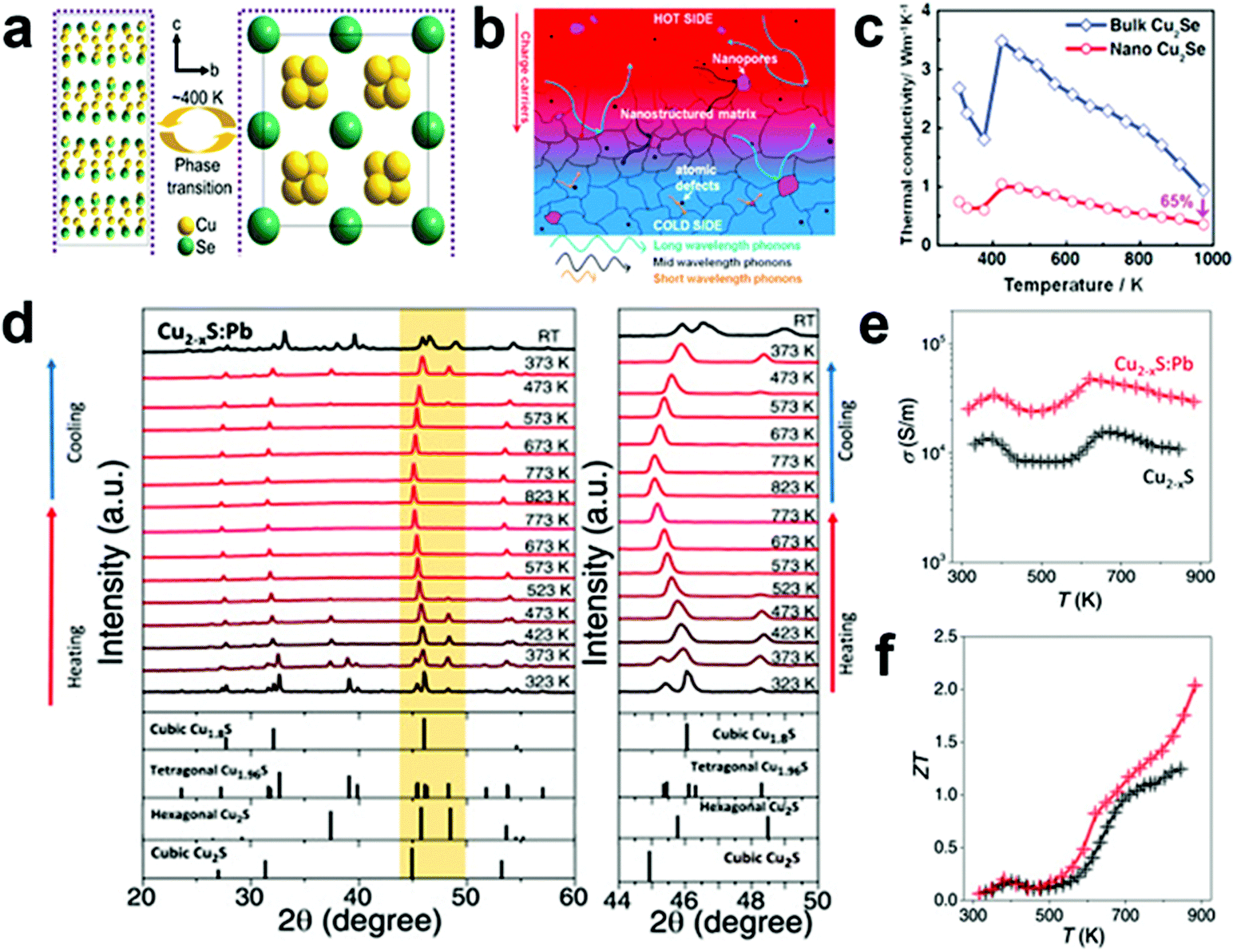 | ||
| Fig. 12 (a) Schematic of reversible structural switching between monoclinic α-Cu2Se and cubic β-Cu2Se; reproduced from ref. 184 with permission from Elsevier, copyright 2020; (b) schematic illustration of phonon scattering associated with nanostructure engineering, (c) 65% reduction in thermal conductivity of Cu2Se through nanostructuring; reproduced from ref. 185 with permission from Elsevier, copyright 2015; (d) doping of Pb in Cu2−xS stabilized the Cu1.8S phase during annealing resulting in (e) higher electrical conductivity and (f) higher thermoelectric figure of merit. Reproduced from ref. 163 with permission from Elsevier, copyright 2021. | ||
5.3. Photocatalysis
The two major applications of photocatalysis are hydrogen fuel production via a water-splitting process utilising sunlight and pollutant dye degradation for wastewater treatment. 2D semiconductor nanomaterials with confined thickness, defined surface area, distinct structural, physical, and chemical characteristics are beneficial in the development of novel photocatalysts. Due to the outstanding visible light absorption properties, non-toxic composition, and environmentally benign nature of different Cu based 2D NCs, they are explored as photocatalysts for harnessing solar energy. Liu et al. reported multinary copper chalcogenides CuGaS2 (CGS) NCs with tunable one dimensional and two-dimensional morphologies for photocatalytic hydrogen production.52 Their research demonstrated that the photocatalytic hydrogen evolution rate (HER) was significantly enhanced by controlling the dimensions. The morphological modification substantially influenced the optical bandgap and energy level of the CGS NCs having 1D and 2D morphologies. These findings are associated with the fraction of exposed {001} and {100} facets. {001} facets were exposed in 2D CGS nanoplates, whereas {100} facets were primarily exposed in 1D wurtzite CGS nanorods (Fig. 13a). In another study, Zhang et al. reported copper phosphosulphides as a highly active and stable photocatalyst for hydrogen evolution reactions.28 Compared to pristine Cu3−xP, copper phosphosulphides have a maximal HER of 2085 mol g−1 h−1, accounting for a five-fold increase in activity. According to the DFT calculations, the phosphosulphide structures push the Gibbs free energies for the intermediate states to be near 0 eV and increase the number of active sites for hydrogen adsorption, resulting in significantly enhanced catalytic activity toward the HER.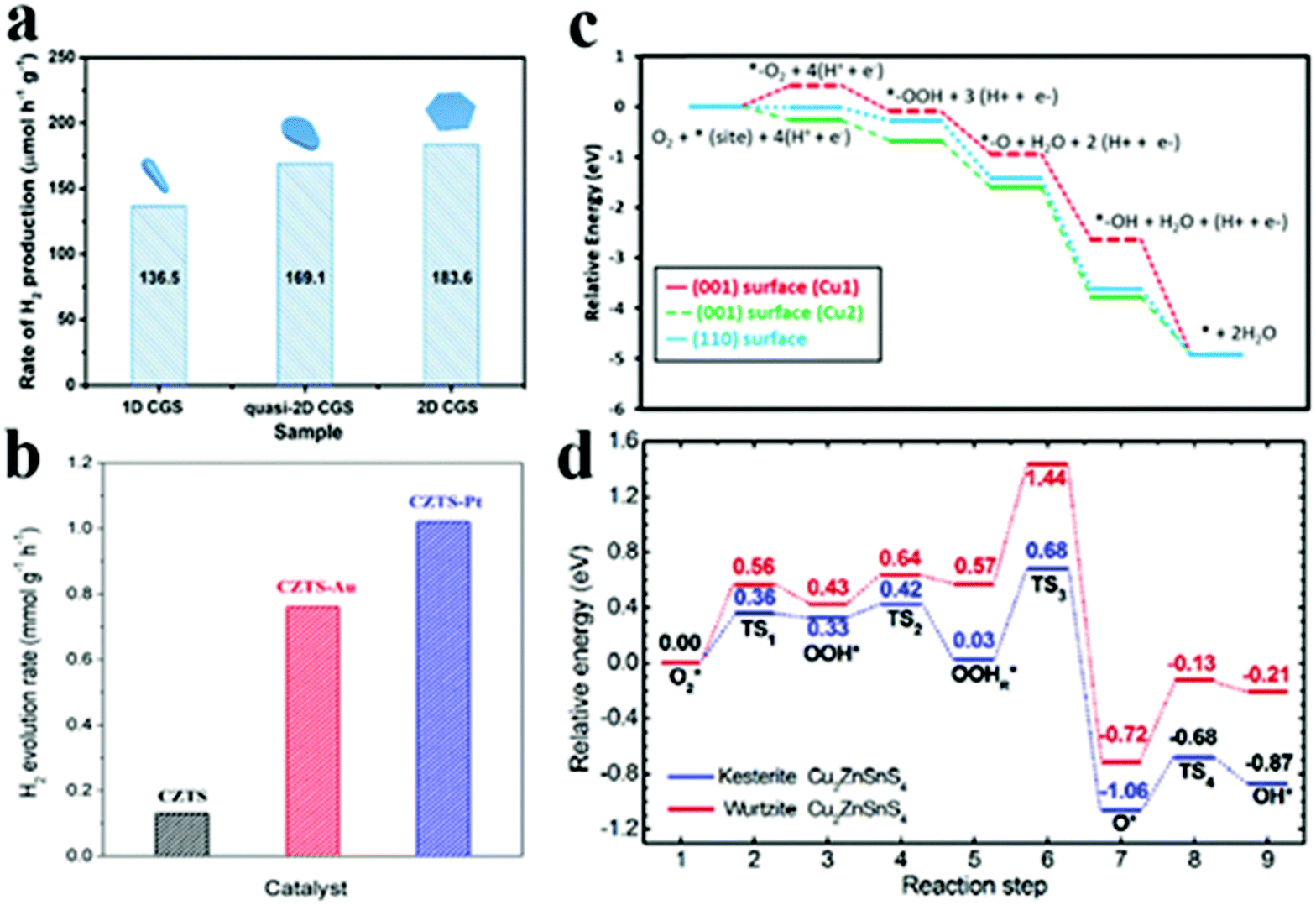 | ||
| Fig. 13 (a) The photocatalytic hydrogen generation rates for 1D CGS nanorods, quasi-2D nanodiscs, and 2D CGS nanoplates. Reproduced from ref. 52 with permission from RSC, copyright 2019; (b) the rate of hydrogen evolution in post-modified CZTS–Au and CZTS–Pt heterostructure nanoparticles, along with pristine CZTS. Reproduced from ref. 196 with permission from ACS, copyright 2016; (c) energy profile diagram of the ORR for Cu (1)-terminated (001) surfaces (red), Cu (2)-terminated (001) surfaces (blue), and (110) surfaces (green). Reproduced from ref. 197 with permission from ACS, copyright 2018. (d) Reaction free-energy diagrams for the ORR on kesterite (blue) and wurtzite CZTS (red). Reproduced from ref. 196 with permission from ACS, copyright 2016. | ||
Post synthetic modification of copper-based NCs with noble metals has shown enhancement in the catalytic activity. One such example is Cu2ZnSnS4 (CZTS) nanoplates used as templates to synthesise CZTS–Au and CZTS–Pt heterostructure nanoparticles for hydrogen generation by water splitting and degradation of water pollutants (Fig. 13b).196 Through effective charge transfer of photogenerated carriers from the semiconductor to metal co-catalysts promoted solar energy conversion. The co-catalyst decreased the overpotential of the reaction, lowering the charge carrier transfer interface barrier and enhancing surface redox processes such as water splitting and dye degradation. Cui et al. demonstrated Cu7S4@Pd hetero nanostructures for high-efficiency photocatalytic reactions such as the Suzuki coupling reaction, nitrobenzene hydrogenation, and benzyl alcohol oxidation. Cu7S4@Pd enabled hot-carrier transfer from Cu7S4 to Pd, which accelerated catalytic processes on the Pd metallic surface.29 For all three types of organic reactions, hetero nanostructures exhibited remarkable catalytic activity, with over 80–100% conversion accomplished in less than 2 hours.
Cu chalcogenides’ plasmonic characteristics have also been explored to enhance the photocatalytic degradation of contaminants and dyes. Shao et al., for example, illustrated the use of such plasmonic Cu2−xS nanoparticles for rhodamine B and methyl orange degradation.198 Their study shows that photocatalytic activity could be regulated and controlled by modulating carrier concentrations. For example, Cu1.94S NCs with the highest free carrier density exhibited the best photocatalytic activity compared to Cu1.2S NC. Cu1.94S NCs degraded over 80% of the dye molecules in the first 20 minutes, compared to Cu1.2S NCs, which degraded only 57% of the dye molecules after 120 minutes of UV exposure. In another instance, Cu2−xSySe1−y NCs have been demonstrated to be a potent photo-activated catalyst for the Huisgen [3 + 2] cycloaddition process.199 Here, the NCs serve as an efficient photocatalyst by generating Cu(I) into the solution when irradiated, providing fascinating new avenues for doing such archetypal click chemistry.
Post-modification of copper phosphide has become an avenue for the plethora of efficient photo and photo electrocatalysts. Dutta et al. described an oriented attachment of Au and Cu3−xP to develop a novel ring-on-disc heterostructure.200 The addition of phosphine gas as a bridging agent to the reaction mixture guided the pre synthesised Au nanoparticles onto the edges of Cu3−xP discs, resulting in a unique heterostructure. Furthermore, the Au–Cu3−xP has been employed as a photocatalyst for the evolution of hydrogen from water. Since the coupling of exciton and plasmon inhibits excitonic recombination and thus promotes the photo-electron transfer process, making Au–Cu3−xP a better photocatalyst than pristine Cu3−xP. In another instance, thin Au nanowires grown in a stripy patterned pattern on Cu3−xP platelets are investigated as photoanode materials for the catalytic oxidation of an essential biomolecule, nicotinamide adenine dinucleotide (NADH).32 In the presence of NADH, Au–Cu3−xP exhibited a more than 30-fold rise in current density compared to its absence. Thus, the functionalisation of the 2D NC surface with various capping agents and metal NCs has proven to be highly significant for enhancing catalytic activity.
5.4. Electrocatalysis
Electrocatalysis has recently received much attention due to its application in fuel cells, electrolysis, and other energy storage and conversion reactions.25 In general, charge transport at electrode/electrolyte interfaces has a substantial influence on electrocatalytic activity.162 Pt-free electrocatalysts composed of 2D Cu-based NCs offer potential benefits such as low cost, comparative electrochemical activity to precious metals, and adjustable physical and chemical characteristics. Implementing a 2D ultrathin nanostructure can enhance accessible active sites and improve the electronic conductivity in electrocatalysis. Cu deficient Cu2−xS nanoplates are demonstrated as active oxygen reduction reaction (ORR) electrocatalysts with increased ORR activity when the free hole concentration in Cu2−xS was increased.68 Additionally, post-modified Cu2−xS nanoplates with carbon black demonstrated a significant increase in activity compared to Cu2−xS alone due to increased electrical conductivity and synergistic effect between Cu2−xS and graphene. The electrochemical performance of anisotropic size-tunable 2D CuS nanoplatelets has been comprehensively studied by Liu et al.197 CuS nanoplatelets with a uniform thickness, but a broadly variable diameter demonstrated electrochemical activity for the oxygen reduction reaction in alkaline solution. The activity of the oxygen reduction process in alkaline solution enhanced as the diameter was increased (Fig. 13c). Cu2−xS NC's electrochemical activity was found to be strongly facet-dependent and anisotropic. Increasing the diameter of NCs promotes charge carrier transport through the electrocatalyst layer while maintaining the specific surface area of the most electrochemically active facets. Yu et al. reported two phases of Cu2ZnSnS4 (CZTS) NCs and examined their catalytic activity targeting the ORR.196 Higher oxidation states, such as Zn2+ and Sn4+, give more energetically favourable sites for oxygen adsorption and subsequent hydroxyl production. CZTS NCs demonstrated ORR activity comparable to commercial Pt/C electrocatalysts, with high current densities (5.45 mA cm2 at 0.1 V vs. RHE) and low onset reduction potential (0.89 V vs. RHE). Sn4+ and Zn2+ sites offered low energy barriers for the rate-determining phase and enhanced electrocatalytic activity. The catalytic reactivity is influenced mainly by the shape and primarily exposed crystal planes. The two exposed planes in 2D materials (Fig. 13d) can have distinct electrocatalytic reactions. Zhao et al. successfully synthesised ultrathin CuCo2S4 nanosheets (NSs) with predominantly exposed {111}, {022}, and {004} planes.201 In terms of the oxygen reduction reaction/oxygen evolution reaction (ORR/OER), the two exposed planes (022) and (004) have distinct preferences. Therefore, the CuCo2S4 NCs display outstanding bifunctional catalytic capabilities toward both the ORR and OER in alkaline solutions and CuCo2S4 nanosheets outperform commercial Pt/C electrocatalysts well as CuCo2S4 nanoparticles in electrochemical activity. Park et al. synthesised Pd13Cu3S7 nanoplates via partial cation exchange of Cu1.81S with the Pd precursor and investigated their electrocatalytic activity for the HER.30 This Pd13Cu3S7 NCs showed an overpotential of only 64.0 mV at −10 mA cm−2 and a Tafel slope of 49.6 mV dec−1, making them one of the finest sulphide-based catalysts for the HER comparable to the commercial Pd/C catalysts. In another report, Li et al. investigated Co-doped Cu7S4 nanodiscs as highly efficient electrocatalysts for oxygen evolution reactions.202 In comparison to bare Cu7S4, the developed catalyst exhibited a low overpotential of 270 mV to reach a current density of 10 mA cm−2 and with a reduced Tafel slope and improved turnover frequencies. Thus, the heterostructure formation and cationic doping have proven significant for enhancing the catalytic activity of the semiconductor NCs. The copper–metal alloy nanocrystals have the potential to replace commercial Pt/C for various electrocatalytic reactions. Alloying noble elements with Earth-abundant Cu becomes a realistic, simple, and successful method for lowering the cost of electrocatalysts while increasing their catalytic performance. Several 2D copper-based alloys have been utilized as a catalyst for electrocatalytic reactions. For example, the composition-dependent electrocatalytic activities of CuAg alloy nanocrystals in ORR have been reported.141 CuAg NCs outperform pristine Ag by about 1.5 times in ORR activity. Furthermore, with increased Cu content, electrochemical impedance spectroscopy revealed lower charge transfer resistance. The optimal composition was discovered to be Cu40Ag60, and its increased activity was attributed to changes in the electrical structure caused by alloying. In another study, in alkaline circumstances, PdPtCu nanorings have been employed as efficient and long-lasting electrocatalysts for the ORR.85 This catalyst achieves an ultrahigh electrochemical active surface area, significantly more than the commercial Pt/C catalyst, leading to an enhanced ORR activity. Furthermore, the catalyst demonstrated consistent endurance, with an activity retention of 86% after 30000 cycles. Post-modified PdCu alloy nanosheets have shown a high activity towards formic acid oxidation.161 The elevated activity has been attributed to the ultrathin morphology, a synergistic impact between Pd and Cu, and post-treatment.
Despite many theoretical calculations and ex situ observations, no firm conclusions on determining the actual active sites for catalytic processes have been reached yet. As a result, a comprehensive in situ study is required to elucidate the unique active sites for various catalytic processes. Considering the high potential of Cu based catalysts, there is still a gap in meeting commercial needs, offering an opportunity to increase the catalytic activity further. Understanding facet-specific binding with in situ X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) and simulation of crystal facets with active species will be beneficial to enhance catalytic activity further.
5.5. Other applications
The ubiquity of the application of 2D Cu based NCs has also been extended to electrodes for batteries and sensors. One of their notable applications is in lithium-ion batteries (LIBs), where the Cu2−xE based anode undergoes a conversion mechanism forming Li2E and Cu (Fig. 14a). In this regard, the copper telluride nanostructure with different morphologies ranging from nanocubes, nanosheets, and nanoparticles have been tested as an anode in lithium-ion batteries.31 Among all the nanosheet, nanocube, or nanoparticle electrodes, the Cu2xTe nanosheet electrode offered the highest capacity and best rate capability (Fig. 14b). The superior electrical contact and homogeneity of nanosheets over nanoparticles or nanocubes contributed to the outstanding performance of the nanosheet anode.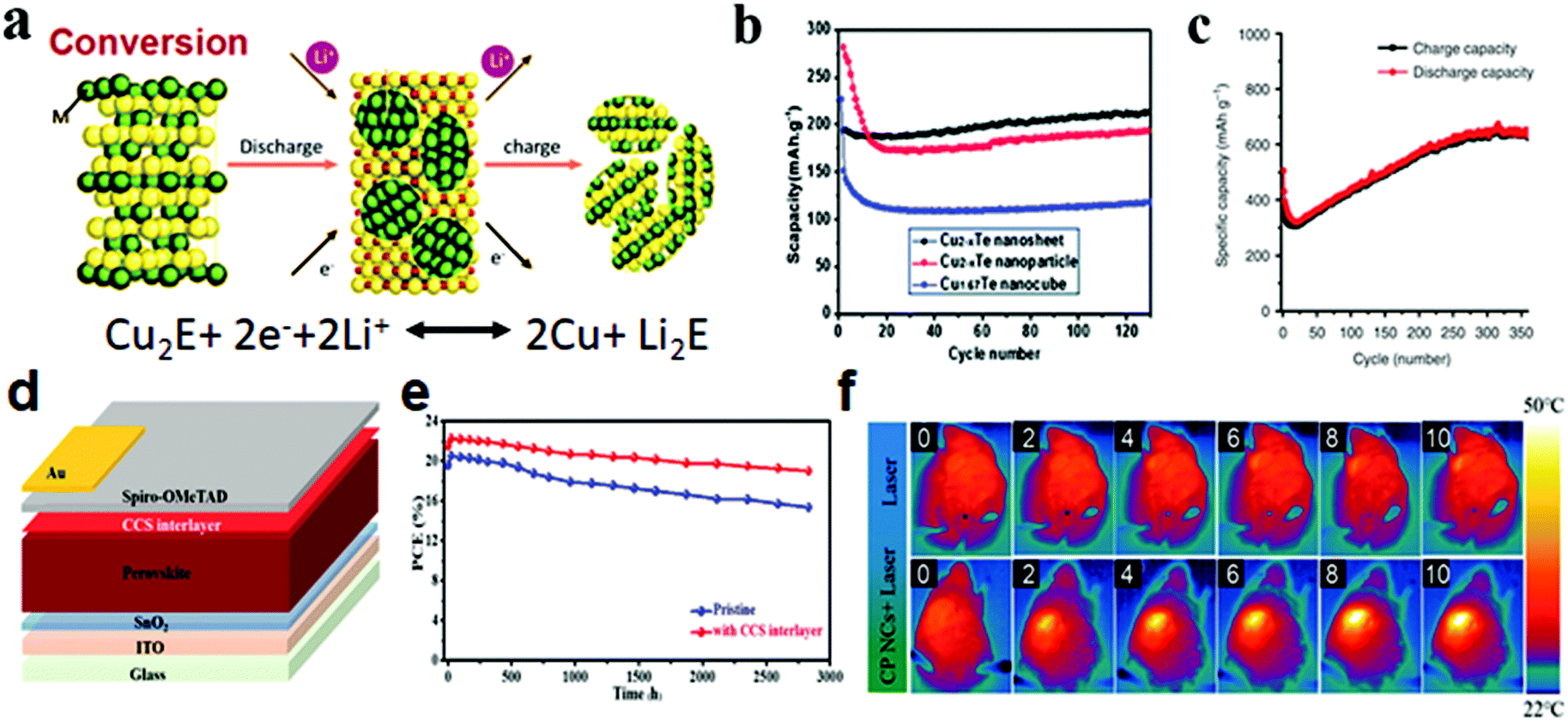 | ||
| Fig. 14 (a) Schematic of the conversion reaction of copper chalcogenide anodes (E = S, Se, Te) in lithium ion batteries (LIB); (b) specific capacity at a rate of 0.5C for three different Cu2−xTe based anodes in LIB; reproduced from ref. 31 with permission from RSC, copyright 2014; (c) specific capacity at a current density of 0.2 A g−1 of CuS nanosheet based anode in LIB; reproduced from ref. 74 with permission from Nature, copyright 2012; (d) a schematic cross-sectional representation of the device architecture consisting of the CsCu5S3 NC interlayer, (e) stability of unencapsulated PSCs without and with a CsCu5S3 interlayer. Reproduced from ref. 65 with permission from Elsevier, copyright 2021; (f) in vivo thermal images of tumor-bearing mice. Reproduced from ref. 203 with permission from Wiley, copyright 2019. | ||
Furthermore, the assembled cell composed of Cu2xTe NCs displayed extremely high cycling stability (up to 5000 cycles). This example clearly demonstrates the significance of regulating the morphology for improved performance. Kim et al. studied the electrochemical performance of CuS nanosheets as an electrode for LIBs. They obtained a specific capacity of 642 mA h g−1 after 270 cycles at a current density of 0.2 A g−1 (Fig. 14c).74 Being a conversion material, CuS shows a theoretical capacity of 560 mA h g−1 for Li ion batteries. However, the expulsion of Cu from CuS upon Li2S formation disassembled the nanosheets and increased the exposed surface area for more Li reactivity, resulting in higher capacity than usual CuS based electrodes.
Apart from application in batteries, CuSe nanosheets have been employed for flexible electronics by making conductive films.204 Films coated with CuSe hexagonal nanosheets exhibited ohmic activity, with a conductivity of 535 S m−1 that sustained for several months in the air. In another study, Sun and co-workers reported the integration of graphene with colloidal Cu3−xP 2D quantum dots for optoelectronic applications.178 These hybrids demonstrated high broadband operation capability as well as good flexibility. Recently, hexagonal CsCu5S3 NCs were employed as an interlayer between the perovskite film and the hole transport layer in perovskite solar cells (Fig. 14d).65 This interlayer enhanced the hole transport while acting as an energy barrier to minimise carrier recombination, resulting in a power conversion efficiency of 22.29% (Fig. 14e). In another instance, a high-energy Q-switched laser using plasmonic Cu3−xP NCs as effective nonlinear absorbers has been reported.50 Cu3−xP NCs exhibit comparable nonlinear optical characteristics and device performance to other Q-switchers and improved compatibility with optical components.
In recent years, copper chalcogenide based 2D nanomaterials have shown great potential in the biomedical field. Copper chalcogenide-based 2D nanomaterials have recently emerged as effective photothermal agents for photothermal treatment (PTT) owing to their attractive low cost, low toxicity, biodegradability and high absorption of near-infrared (NIR) light across a relatively wide wavelength range.122,205 He et al. reported plasmonic CuS nanodisc based composite nanocapsules for chemo-photothermal cancer therapy.206 In another study, CuTe NCs spanning from 1D to 3D morphologies were explored as photothermal agents and surface-enhanced Raman scattering probes. With enhancement factors of 1.5 × 106 and 106, respectively, CuTe nanocubes and nanoplates produced excellent signal-to-noise spectra, whereas CuTe nanorods did not yield any SERS signal. Recently, Wang and co-workers investigated photoacoustic ovarian tumour imaging with CuS nanodiscs and nanoprisms. CuS nanodiscs and nanoprisms both generated sensitive photoacoustic signals in the picomolar range.67 Liu and co-workers reported a Cu3−xP NCs based Fenton agent for in situ self-generation T1-MRI-guided PTT and PT-enhanced CDT.203 Cu3−xP NCs potentially generated a high quantity of toxic ˙OH in response to over-expressed H2O2 in tumour tissue. Furthermore, the tumour's excess GSH can decrease Cu(II) generated in the Fenton-like reaction to Cu(I), boosting the rate of ˙OH production. (Fig. 14f). This study shed insights into the approach towards generating more Fenton-like agents that are both revolutionary and efficient.
6. Challenges and future directions
In summary, we have highlighted the research progress made on the synthesis and applications of 2D Cu based NCs, Cu-chalcogenides, phosphides, halides, and Cu–metal alloys. Though these NCs are promising due to their cost-effective and environmentally benign nature, they are still lagging behind heavy chalcogenides in terms of optoelectronic properties due to limitations associated with ultrathin NC formation. To date, ultrathin nanosheets of Cu2−xS and In-poor CuInS have been successfully synthesized based on soft templated growth and self-organization, respectively. However, transformation into multinary composition in most other instances is dependent on cation exchange. Hence, future exploration is necessary for the direct growth of ultrathin nanosheets with multinary compositions. Here, the real-time study of nucleation and growth kinetics such as liquid cell in situ TEM, small-angle X-ray scattering (SAXS), X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy for these existing NCs will provide insights into the design of ultrathin NCs of complex multinary composition including Cu3−xP, Cu–metal alloys, and Cu-halides.207 The Cu+ mobility in the Cu3−xP template is similar to the Cu-chalcogenides. However, the formation of multinary NCs based on Cu3−xP has not been explored except for Cu3P1−xSx. The Cu-halides, which have shown more stability than lead based perovskites, self-trapped emissions, longer lifetime, and wide bandwidths, are promising for lighting devices. Nevertheless, the moisture stability is not good enough for large scale device fabrications. Here the shell formation of metal chalcogenides such as ZnS will enhance the stability.208The layered 2D Cu-based NCs are expected to show low thermal conductivities resulting from low phonon group velocity and lattice anharmonicity from weak interaction between layers and heterogeneity in chemical bonding. However, each monolayer exhibits quantum confinement of charge carriers resulting in a high Seebeck coefficient, ideal for efficient thermoelectric performance. Here, apart from CuCrSe2, other layered structures such as CuME2 (M = Mo, W, E = S, Se) and ACuME2 (A = Na, K, Cs, Rb, M = Mn, Fe, Co, Ni, Zn) remain unexplored as NCs in thermoelectrics. Furthermore, the Kirkendall effect is another less explored yet effective tool for synthesizing hollow morphologies with more active sites for electrochemical reactions. Therefore, the Cu-based 2D NC family, which comprises a massive library of NCs with different phases and compositions, holds great promise as future 2D materials for energy applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NK acknowledges financial support of the Irish Research Council (IRC) under Grant Number IRCLA/2017/285. NP acknowledges financial support from the Department of Chemical Sciences, University of Limerick. The authors also acknowledge Science Foundation Ireland (SFI) under the Principal Investigator Program under contract no. 16/IA/4629 and under grant no. SFI 16/M-ERA/3419.Notes and references
- P. S. Zhou, P. Schiettecatte, M. Vandichel, A. Rousaki, P. Vandenabeele, Z. Hens and S. Singh, Cryst. Growth Des., 2021, 21, 1451 CrossRef CAS.
- Y. F. Sun, M. Terrones and R. E. Schaak, Acc. Chem. Res., 2021, 54, 1517 CrossRef CAS PubMed.
- P. S. Zhou, G. Collins, Z. Hens, K. M. Ryan, H. Geaney and S. Singh, Nanoscale, 2020, 12, 22307 RSC.
- Y. F. Sun, Y. X. Wang, J. Y. C. Chen, K. Fujisawa, C. F. Holder, J. T. Miller, V. H. Crespi, M. Terrones and R. E. Schaak, Nat. Chem., 2020, 12, 284 CrossRef CAS PubMed.
- M. S. Sokolikova and C. Mattevi, Chem. Soc. Rev., 2020, 49, 3952 RSC.
- A. C. Berends and C. D. Donega, J. Phys. Chem. Lett., 2017, 8, 4077 CrossRef CAS PubMed.
- M. Nasilowski, B. Mahler, E. Lhuillier, S. Ithurria and B. Dubertret, Chem. Rev., 2016, 116, 10934 CrossRef CAS PubMed.
- J. S. Son, J. H. Yu, S. G. Kwon, J. Lee, J. Joo and T. Hyeon, Adv. Mater., 2011, 23, 3214 CrossRef CAS PubMed.
- D. D. Vaughn, R. J. Patel, M. A. Hickner and R. E. Schaak, J. Am. Chem. Soc., 2010, 132, 15170 CrossRef CAS PubMed.
- F. Li, M. M. R. Moayed, E. Klein, R. Lesyuk and C. Klinke, J. Phys. Chem. Lett., 2019, 10, 993 CrossRef CAS PubMed.
- S. Jeong, D. Yoo, J. T. Jang, M. Kim and J. Cheon, J. Am. Chem. Soc., 2012, 134, 18233 CrossRef CAS PubMed.
- J. H. Han, S. Lee and J. Cheon, Chem. Soc. Rev., 2013, 42, 2581 RSC.
- L. W. Dai, C. Strelow, T. Kipp, A. Mews, I. Benkenstein, D. Eifler, T. H. Vuong, J. Rabeah, J. McGettrick, R. Lesyuk and C. Klinke, Chem. Mater., 2021, 33, 275 CrossRef CAS.
- L. Sonntag, V. Shamraienko, X. L. Fan, M. S. Khoshkhoo, D. Kneppe, A. Koitzsch, T. Gemming, K. Hiekel, K. Leo, V. Lesnyak and A. Eychmuller, Nanoscale, 2019, 11, 19370 RSC.
- T. Galle, M. S. Khoshkhoo, B. Martin-Garcia, C. Meerbach, V. Sayevich, A. Koitzsch, V. Lesnyak and A. Eychmuller, Chem. Mater., 2019, 31, 3803 CrossRef CAS.
- S. Delikanli, G. N. Yu, A. Yeltik, S. Bose, T. Erdem, J. H. Yu, O. Erdem, M. Sharma, V. K. Sharma, U. Quliyeva, S. Shendre, C. Dang, D. H. Zhang, Z. C. Sum, W. J. Fan and H. V. Demir, Adv. Funct. Mater., 2019, 29, 1901028 CrossRef.
- L. W. Dai, R. Lesyuk, A. Karpulevich, A. Torche, G. Bester and C. Klinke, J. Phys. Chem. Lett., 2019, 10, 3828 CrossRef CAS PubMed.
- E. Izquierdo, A. Robin, S. Keuleyan, N. Lequeux, E. Lhuillier and S. Ithurria, J. Am. Chem. Soc., 2016, 138, 10496 CrossRef CAS PubMed.
- V. Dzhagan, A. G. Milekhin, M. Y. Valakh, S. Pedetti, M. Tessier, B. Dubertret and D. R. T. Zahn, Nanoscale, 2016, 8, 17204 RSC.
- S. Singh, R. Tomar, S. ten Brinck, J. De Roo, P. Geiregat, J. C. Martins, I. Infante and Z. Hens, J. Am. Chem. Soc., 2018, 140, 13292 CrossRef CAS PubMed.
- A. C. Berends, J. D. Meeldijk, M. A. van Huis and C. D. Donega, Chem. Mater., 2017, 29, 10551 CrossRef CAS PubMed.
- K. Ramasamy, H. Sims, W. H. Butler and A. Gupta, J. Am. Chem. Soc., 2014, 136, 1587 CrossRef CAS PubMed.
- L. De Trizio, A. Figuerola, L. Manna, A. Genovese, C. George, R. Brescia, Z. Saghi, R. Simonutti, M. Van Huis and A. Falqui, ACS Nano, 2012, 6, 32 CrossRef CAS PubMed.
- H. Z. Guo, Y. Z. Chen, H. M. Ping, L. S. Wang and D. L. Peng, J. Mater. Chem., 2012, 22, 8336 RSC.
- Y. T. Guntern, V. Okatenko, J. Pankhurst, S. B. Varandili, P. Iyengar, C. Koolen, D. Stoian, J. Vavra and R. Buonsanti, ACS Catal., 2021, 11, 1248 CrossRef CAS.
- P. Reiss, M. Carriere, C. Lincheneau, L. Vaure and S. Tamang, Chem. Rev., 2016, 116, 10731 CrossRef CAS PubMed.
- K. Ramasamy, R. K. Gupta, H. Sims, S. Palchoudhury, S. Ivanov and A. Gupta, J. Mater. Chem. A, 2015, 3, 13263 RSC.
- X. Zhang, K. A. Min, W. Zheng, J. Hwang, B. Han and L. Y. S. Lee, Appl. Catal., B, 2020, 273, 118927 CrossRef CAS.
- J. B. Cui, Y. J. Li, L. Liu, L. Chen, J. Xu, J. W. Ma, G. Fang, E. B. Zhu, H. Wu, L. X. Zhao, L. Y. Wang and Y. Huang, Nano Lett., 2015, 15, 6295 CrossRef CAS PubMed.
- J. Park, H. Jin, J. Lee, A. Oh, B. Kim, J. H. Kim, H. Baik, S. H. Joo and K. Lee, Chem. Mater., 2018, 30, 6884 CrossRef CAS.
- C. Han, Z. Li, W. J. Li, S. L. Chou and S. X. Dou, J. Mater. Chem. A, 2014, 2, 11683 RSC.
- A. Dutta, A. K. Samantara, S. Das Adhikari, B. K. Jena and N. Pradhan, J. Phys. Chem. Lett., 2016, 7, 1077 CrossRef CAS PubMed.
- P. Vashishtha, T. J. N. Hooper, Y. N. Fang, D. Kathleen, D. Giovanni, M. Klein, T. C. Sum, S. G. Mhaisalkar, N. Mathews and T. White, Nanoscale, 2021, 13, 59 RSC.
- P. F. Cheng, L. Sun, L. Feng, S. Q. Yang, Y. Yang, D. Y. Zheng, Y. Zhao, Y. B. Sang, R. L. Zhang, D. H. Wei, W. Q. Deng and K. L. Han, Angew. Chem., Int. Ed., 2019, 58, 16087 CrossRef CAS PubMed.
- Y. K. Jung, I. T. Han, Y. C. Kim and A. Walsh, npj Comput. Mater., 2021, 7, 51 CrossRef CAS.
- N. Kapuria, U. V. Ghorpade, M. Zubair, M. Mishra, S. Singh and K. M. Ryan, J. Mater. Chem. C, 2020, 8, 13868 RSC.
- Y. Liu, M. X. Liu, D. Q. Yin, W. Wei, P. N. Prasad and M. T. Swihart, Chem. Mater., 2017, 29, 3555 CrossRef CAS.
- H. R. Ning, Y. Zeng, S. W. Zuo, S. V. Kershaw, Y. Hou, Y. Y. Li, X. N. Li, J. Zhang, Y. P. Yi, L. H. Jing, J. Li and M. Y. Gao, ACS Nano, 2021, 15, 873 CrossRef CAS PubMed.
- E. Lhuillier, S. Pedetti, S. Ithurria, B. Nadal, H. Heuclin and B. Dubertret, Acc. Chem. Res., 2015, 48, 22 CrossRef CAS PubMed.
- C. Coughlan, M. Ibanez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865 CrossRef CAS PubMed.
- X. Q. Chen, J. P. Yang, T. Wu, L. Li, W. Luo, W. Jiang and L. J. Wang, Nanoscale, 2018, 10, 15130 RSC.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012 CrossRef CAS PubMed.
- Y. Chen, Z. X. Fan, Z. C. Zhang, W. X. Niu, C. L. Li, N. L. Yang, B. Chen and H. Zhang, Chem. Rev., 2018, 118, 6409 CrossRef CAS PubMed.
- Y. A. Yang, H. M. Wu, K. R. Williams and Y. C. Cao, Angew. Chem., Int. Ed., 2005, 44, 6712 CrossRef CAS PubMed.
- X. G. Peng, J. Wickham and A. P. Alivisatos, J. Am. Chem. Soc., 1998, 120, 5343 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- A. C. Berends, W. van der Stam, Q. A. Akkerman, J. D. Meeldijk, J. van der Lit and C. D. Donega, Chem. Mater., 2018, 30, 3836 CrossRef CAS PubMed.
- W. Bryks, M. Wette, N. Velez, S. W. Hsu and A. R. Tao, J. Am. Chem. Soc., 2014, 136, 6175 CrossRef CAS PubMed.
- W. van der Stam, F. T. Rabouw, J. J. Geuchies, A. C. Berends, S. O. M. Hinterding, R. G. Geitenbeek, J. van der Lit, S. Prevost, A. V. Petukhov and C. D. Donega, Chem. Mater., 2016, 28, 6381 CrossRef CAS.
- Z. K. Liu, H. R. Mu, S. Xiao, R. B. Wang, Z. T. Wang, W. W. Wang, Y. J. Wang, X. X. Zhu, K. Y. Lu, H. Zhang, S. T. Lee, Q. L. Bao and W. L. Ma, Adv. Mater., 2016, 28, 3535 CrossRef CAS PubMed.
- S. Das Adhikari, A. Dutta, G. Prusty, P. Sahu and N. Pradhan, Chem. Mater., 2017, 29, 5384 CrossRef.
- Z. M. Liu, J. Liu, Y. B. Huang, J. Li, Y. Yuan, H. H. Ye, D. X. Zhu, Z. J. Wang and A. W. Tang, Nanoscale, 2019, 11, 158 RSC.
- J. J. Wang, Y. Q. Wang, F. F. Cao, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2010, 132, 12218 CrossRef CAS PubMed.
- C. Coughlan, Y. N. Guo, S. Singh, S. Nakahara and K. M. Ryan, Chem. Mater., 2018, 30, 8679 CrossRef CAS.
- C. Behera, R. Samal, C. S. Rout, R. S. Dhaka, G. Sahoo and S. L. Samal, Inorg. Chem., 2019, 58, 15291 CrossRef CAS PubMed.
- S. Bera, S. Shyamal, S. Sen and N. Pradhan, J. Phys. Chem. C, 2020, 124, 15607 CrossRef CAS.
- S. Lee, S. Baek, J. P. Park, J. H. Park, D. Y. Hwang, S. K. Kwak and S. W. Kim, Chem. Mater., 2016, 28, 3337 CrossRef CAS.
- J. F. L. Lox, Z. Y. Dang, M. L. Anh, E. Hollinger and V. Lesnyak, Chem. Mater., 2019, 31, 2873 CrossRef CAS.
- J. J. Wang, P. Liu, C. C. Seaton and K. M. Ryan, J. Am. Chem. Soc., 2014, 136, 7954 CrossRef CAS PubMed.
- X. Y. Zhang, Y. Xu, J. J. Zhang, S. Dong, L. M. Shen, A. Gupta and N. Z. Bao, Sci. Rep., 2018, 8, 248 CrossRef PubMed.
- Y. Liu, D. Q. Yin and M. T. Swihart, Chem. Mater., 2018, 30, 1399 CrossRef CAS.
- P. Vashishtha, G. V. Nutan, B. E. Griffith, Y. A. Fang, D. Giovanni, M. Jagadeeswararao, T. C. Sum, N. Mathews, S. G. Mhaisalkar, J. V. Hanna and T. White, Chem. Mater., 2019, 31, 9003 CrossRef CAS.
- M. G. Li, F. Y. Tian, T. S. Lin, L. Tao, X. Guo, Y. G. Chao, Z. Q. Guo, Q. H. Zhang, L. Gu, W. W. Yang, Y. S. Yu and S. J. Guo, Small Methods, 2021, 5, 2100154 CrossRef CAS PubMed.
- Y. Hong, T. Kim, J. Jo, B. Kim, H. Jin, H. Baik and K. Lee, ACS Nano, 2020, 14, 11205 CrossRef CAS PubMed.
- C. Yang, Z. J. Wang, Y. H. Lv, R. H. Yuan, Y. H. Wu and W. H. Zhang, Chem. Eng. J., 2021, 406, 126855 CrossRef CAS.
- S. W. Hsu, C. Ngo, W. Bryks and A. R. Tao, Chem. Mater., 2015, 27, 4957 CrossRef CAS.
- W. H. Li, R. Zamani, P. R. Gil, B. Pelaz, M. Ibanez, D. Cadavid, A. Shavel, R. A. Alvarez-Puebla, W. J. Parak, J. Arbiol and A. Cabot, J. Am. Chem. Soc., 2013, 135, 7098 CrossRef CAS PubMed.
- S. Deka, A. Genovese, Y. Zhang, K. Miszta, G. Bertoni, R. Krahne, C. Giannini and L. Manna, J. Am. Chem. Soc., 2010, 132, 8912 CrossRef CAS PubMed.
- A. E. Henkes and R. E. Schaak, Chem. Mater., 2007, 19, 4234 CrossRef CAS.
- B. Koo, R. N. Patel and B. A. Korgel, Chem. Mater., 2009, 21, 1962 CrossRef CAS.
- G. S. Shanker, A. Swarnkar, A. Chatterjee, S. Chakraborty, M. Phukan, N. Parveen, K. Biswas and A. Nag, Nanoscale, 2015, 7, 9204 RSC.
- Z. W. Wang, C. Schliehe, T. Wang, Y. Nagaoka, Y. C. Cao, W. A. Bassett, H. M. Wu, H. Y. Fan and H. Weller, J. Am. Chem. Soc., 2011, 133, 14484 CrossRef CAS PubMed.
- C. Schliehe, B. H. Juarez, M. Pelletier, S. Jander, D. Greshnykh, M. Nagel, A. Meyer, S. Foerster, A. Kornowski, C. Klinke and H. Weller, Science, 2010, 329, 550 CrossRef CAS PubMed.
- Y. P. Du, Z. Y. Yin, J. X. Zhu, X. Huang, X. J. Wu, Z. Y. Zeng, Q. Y. Yan and H. Zhang, Nat. Commun., 2012, 3, 1177 CrossRef PubMed.
- F. D. Wang, Y. Y. Wang, Y. H. Liu, P. J. Morrison, R. A. Loomis and W. E. Buhro, Acc. Chem. Res., 2015, 48, 13 CrossRef CAS PubMed.
- J. Lauth, F. E. S. Gorris, M. S. Khoshkhoo, T. Chasse, W. Friedrich, V. Lebedeya, A. Meyer, C. Klinke, A. Komowsld, M. Scheele and H. Weller, Chem. Mater., 2016, 28, 1728 CrossRef CAS.
- M. X. Liu, Y. Liu, B. B. Gu, X. B. Wei, G. X. Xu, X. M. Wang, M. T. Swihart and K. T. Yong, Chem. Soc. Rev., 2019, 48, 4950 RSC.
- C. L. Johnson, E. Snoeck, M. Ezcurdia, B. Rodriguez-Gonzalez, I. Pastoriza-Santos, L. M. Liz-Marzan and M. J. Hytch, Nat. Mater., 2008, 7, 120 CrossRef CAS PubMed.
- Z. R. Guo, Y. Zhang, L. Huang, M. Wang, J. Wang, J. F. Sun, L. N. Xu and N. Gu, J. Colloid Interface Sci., 2007, 309, 518 CrossRef CAS PubMed.
- I. Pastoriza-Santos, R. A. Alvarez-Puebla and L. M. Liz-Marzan, Eur. J. Inorg. Chem., 2010, 4288, DOI:10.1002/ejic.201000575.
- T. C. R. Rocha, H. Winnischofer, E. Westphal and D. Zanchet, J. Phys. Chem. C, 2007, 111, 2885 CrossRef CAS.
- R. Tan, Y. C. Yuan, Y. Nagaoka, D. Eggert, X. D. Wang, S. Thota, P. Guo, H. R. Yang, J. Zhao and O. Chen, Chem. Mater., 2017, 29, 4097 CrossRef CAS.
- L. Manna, D. J. Milliron, A. Meisel, E. C. Scher and A. P. Alivisatos, Nat. Mater., 2003, 2, 382 CrossRef CAS PubMed.
- S. Ghosh, R. Gaspari, G. Bertoni, M. C. Spadaro, M. Prato, S. Turner, A. Cavalli, L. Manna and R. Brescia, ACS Nano, 2015, 9, 8537 CrossRef CAS PubMed.
- N. L. Yang, Z. C. Zhang, B. Chen, Y. Huang, J. Z. Chen, Z. C. Lai, Y. Chen, M. Sindoro, A. L. Wang, H. F. Cheng, Z. X. Fan, X. Z. Liu, B. Li, Y. Zong, L. Gu and H. Zhang, Adv. Mater., 2017, 29, 1700769 CrossRef PubMed.
- A. W. Tang, Z. L. Hu, Z. Yin, H. H. Ye, C. H. Yang and F. Teng, Dalton Trans., 2015, 44, 9251 RSC.
- D. H. Li, J. Sun, X. Zhao and X. R. Yang, Nanoscale, 2015, 7, 13191 RSC.
- Z. Tang, H. Kwon, M. Y. Yi, K. Kim, J. W. Han, W. S. Kim and T. Yu, ChemistrySelect, 2017, 2, 4655 CrossRef CAS.
- X. L. Wang, X. Liu, D. Q. Yin, Y. J. Ke and M. T. Swihart, Chem. Mater., 2015, 27, 3378 CrossRef CAS.
- Y. Zhai and M. Shim, Chem. Mater., 2017, 29, 2390 CrossRef CAS.
- W. van der Stam, Q. A. Akkerman, X. X. Ke, M. A. van Huis, S. Bals and C. D. Donega, Chem. Mater., 2015, 27, 283 CrossRef CAS.
- X. M. Li, H. B. Shen, J. Z. Niu, Y. G. Zhang, H. Z. Wang and L. S. Li, J. Am. Chem. Soc., 2010, 132, 12778 CrossRef CAS PubMed.
- L. X. Yi and M. Y. Gao, Cryst. Growth Des., 2011, 11, 1109 CrossRef CAS.
- W. Bryks, E. Lupi, C. Ngo and A. R. Tao, J. Am. Chem. Soc., 2016, 138, 13717 CrossRef CAS PubMed.
- W. Y. Wu, S. Chakrabortty, C. K. L. Chang, A. Guchhait, M. Lin and Y. Chan, Chem. Mater., 2014, 26, 6120 CrossRef CAS.
- V. Lesnyak, C. George, A. Genovese, M. Prato, A. Casu, S. Ayyappan, A. Scarpellini and L. Manna, ACS Nano, 2014, 8, 8407 CrossRef CAS PubMed.
- Y. Liu, D. Q. Yin and M. T. Swihart, Chem. Mater., 2018, 30, 8089 CrossRef CAS.
- Z. Li, M. Saruyama, T. Asaka, Y. Tatetsu and T. Teranishi, Science, 2021, 373, 332 CrossRef CAS PubMed.
- L. De Trizio and L. Manna, Chem. Rev., 2016, 116, 10852 CrossRef CAS PubMed.
- L. J. Mu, F. D. Wang and W. E. Buhro, Chem. Mater., 2017, 29, 3686 CrossRef CAS.
- P. Ramasamy, M. Kim, H. S. Ra, J. Kim and J. S. Lee, Nanoscale, 2016, 8, 7906 RSC.
- A. H. Khan, S. Pal, A. Dalui, B. Pradhan, D. D. Sarma and S. Acharya, J. Mater. Chem. A, 2019, 7, 9782 RSC.
- Y. Liu, C. K. Lim, Z. Fu, D. Q. Yin and M. T. Swihart, Chem. Mater., 2019, 31, 5706 CrossRef CAS.
- J. L. Fenton, B. C. Steimle and R. E. Schaak, Inorg. Chem., 2019, 58, 672 CrossRef CAS PubMed.
- A. E. Powell, J. M. Hodges and R. E. Schaak, J. Am. Chem. Soc., 2016, 138, 471 CrossRef CAS PubMed.
- A. Wolf, T. Kodanek and D. Dorfs, Nanoscale, 2015, 7, 19519 RSC.
- D. Yoon, S. Yoo, K. S. Nam, H. Baik, K. Lee and Q. H. Park, Small, 2016, 12, 5728 CrossRef CAS PubMed.
- J. Park, J. Park, J. Lee, A. Oh, H. Baik and K. Lee, ACS Nano, 2018, 12, 7996 CrossRef CAS PubMed.
- T. Kim, J. Park, Y. Hong, A. Oh, H. Baik and K. Lee, ACS Nano, 2019, 13, 11834 CrossRef CAS PubMed.
- L. J. Mu, F. D. Wang, B. Sadtler, R. A. Loomis and W. E. Buhro, ACS Nano, 2015, 9, 7419 CrossRef CAS PubMed.
- Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711 CrossRef CAS PubMed.
- S. E. Moosavifard, S. Fani and M. Rahmanian, Chem. Commun., 2016, 52, 4517 RSC.
- S. Koh, W. D. Kim, W. K. Bae, Y. K. Lee and D. C. Lee, Chem. Mater., 2019, 31, 1990 CrossRef CAS.
- C. W. Dong, D. Yao, J. Y. Feng, T. T. Huang, X. Y. Hu, Z. N. Wu, Y. Liu, B. Yang and H. Zhang, Chem. Mater., 2016, 28, 9139 CrossRef CAS.
- R. Lesyuk, E. Klein, I. Yaremchuk and C. Klinke, Nanoscale, 2018, 10, 20640 RSC.
- Y. Wang, Y. X. Hu, Q. Zhang, J. P. Ge, Z. D. Lu, Y. B. Hou and Y. D. Yin, Inorg. Chem., 2010, 49, 6601 CrossRef CAS PubMed.
- Y. Xie, L. Carbone, C. Nobile, V. Grillo, S. D'Agostino, F. Della Sala, C. Giannini, D. Altamura, C. Oelsner, C. Kryschi and P. D. Cozzoli, ACS Nano, 2013, 7, 7352 CrossRef CAS PubMed.
- W. H. Li, A. Shavel, R. Guzman, J. Rubio-Garcia, C. Flox, J. D. Fan, D. Cadavid, M. Ibanez, J. Arbiol, J. R. Morante and A. Cabot, Chem. Commun., 2011, 47, 10332 RSC.
- M. X. Liu, X. Z. Xue, C. Ghosh, X. Liu, Y. Liu, E. P. Furlani, M. T. Swihart and P. N. Prasad, Chem. Mater., 2015, 27, 2584 CrossRef CAS.
- X. L. Wang, Y. J. Ke, H. Y. Pan, K. Ma, Q. Q. Xiao, D. Q. Yin, G. Wu and M. T. Swihart, ACS Catal., 2015, 5, 2534 CrossRef CAS.
- Z. T. Deng, M. Mansuripur and A. J. Muscat, J. Mater. Chem., 2009, 19, 6201 RSC.
- X. J. Wu, X. Huang, J. Q. Liu, H. Li, J. Yang, B. Li, W. Huang and H. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5083 CrossRef CAS PubMed.
- R. W. Lord, J. Fanghanel, C. F. Holder, I. Dabo and R. E. Schaak, Chem. Mater., 2020, 32, 10227 CrossRef CAS.
- C. M. Hessel, V. P. Pattani, M. Rasch, M. G. Panthani, B. Koo, J. W. Tunnell and B. A. Korgel, Nano Lett., 2011, 11, 2560 CrossRef CAS PubMed.
- J. Choi, N. Kang, H. Y. Yang, H. J. Kim and S. U. Son, Chem. Mater., 2010, 22, 3586 CrossRef CAS.
- H. B. Li, R. Brescia, M. Povia, M. Prato, G. Bertoni, L. Manna and I. Moreels, J. Am. Chem. Soc., 2013, 135, 12270 CrossRef CAS PubMed.
- E. H. Robinson, K. M. Dwyer, A. C. Koziel, A. Y. Nuriye and J. E. Macdonald, Nanoscale, 2020, 12, 23036 RSC.
- D. X. Zhu, A. W. Tang, Q. H. Kong, B. Zeng, C. H. Yang and F. Teng, J. Phys. Chem. C, 2017, 121, 15922 CrossRef CAS.
- J. Xu, X. Yang, Q. D. Yang, W. J. Zhang and C. S. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 16352 CrossRef CAS PubMed.
- D. Y. Xu, S. L. Shen, Y. J. Zhang, H. W. Gu and Q. B. Wang, Inorg. Chem., 2013, 52, 12958 CrossRef CAS PubMed.
- G. Gabka, P. Bujak, A. Ostrowski, W. Tomaszewski, W. Lisowski, J. W. Sobczak and A. Pron, Inorg. Chem., 2016, 55, 6660 CrossRef CAS PubMed.
- X. T. Lu, Z. B. Zhuang, Q. Peng and Y. D. Li, Chem. Commun., 2011, 47, 3141 RSC.
- H. Ren, Z. Li, Y. W. Sun, P. Gao, C. McCarthy, N. Liu, H. X. Xu and K. M. Ryan, Chem. Mater., 2020, 32, 7254 CrossRef CAS.
- G. Manna, R. Bose and N. Pradhan, Angew. Chem., Int. Ed., 2013, 52, 6762 CrossRef CAS PubMed.
- J. F. Liu, M. Meyns, T. Zhang, J. Arbiol, A. Cabot and A. Shavel, Chem. Mater., 2018, 30, 1799 CrossRef CAS.
- A. G. Rachkov and A. M. Schimpf, Chem. Mater., 2021, 33, 1394 CrossRef CAS.
- T.-H. Le, S. Lee, H. Jo, M. Kim, J. Lee, M. Chang and H. Yoon, ACS Appl. Nano Mater., 2021, 4, 7621 CrossRef CAS.
- J. Wang, X. C. Guo, Y. He, M. J. Jiang and R. Sun, RSC Adv., 2017, 7, 40249 RSC.
- W. Luc, X. B. Fu, J. J. Shi, J. J. Lv, M. Jouny, B. H. Ko, Y. B. Xu, Q. Tu, X. B. Hu, J. S. Wu, Q. Yue, Y. Y. Liu, F. Jiao and Y. J. Kang, Nat. Catal., 2019, 2, 423 CrossRef CAS.
- I. Pastoriza-Santos, A. Sanchez-Iglesias, B. Rodriguez-Gonzalez and L. M. Liz-Marzan, Small, 2009, 5, 440 CrossRef CAS PubMed.
- T. Balkan, H. Kucukkececi, H. Zarenezhad, S. Kaya and O. Metin, J. Alloys Compd., 2020, 831, 154787 CrossRef CAS.
- S. Singh, P. Liu, A. Singh, C. Coughlan, J. J. Wang, M. Lusi and K. M. Ryan, Chem. Mater., 2015, 27, 4742 CrossRef CAS.
- B. A. Tappan and R. L. Brutchey, ChemNanoMat, 2020, 6, 1567 CrossRef CAS.
- D. J. Chakrabarti and D. E. Laughlin, Bull. Alloy Phase Diagrams, 1983, 4, 254 CrossRef.
- Y. He, T. S. Zhang, X. Shi, S. H. Wei and L. D. Chen, NPG Asia Mater., 2015, 7, 210 CrossRef.
- C. Nethravathi, C. R. Rajamathi, M. Rajamathi, R. Maki, T. Mori, D. Golberg and Y. Bando, J. Mater. Chem. A, 2014, 2, 985 RSC.
- J. Xu, X. Yang, Q. D. Yang, X. Huang, Y. B. Tang, W. J. Zhang and C. S. Lee, Chem. – Asian J., 2015, 10, 1490 CrossRef CAS PubMed.
- K. M. Koskela, M. J. Strumolo and R. L. Brutchey, Trends Chem., 2021, 3, 1061 CrossRef.
- J. C. Sarker and G. Hogarth, Chem. Rev., 2021, 121, 6057 CrossRef CAS PubMed.
- A. F. Harper, M. L. Evans and A. J. Morris, Chem. Mater., 2020, 32, 6629 CrossRef CAS PubMed.
- Z. H. Guo, J. Z. Li, R. K. Pan, J. J. Cheng, R. Chen and T. C. He, Nanoscale, 2020, 12, 15560 RSC.
- Y. Dai, S. J. Liu and N. F. Zheng, J. Am. Chem. Soc., 2014, 136, 5583 CrossRef CAS PubMed.
- K. Tanabe, Mater. Lett., 2007, 61, 4573 CrossRef CAS.
- Y. N. Wijaya, J. Kim, W. M. Choi, S. H. Park and M. H. Kim, Nanoscale, 2017, 9, 11705 RSC.
- Z. Q. Shi, J. T. Tang, L. Chen, C. R. Yan, S. Tanvir, W. A. Anderson, R. M. Berry and K. C. Tam, J. Mater. Chem. B, 2015, 3, 603 RSC.
- H. Y. Zhu, S. Zhang, S. J. Guo, D. Su and S. H. Sun, J. Am. Chem. Soc., 2013, 135, 7130 CrossRef CAS PubMed.
- S. Thota, Y. C. Wang and J. Zhao, Mater. Chem. Front., 2018, 2, 1074 RSC.
- J. Monzo, Y. Malewski, R. Kortlever, F. J. Vidal-Iglesias, J. Solla-Gullon, M. T. M. Koper and P. Rodriguez, J. Mater. Chem. A, 2015, 3, 23690 RSC.
- J. Jiang, S. Yoon and L. Piao, CrystEngComm, 2020, 22, 4386 RSC.
- X. Q. Huang, S. H. Tang, X. L. Mu, Y. Dai, G. X. Chen, Z. Y. Zhou, F. X. Ruan, Z. L. Yang and N. F. Zheng, Nat. Nanotechnol., 2011, 6, 28 CrossRef CAS PubMed.
- K. W. Zhang, T. X. Song, C. Wang, H. M. You, B. Zou, S. Y. Guo, Y. K. Du and S. J. Li, Inorg. Chem., 2021, 60, 7527 CrossRef CAS PubMed.
- H. Y. Jin, C. X. Guo, X. Liu, J. L. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118, 6337 CrossRef CAS PubMed.
- Y. Zhang, C. C. Xing, Y. Liu, M. C. Spadaro, X. Wang, M. Y. Li, K. Xiao, T. Zhang, P. Guardia, K. H. Lim, A. O. Moghaddam, J. Llorca, J. Arbiol, M. Ibanez and A. Cabot, Nano Energy, 2021, 85, 105991 CrossRef CAS.
- Y. Liu, X. K. Zeng, X. Y. Hu, J. Hu and X. W. Zhang, J. Chem. Technol. Biotechnol., 2019, 94, 22 CrossRef CAS.
- D. H. Deng, K. S. Novoselov, Q. Fu, N. F. Zheng, Z. Q. Tian and X. H. Bao, Nat. Nanotechnol., 2016, 11, 218 CrossRef CAS PubMed.
- Z. X. Zhang, K. P. Zhao, T. R. Wei, P. F. Qiu, L. D. Chen and X. Shi, Energy Environ. Sci., 2020, 13, 3307 RSC.
- P. F. Qiu, X. Shi and L. D. Chen, Energy Storage Mater., 2016, 3, 85 CrossRef.
- J. Wu, Y. B. Chen, J. Q. Wu and K. Hippalgaonkar, Adv. Electron. Mater., 2018, 4, 1800248 CrossRef.
- Q. Wang, J. H. Li and J. J. Li, Phys. Chem. Chem. Phys., 2018, 20, 1460 RSC.
- B. Peng, H. Zhang, H. Z. Shao, K. Xu, G. Ni, J. Li, H. Y. Zhu and C. M. Soukoulis, J. Mater. Chem. A, 2018, 6, 2018 RSC.
- M. Q. Sun, N. Kreis, K. X. Chen, X. Q. Fu, S. Guo and H. Wang, Chem. Mater., 2021, 33, 8546 CrossRef CAS.
- R. M. Cordova-Castro, M. Casavola, M. van Schilfgaarde, A. V. Krasavin, M. A. Green, D. Richards and A. V. Zayats, ACS Nano, 2019, 13, 6550 CrossRef CAS PubMed.
- T. X. Wei, Y. F. Liu, W. J. Dong, Y. Zhang, C. Y. Huang, Y. Sun, X. Chen and N. Dai, ACS Appl. Mater. Interfaces, 2013, 5, 10473 CrossRef CAS PubMed.
- G. N. Liu, S. P. Qi, J. X. Chen, Y. B. Lou, Y. X. Zhao and C. Burda, Nano Lett., 2021, 21, 2610 CrossRef CAS PubMed.
- X. Liu and M. T. Swihart, Chem. Soc. Rev., 2014, 43, 3908 RSC.
- A. Comin and L. Manna, Chem. Soc. Rev., 2014, 43, 3957 RSC.
- J. A. Faucheaux, A. L. D. Stanton and P. K. Jain, J. Phys. Chem. Lett., 2014, 5, 976 CrossRef CAS PubMed.
- T. Sun, Y. J. Wang, W. Z. Yu, Y. S. Wang, Z. G. Dai, Z. K. Liu, B. N. Shivananju, Y. P. Zhang, K. Fu, B. Shabbir, W. L. Ma, S. J. Li and Q. L. Bao, Small, 2017, 13, 1701881 CrossRef PubMed.
- S. W. Hsu, K. On and A. R. Tao, J. Am. Chem. Soc., 2011, 133, 19072 CrossRef CAS PubMed.
- L. H. Chen, H. F. Hu, Y. Z. Chen, J. Gao and G. H. Li, Mater. Adv., 2021, 2, 907 RSC.
- Y. Liu, M. Liu and M. T. Swihart, J. Phys. Chem. C, 2017, 121, 13435 CrossRef CAS.
- C. Hu, W. H. Chen, Y. Xie, S. K. Verma, P. Destro, G. Zhan, X. Z. Chen, X. J. Zhao, P. J. Schuck, I. Kriegel and L. Manna, Nanoscale, 2018, 10, 2781 RSC.
- Y. Yao, A. Bhargava and R. D. Robinson, Chem. Mater., 2021, 33, 608 CrossRef CAS.
- W. D. Liu, L. Yang and Z. G. Chen, Nano Today, 2020, 35, 100938 CrossRef CAS.
- B. Gahtori, S. Bathula, K. Tyagi, M. Jayasimhadri, A. K. Srivastava, S. Singh, R. C. Budhani and A. Dhar, Nano Energy, 2015, 13, 36 CrossRef CAS.
- Y. Liu, Y. Zhang, S. Ortega, M. Ibanez, K. H. Lim, A. Grau-Carbonell, S. Marti-Sanchez, K. M. Ng, J. Arbiol, M. V. Kovalenko, D. Cadavid and A. Cabot, Nano Lett., 2018, 18, 2557 CrossRef CAS PubMed.
- S. Ortega, M. Ibanez, Y. Liu, Y. Zhang, M. V. Kovalenko, D. Cadavid and A. Cabot, Chem. Soc. Rev., 2017, 46, 3510 RSC.
- R. Freer and A. V. Powell, J. Mater. Chem. C, 2020, 8, 441 RSC.
- K. P. Zhao, P. F. Qiu, Q. F. Song, A. B. Blichfeld, E. Eikeland, D. D. Ren, B. H. Ge, B. B. Iversen, X. Shi and L. D. Chen, Mater. Today Phys., 2017, 1, 14 CrossRef.
- L. Yang, Z. G. Chen, G. Han, M. Hong, Y. C. Zou and J. Zou, Nano Energy, 2015, 16, 367 CrossRef CAS.
- W. W. Liao, L. Yang, J. Chen, D. L. Zhou, X. L. Qu, K. Zheng, G. Han, J. B. Zhou, M. Hong and Z. G. Chen, Chem. Eng. J., 2019, 371, 593 CrossRef CAS.
- Y. He, T. Day, T. S. Zhang, H. L. Liu, X. Shi, L. D. Chen and G. J. Snyder, Adv. Mater., 2014, 26, 3974 CrossRef CAS PubMed.
- J. B. Yu, T. W. Li, G. Nie, B. P. Zhang and Q. Sun, Nanoscale, 2019, 11, 10306 RSC.
- T. Mao, P. F. Qiu, P. Hu, X. L. Du, K. P. Zhao, T. R. Wei, J. Xiao, X. Shi and L. D. Chen, Adv. Sci., 2020, 7, 1901598 CrossRef CAS PubMed.
- M. Y. Li, Y. Liu, Y. Zhang, X. Han, T. Zhang, Y. Zuo, C. Y. Xie, K. Xiao, J. Arbiol, J. Llorca, M. Ibanez, J. F. Liu and A. Cabot, ACS Nano, 2021, 15, 4967 CrossRef CAS PubMed.
- X. L. Yu, D. Wang, J. L. Liu, Z. S. Luo, R. F. Du, L. M. Liu, G. J. Zhang, Y. H. Zhang and A. Cabot, J. Phys. Chem. C, 2016, 120, 24265 CrossRef CAS.
- Y. Liu, H. G. Zhang, P. K. Behara, X. Y. Wang, D. W. Zhu, S. Ding, S. P. Ganesh, M. Dupuis, G. Wu and M. T. Swihart, ACS Appl. Mater. Interfaces, 2018, 10, 42417 CrossRef CAS PubMed.
- X. Shao, T. Y. Zhang, B. Li, Y. Wu, X. Y. Ma, J. C. Wang and S. Jiang, CrystEngComm, 2020, 22, 678 RSC.
- H. Y. Zou, M. X. Gao, T. Yang, Q. L. Zeng, X. X. Yang, F. Liu, M. T. Swihart, N. Li and C. Z. Huang, Phys. Chem. Chem. Phys., 2017, 19, 6964 RSC.
- A. Dutta, S. K. Dutta, S. K. Mehetor, I. Mondal, U. Pal and N. Pradhan, Chem. Mater., 2016, 28, 1872 CrossRef CAS.
- S. L. Zhao, Y. Wang, Q. H. Zhang, Y. F. Li, L. Gu, Z. H. Dai, S. L. Liu, Y. Q. Lan, M. Han and J. C. Bao, Inorg. Chem. Front., 2016, 3, 1501 RSC.
- Q. Li, X. F. Wang, K. Tang, M. F. Wang, C. Wang and C. L. Yan, ACS Nano, 2017, 11, 12230 CrossRef CAS PubMed.
- Y. Liu, J. D. Wu, Y. H. Jin, W. Y. Zhen, Y. H. Wang, J. H. Liu, L. H. Jin, S. T. Zhang, Y. Zhao, S. Y. Song, Y. Yang and H. J. Zhang, Adv. Funct. Mater., 2019, 29, 1904678 CrossRef CAS.
- S. Vikulov, F. Di Stasio, L. Ceseracciu, P. L. Saldanha, A. Scarpellini, Z. Y. Dang, R. Krahne, L. Manna and V. Lesnyak, Adv. Funct. Mater., 2016, 26, 3670 CrossRef CAS.
- D. Yoon, H. Jin, S. Ryu, S. Park, H. Baik, S. J. Oh, S. Haam, C. Joo and K. Lee, CrystEngComm, 2015, 17, 4627 RSC.
- J. He, L. S. Ai, X. Liu, H. Huang, Y. B. Li, M. G. Zhang, Q. R. Zhao, X. G. Wang, W. Chen and H. S. Gu, J. Mater. Chem. B, 2018, 6, 1035 RSC.
- J. Just, C. Coughlan, S. Singh, H. Ren, O. Muller, P. Becker, T. Unold and K. M. Ryan, ACS Nano, 2021, 15, 6439 CrossRef CAS PubMed.
- V. K. Ravi, S. Saikia, S. Yadav, V. V. Nawale and A. Nag, ACS Energy Lett., 2020, 5, 1794 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |