
An extremely superhydrophobic and intrinsically stable Si/fluorocarbon energetic composite based on upright nano/submicron-sized Si wire arrays†
Abstract
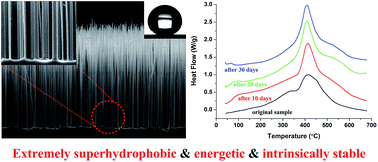
Si is chemically stable compared with many highly reactive metallic elements when used as a fuel for energetic formulations, and thus Si-based energetic composites can be intrinsically suitable for long-term storage. In this study upright nano/submicron-sized Si wire arrays are fabricated by a mask-free deep reactive ion etching method. A fluorocarbon layer is then sputter-deposited onto the Si wire arrays to form a Si/fluorocarbon energetic composite. The morphological and compositional information of the composites is characterized by various techniques such as scanning electron microscopy, X-ray photoelectron spectroscopy, Fourier transform infrared spectroscopy, and X-ray diffraction. The energetic property is studied by thermal analysis. Orderly Si wires are obtained by tuning the passivation/etching ratio in a processing cycle. As the total processing time increases, the number density of Si wires decreases while the length increases. The effects of power density and Ar flow rate on the chemical composition of the sputtered fluorocarbon are also investigated, but neither of them show appreciable impact within the ranges studied. The Si/fluorocarbon surface is so superhydrophobic that the water drops roll easily and the contact angles can hardly be measured. Si wires and fluorocarbon react exothermically and much gas products are generated. After a prolonged accelerated aging test, the Si/fluorocarbon energetic composites show almost no decay of the energy density. Si wire arrays can also be used as scaffolds to prepare other highly superhydrophobic energetic composites.
 Please wait while we load your content...
Please wait while we load your content...

