
A low-cost and one-step synthesis of a novel hierarchically porous Fe3O4/C composite with exceptional porosity and superior Li+ storage performance†
Luyu Yang,
Wei Liu*,
Huanlei Wang,
Shuang Liu,
Jifei Wang and
Jiaxin Chen
Institute of Materials Science and Engineering, Ocean University of China, Qingdao, 266100 China. E-mail: weiliu@ouc.edu.cn; Tel: +86 532 66781906
First published on 23rd November 2015
Abstract
A hierarchical pore structure is believed to be an excellent architecture for metal oxide anode materials of lithium-ion batteries (LIBs). However, the difficulty and complication in synthesis limit their further application. In this work, a novel Fe3O4/C composite with a hierarchical pore carbon (HPC) network has been synthesized simply by one-step pyrolysis synthesis using ferrous gluconate as the precursor. This hierarchical porous framework derived from a loose assembly of the intersecting porous carbon rods presents a wide range of pore sizes and a rather large surface area (>226 cm2 g−1). Embedded with well-crystalline Fe3O4 particles, the resulting Fe3O4@HPC composite exhibits a high capacity of 1112 mA h g−1 at the end of the 100th cycle and an enhanced rate performance of above 600 mA h g−1 at a high current density of 2000 mA g−1. This might derive from the unique hierarchical pore system with a high overall porosity, which can not only facilitate the electrolyte diffusion but also alleviate severe volume variation efficiently in lithium ion insertion/extraction. More importantly, this provides us with a successful example to fabricate advanced anode materials simply by one-step pyrolysis of cheap organic–inorganic hybrids at low-cost.
1. Introduction
To meet the demands of next generation electric devices including plug-in hybrid electric vehicles and so on, the preparation of high performance electrode materials for Li-ion batteries becomes more and more urgent. Recently, as a new class of negative electrode materials, metal oxides have attracted much attention because of their higher theoretical capacity (500–1000 mA h g−1) compared with commercialized graphite (372 mA h g−1).1–6 However, the intrinsic disadvantages of severe volume variation during cycling and low electrical conductivity make them suffer from poor capacity retention upon cycling and/or poor rate capability, which limits their application as commercial anode materials.7–16 Among various methods to conquer these problems, fabricating porous architecture is a promising strategy owing to its advantages of providing the buffer room for volume changes and increasing the contact area of active materials with electrolyte.17–22 In these single porous structures, the voids are utilized generally as both the buffer room for volume expansion and the diffusing channels for electrolytes. Nevertheless, this inevitably causes an undesirable phenomenon that the diffusing channels of electrolytes are always narrowed by the volume expansion of active materials during Li+ insert process, and thereby undermine their electrochemical property greatly. Fortunately, the hierarchical porous structure provides a feasible way to solve this problem by providing independent space for active materials and electrolytes, respectively, avoiding their spatial competition efficiently. Moreover, their larger pore volume and surface area also result in a better accessibility of electrolyte and active materials, leading to higher storage capacity. Therefore, as a kind of ideal architecture of anode materials for LIBs, the hierarchically porous networks attract fast-growing attention in recent years.23–30 Nevertheless, the preparation of hierarchical pore structures for anode material is still limited due to the difficulty and complication of synthetic process, so searching for a facile and green method for preparation is an urgent matter.In recent years, an immense upsurge of interest towards the utilization of MOFs as precursors to prepare novel porous structures appears.31–35 Particularly, several hierarchical porous structures have been fabricated successfully in this way. For example, the hierarchical porous carbon sponges were prepared from MOFs and further combined with Se to be used as anode materials of Li–Se cells;36 a hierarchical mesoporous Fe2O3/carbon nanocomposites were synthesized by the pyrolysis of MIL-100(Fe) as cathode catalyst in Li–O2 batteries;37 MOF-derived hierarchical porous carbon with a high overall porosity exhibited exceptional H2 storage capacities.38 On the one hand, MOF templates often have porous channel frameworks, which can maintain the porous framework after pyrolysis process. On the other hand, mesopores or micropores can be easily formed during the carbonization and graphitization process of organic phase. Based on the two advantages of this method mentioned above, lots of novel materials with various hierarchical pore structures have been synthesized from MOFs. In situ pyrolysis of MOFs has become a feasible way to produce 3D hierarchical porous MOx/C composite materials. But most of the precursors introduced are expensive and the synthetic processes are also complex, time consuming, low-yield and highly toxic. Despite intensive efforts exerted to explore cheap and green candidates, the suitable MOFs with low price and low toxicity are still rare. Very recently, our group reported a porous Fe3O4/C composite with a simple organic–inorganic layered hybrid as templates, exhibiting an enhanced electrochemical property.39 This successful example wakes us that some simple and cheap hybrids can also be utilized to fabricate anode materials with novel architectures, which thereby inspires us make more attempts in this aspect.
Herein, we introduce a simple organic–inorganic hybrid, ferrous gluconate, as the precursor to prepare high-performance anode materials. In this work, the commercialized ferrous gluconate has been directly used as template without further treatment. Only by one-step pyrolysis process, a novel Fe3O4/C composite with complex hierarchically porous carbon (HPC) framework and a large surface area has been elaborately fabricated. As expected, the as-obtained composites demonstrate an excellent electrochemical performance in terms of specific capacity, rate performance and long-term cycling stability, which can be served as a kind of potential commercial anode materials. Moreover, considering the merits of ferrous gluconate including low-cost, accessibility and non-toxic, this work also provides a feasible avenue to fabricate Fe3O4@HPC anode materials toward practical industrial manufacture in large-scale.
2. Experimental
Materials synthesis
Ferrous gluconate was purchased from commercial sources (Aladdin, 99.9%) and used as precursor without further treatment. The precursor was grounded by agate mortar, transferred into the tube furnace and calcined at 750 °C for 2 hours under an Ar atmosphere. Then the obtained powder was washed and dried at 80 °C for 24 h to obtain the Fe3O4@HPC composites.Materials characterization
The power X-ray diffraction (XRD) measurement was carried out on a Shimadzu-6000 X-ray diffractometer with Cu Kα radiation operated at 40 kV and 50 mA. The morphology of the as-prepared samples were investigated by scanning electron microscope (SEM, S-4800N) and transmission electron microscope (TEM, JEM-2010). High-resolution transmission electron microscope (HRTEM, JEM-2010) was carried out to determine the detailed microstructures. The specific surface areas and the pore size distributions were measured by the Brunauer–Emmett–Teller nitrogen adsorption–desorption measurement (Belsorp-max surface area detecting instrument) and Barret–Joyner–Halenda (BJH) gas adsorption method, respectively. Raman spectrum was recorded on a Renishaw Invia Raman spectrometer. To obtain the carbon content of the as prepared samples, thermal gravimetric analysis (TGA) was performed on an NETZSCH TG 209 apparatus with a heating rate of 5 °C per minute from room temperature to 800 °C in air atmosphere.Electrochemical measurement
The electrochemical behavior was tested using the CR2032 coin cells. Pure lithium foil was used as both the counter and the reference electrode. The working electrode was fabricated by mixing the active powder, conductive carbon (acetylene black) and binder (polyvinylidene fluoride, PVDF) in a ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10, followed by dispersing them in N-methyl-2-pyrrolidone. The resultant slurry was then pasted on Cu foils. The electrolyte was prepared by dissolving 1 M LiPF6 in a 50:50 (w/w) mixture of ethylene carbonate (EC) and diethyl carbonate (DMC). Celgard 2400 microporous polypropylene membrane was used as separator. On a Zahner electrochemical workstation, cycle voltammetry (CV) was performed at the range of 0.005–3.0 V with the scan rate of 0.1 mV s−1. The electrochemical impedance spectroscopy (EIS) tests were carried out in the frequency ranging from 100 kHz to 0.01 Hz with an AC perturbation of 5 mV. The discharge–charge cycle tests were measured at various current densities in the voltage range of 0.05–3.0 V on a battery testing system (LAND CT 2001A, China).
:20:10, followed by dispersing them in N-methyl-2-pyrrolidone. The resultant slurry was then pasted on Cu foils. The electrolyte was prepared by dissolving 1 M LiPF6 in a 50:50 (w/w) mixture of ethylene carbonate (EC) and diethyl carbonate (DMC). Celgard 2400 microporous polypropylene membrane was used as separator. On a Zahner electrochemical workstation, cycle voltammetry (CV) was performed at the range of 0.005–3.0 V with the scan rate of 0.1 mV s−1. The electrochemical impedance spectroscopy (EIS) tests were carried out in the frequency ranging from 100 kHz to 0.01 Hz with an AC perturbation of 5 mV. The discharge–charge cycle tests were measured at various current densities in the voltage range of 0.05–3.0 V on a battery testing system (LAND CT 2001A, China).
3. Results and discussion
The synthetic procedure based on the simple pyrolysis method is schematically illustrated in Fig. 1. As shown in Fig. 1a, each ferrous gluconate molecule with low molecular mass contains one ferrous ion in octahedral coordination surrounded by two long-chain gluconates and two water molecules. In the crystalline state, the strong hydrogen bonding interaction promote these molecules to form a uniform 3D arrangement. Meanwhile, due to the low molting point (Fig. S1†), ferrous gluconate can be melted to a liquid phase with high viscosity at the early stage of pyrolysis process. The subsequent gases (H2O, CO2, CO, etc.) produced through the dehydration and decomposition of the precursors will be trapped in this viscous liquid, resulting in a “melting foaming” phenomenon as illustrated by Fig. 1b. As the temperature decreases, the liquid phase is solidified gradually to rigid framework and then the bubbles inside are also saved in the form of voids (Fig. 1c). In this sense, these bubbles can be considered as templates to get the initial 3D porous framework. Furthermore, the synergistic effect of carbonization and graphitization of the organic phase and the oxidation reaction of ferrous mass creates the mesopores or micropores structure with nanosized Fe3O4 particles embedded in (Fig. 1c right). As a result, the final hierarchically porous structure are successfully fabricated (Fig. 1c left). | ||
| Fig. 1 The proposed formation mechanism of Fe3O4@HPC composites. | ||
As shown in Fig. 2a, the X-ray diffraction pattern of the as-prepared sample match well with the standard card of Fe3O4. Meanwhile, the existence of graphite can be proved by the wide peak centered at about 23°. Thermogravimetric analysis was conducted to investigate the carbon content of the Fe3O4/C composite. As shown in Fig. 2b, the weight loss because of carbon oxidation was observed between 365 °C and 630 °C. Based on the residual weight of Fe2O3, the carbon content of Fe3O4/C can be evaluated to be 36.1%. Furthermore, Raman measurement was carried out to analyze the structure of carbon. Two peaks at 1581.28 and 1339.61 cm−1 corresponding to the G and D band were observed in Fig. 2c. The integrated intensity ratio (IG/ID) of 0.85 indicates a relative high graphitization degree of carbon, which is favorable to the electrical conductivity.
 | ||
| Fig. 2 (a) XRD pattern of the as-obtained Fe3O4@HPC composites and the standard pattern of Fe3O4; (b) TGA curve of the Fe3O4@HPC composites and (c) Raman spectrum of the Fe3O4@HPC composites. | ||
Compared with the crystalline powder of ferrous gluconate, the calcined products are looser and higher porosity with the change of the color from yellow-green to black. The morphology of the calcined sample is characterized by SEM. As shown in Fig. 3a, the calcined particles can retain their original shape except for a large amount of macropores on their crude surface. Obviously, differing from carbon networks in previous literatures, the carbon framework here can be depicted as the loose assembly of the intersecting carbon rods with the diameter of ca. 500 nm (Fig. 3b). Like tree branches, these rods stretch and interlink each other to form a fantastic 3D carbon framework with full of interconnected voids. It is worth noting that the surface of these carbon rods is not compact but has lots of irregular pores with a 200–300 nm gap in width (Fig. 3c). Such pores extend into the rods and create a sponge-like porous structure wrapped by graphene-like carbon sheets, which can also be confirmed by the cross-section SEM image (Fig. 3d). This porous structure can still be observed in the SEM image of the prepared electrode film on the copper foil, which demonstrates the good rigidity of sponge-like porous structure (Fig. S2†). As a consequence, the combination of intersecting voids surround by carbon rods and the sponge-like inter porous structure inside rods creates an unique hierarchical porous structure. Interestingly, well-crystalline Fe3O4 particles with octahedron or cuboid shape are all existed in the carbon rods, and not packed in interconnected voids (Fig. S3†), thus providing the relatively independent diffusing space for electrolytes. Moreover, Fe3O4 particles are embedded in the sponge-like carbon of the rods and spontaneously separated by porous carbon layer (Fig. 3e, f and S4†). Like air mattresses, evidently, this kind of porous carbon among Fe3O4 particles can not only improve the electron conductivity, facilitate the penetrating of electrolyte but also efficiently relieve the mechanical strains during cycling by providing enough buffer room, which is an ultrastable framework for the delithiation–lithiation process.
 | ||
| Fig. 3 (a–f) SEM images of the synthesized Fe3O4@HPC composites at various magnification, displaying the unique hierarchical porous structure and their close contact with Fe3O4 particles. | ||
TEM analysis further reveals the internal microstructure and morphology of Fe3O4@HPC composites. The relative uniform distribution of Fe3O4 nanoparticles in porous carbon matrix can be clearly demonstrated in the low-magnification TEM images of the products (Fig. S5†). In the magnifying TEM image, the typical capsule-like morphology in the carbon rods can be clearly observed rather than the graphene-like sheets described in SEM analysis (Fig. 4a). When it comes to the isolated carbon capsules, we can clearly see that most of the capsules are so open that they appear to be hollow hemispherical structure and the edges of these capsules are curled in some extent with the thickness of less than 10 nm (Fig. 4b). The numerous carbon capsules with various sizes aggregate together and form the sponge-like carbon network of the rods. The Fe3O4 particles are not encapsulated but wrapped around by these carbon capsules as depicted in Fig. 4c, which can lead to a close combination between the carbon network and Fe3O4 particles. The high-magnification TEM (HRTEM) image (Fig. 4d) further displays the tight contact between Fe3O4 particles and carbon capsules. Furthermore, the clear shell lattice fringes with d-spacing of 0.34 nm of carbon capsules present the high degree of graphitization, which agrees well with the Raman result. Besides, Fe3O4 particles are also highly crystallized and the lattice fringes with interplanar spacing of 0.48 nm are consistent with the (111) plane of Fe3O4, which is also confirmed by XRD measurement. Fig. 4e shows the HRTEM image taken from the edge of an individual Fe3O4 nanoparticle, displaying that Fe3O4 particles are well wrapped by an ultrathin carbon shell with a thickness of about 5 nm. Evidently, this unique aggregation of the carbon capsules and their close contact with Fe3O4 particles facilitate the electron conduction and the structural stability during the charge–discharge process, which endows the composites with an excellent electrochemical property.
 | ||
| Fig. 4 (a–c) TEM images of Fe3O4@HPC composites and (d and e) HRTEM images, displaying the close contact between Fe3O4 particles and carbon capsules. | ||
In order to investigate the porous characteristics and Brunauer–Emmett–Teller (BET) specific surface area of the as-obtained Fe3O4@HPC composites, N2 adsorption/desorption isotherms were carried out. As shown in Fig. 5a, the isotherm profile of the sample can be categorized as type IV with a large hysteresis loop observed at a relative pressure of p/p0 ≈ 0.448, implying a large number of mesopores and a narrow pore diameter distribution. In addition, the N2 absorption can be observed at a relatively high pressure of 0.984 due to the presence of macropores, which is corresponding well with the hierarchical pore structure observed above. As expected, this hierarchically porous structure exhibits a large BET surface area of 226.3 m2 g−1, which is much higher than most of the MOx/C composites reported previously.40–44 Based on the BET data, the specific pore volume of Fe3O4@HPC is calculated to be 0.352 cm3 g−1. As shown in Fig. 5b, the pores of the as-obtained composites show a broad size distribution in the range from 1 nm to 1000 nm besides a sharp peak at 38–43 nm (Fig. 5b inset), suggesting a complex meso/macroporous structure in the structure. With respect to the hierarchical pore structure, these mesopores may come from the voids of carbon capsules, while the macropores arise from the micron-size channels constructed by carbon rods as depicted in above SEM and TEM images.
 | ||
| Fig. 5 (a) N2 adsorption–desorption isotherms of the Fe3O4@HPC composites and (b) the pore-size distribution calculated from the desorption branch (inset is the local enlarged image). | ||
The electrochemical performance of the as-prepared Fe3O4@HPC composite materials is evaluated by standard coin-cells assembled using a Li-metal circular foil as the counter and reference electrode for Fe3O4/Li half-cell. Fig. 6a shows the first five cyclic voltammogram (CV) curves of the Fe3O4@HPC composite electrode at room temperature between 0.005 and 3.0 V at a scan rate of 0.1 mV s−1. It is clear that the CV curve of the first cycle is quite different from those of subsequent cycles, especially for the discharge branch. As plotted, two well-defined peaks are observed at 0.90 and 0.71 V because of the two-step electrochemical reduction of Fe3O4 (Fe3O4 + xLi+ + xe− → LixFe3O4; LixFe3O4 + (8 − x)Li+ + (8 − x)e− → 4Li2O + 3Fe0), as well as side reactions between electrode and electrolyte.45,46 In the anodic sweep, the peeks observed at the range of 1.64 V and 1.84 V correspond to the two-step reversible reaction from Fe0 to Fe2+ and Fe2+ to Fe3+. From the second cycle, the cathodic peak at 0.73 V disappears while the other reduction peaks transfer to the higher value of 1.02 V, and then keep stable at 0.92 V since the third cycle. In addition, a shift of two oxidation peaks from 1.64 V to 1.70 V and from 1.84 V to 2.02 V can be observed, respectively, owing to the occurrence of an irreversible phase transformation during the lithiation/delithiation process in the first circle. After the second cycle, the voltage current curves almost overlap, demonstrating the good reversibility of the electrochemical reaction.
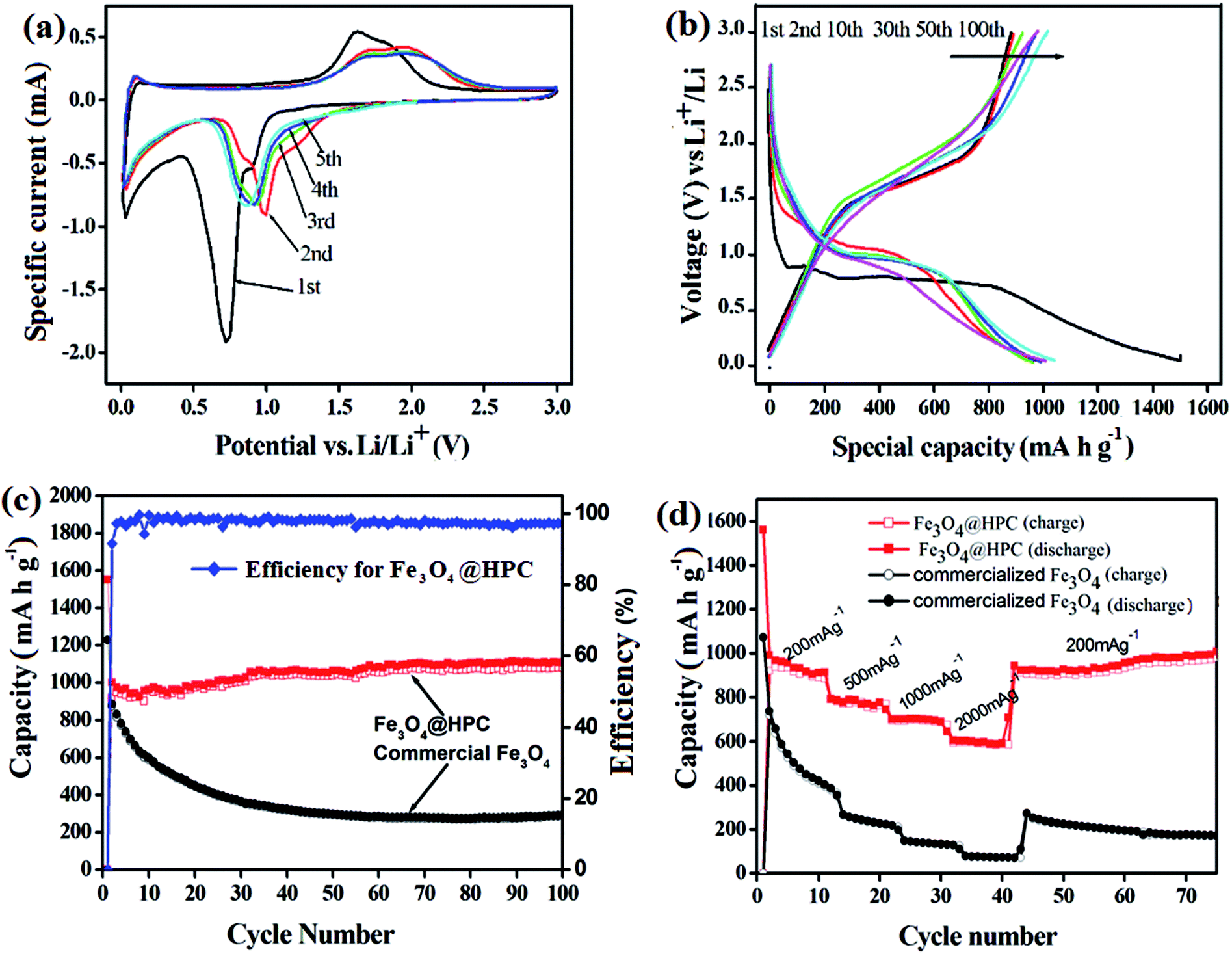 | ||
| Fig. 6 (a) Cyclic voltammograms of the as-prepared samples; (b) the charge–discharge profiles of the electrode at a current density of 100 mA g−1; (c) the cycling performance of the Fe3O4@HPC electrode compared with that of pure Fe3O4 electrode; (d) rate performance of the Fe3O4@HPC composites and commercial Fe3O4 powder at different current densities. | ||
Fig. 6b shows the typical charge (delithiation) and discharge (lithiation) voltage profile of Fe3O4@HPC composites at a current density of 100 mA g−1. The initial discharge and charge capacities are 1550 and 915 mA h g−1 with the coulombic efficiency of 59%. Such high capacity losses may be ascribed to the inevitable formation of the SEI layer and the decomposition of the electrolyte, which is a common phenomenon in the Fe-based anode materials. Interestingly, the capacity exhibits a gradual increase from 941 mA h g−1 (5th cycle) to 1112 mA h g−1 (100th cycle), accompanied by the increase of the coulombic efficiency from 59% to over 97% (Fig. 6c). As a comparison, pure commercialized Fe3O4 also has been investigated under the same conditions. Clearly, the as-prepared Fe3O4@HPC electrode exhibits better cycle retention and higher reversible capacity than pure Fe3O4 electrode after 100 cycles, revealing the excellent electrochemical lithium storage performance. Fig. 6d depicts the rate performance of Fe3O4@HPC composites and pure Fe3O4 sample with the charge/discharge current densities increasing from 200 to 2000 mA g−1, respectively. It can be seen clearly that Fe3O4@HPC composites deliver the high and stable capacities at different current densities except for a slow capacities drops with the increase of current density. Even at a high current density of 2000 mA g−1, the as-prepared Fe3O4@HPC composites can still maintain a stable reversible capacity of above 600 mA h g−1, which is about 65% of the initial reversible capacity. When returns to the initial rate of 200 mA g−1, the capacity can recover quickly to 940 mA h g−1 even after the high-rate measurement. It is noticed that the capacity still displays an increasing trend, indicating the remarkable dynamic performance of Fe3O4@HPC samples. Nevertheless, at the same current density, the capacities of pure Fe3O4 sample exhibit noticeably poor rate performance.
The above electrochemical comparison between Fe3O4@HPC composites and bare Fe3O4 fully demonstrates the great advantages of this hierarchical pore structure in improving electrochemical properties as anode materials. The main contribution for this superior performance may be derived from the unique hierarchical pore structure providing independent diffusing space for electrolyte and buffer room for volume expansion during cycling, respectively. That is to say, interconnected macropores surround by carbon rods behave as efficient diffusing channels for electrolyte to promote the transfer of conductive ions, while mesopores distributed around Fe3O4 particles in the carbon rods not only serve as absorbers for electrolyte to ensure sufficient contact between the active materials and electrolyte, but also act as buffered spaces to cushion large volume changes during cycling.
More interestingly, even though the Fe3O4 particles are disintegrated into small fragments in cycling, this 3D hierarchical carbon network can grasp and fix them, which thus not only avoid the exfoliation of the fragments from electrodes but also enlarge the contact surface between active materials and electrolytes. This is also the reason of the gradual capacity increase of the materials with cycles in this case. As a consequence, the as-prepared materials are endowed with an ideal structure for the fully reaction of active material and an ultrastable electrochemical environment for the delithiation–lithiation process. Moreover, the integrate carbon networks consisted of well graphitized opened carbon capsules and their close contact with Fe3O4 lead to an enhanced electron conductivity. The electrochemical impedance spectra (EIS) of Fe3O4@HPC and bare Fe3O4 electrodes were investigated in Fig. S6.† The Fe3O4@HPC composites exhibit the lower equivalent series resistance (1.2 Ω) and the smaller diameter of the semicircle (19.2 Ω) in the high-medium frequency region than those of bare Fe3O4 particles (3.8 Ω and 98.5 Ω), demonstrating the lower contact and charge-transfer resistances of Fe3O4@HPC composites, which is beneficial to achieve the excellent electrochemical performance of Fe3O4@HPC composites, especially the high rate performance at high current density.
4. Conclusions
In summary, we describe a facile method to prepare a novel Fe3O4@HPC composite with a fantastic hierarchical porous structure using ferrous gluconate as the precursors without complicated process and environmental burden. Microstructural analysis reveals that Fe3O4@HPC has a remarkable hierarchical porous framework with macropores deriving from the assembly of intersecting carbon rods and mesopores situated in the sponge-like carbon network inside rods. Embedded with well-crystalline Fe3O4 nanoparticles, this hierarchical pore carbon structure presents a superior capacity retention and excellent rate capability as anode materials for LIBs. More importantly, this unique hierarchical porous architecture can automatically form only through one-step pyrolysis process using the simple and cheap ferrous gluconate as the precursors, which provides a low-cost avenue to fabricate high-performance Fe3O4@HPC anode materials for LIBs in large-scale. In particular, this work is the first successful example to fabricate hierarchically porous architecture at the “melting foaming” effect of organic–inorganic hybrids, which could be applied readily in other material systems for exploring advanced anode materials in large-scale.Acknowledgements
This work was supported by the national natural science foundation of China (51202230, 21271161, 21471139), the Program for New Century Excellent Talents in University (NCET-13-0530) and Shandong Province Outstanding Youth Scientist Foundation Plan (No. BS2014CL024).References
- C. Wang, D. Higgins, F. F. Wang, D. Li, R. Q. Liu, G. F. Xia, N. Li, Q. Li, H. Xu and G. Wu, Nano Energy, 2014, 9, 334 CrossRef CAS.
- W. Y. Li, D. Yoon, J. Hwang, W. Chang and J. Kim, J. Power Sources, 2015, 293, 1024 CrossRef CAS.
- J. F. Yu, L. Zhu, C. Fan, C. Zan, L. Hu, S. H. Yang, Q. Zhang, W. C. Zhu, L. Shi and F. Wei, Particuology, 2015, 22, 89 CrossRef CAS.
- S. E. Kim, K. W. Kim, S. W. Lee, S. O. Kim, J. S. Kim and J. K. Lee, Curr. Appl. Phys., 2013, 13, 1923 CrossRef.
- P. P. Lv, H. L. Zhao, Z. P. Zeng, C. H. Gao, X. Liu and T. H. Zhang, Appl. Surf. Sci., 2015, 329, 301 CrossRef CAS.
- Z. L. Hu and H. D. Liu, Ceram. Int., 2015, 41, 8257 CrossRef CAS.
- Y. Z. Wan, Z. W. Yang, G. Y. Xiong, R. S. Guo, Z. Liu and H. L. Luo, J. Power Sources, 2015, 294, 414 CrossRef CAS.
- G. X. Pan, X. H. Xia, F. Cao, J. Chen and Y. J. Zhang, J. Power Sources, 2015, 293, 585 CrossRef CAS.
- Y. T. Xu, Y. Guo, C. Li, X. Y. Zhou, M. C. Tucker, X. Z. Fu, R. Sun and C. P. Wong, Nano Energy, 2015, 11, 38 CrossRef CAS.
- M. Lübke, I. Johnson, N. M. Makwana, D. Brett, P. Shearing, Z. L. Liu and J. A. Darr, J. Power Sources, 2015, 294, 94 CrossRef.
- J. W. Qin, Q. Zhang, Z. Y. Cao, X. Li, C. W. Hu and B. Q. Wei, Nano Energy, 2013, 2, 733 CrossRef CAS.
- X. Y. Chen, B. H. Liu and Z. P. Li, Solid State Ionics, 2014, 261, 45 CrossRef CAS.
- Y. Ding, J. Du, L. G. Guo, H. B. Zhou, H. P. Yang and F. Wang, Electrochim. Acta, 2015, 170, 9 CrossRef CAS.
- S. Khamlich, Z. Y. Nuru, A. Bello, M. Fabiane, J. K. Dangbegnon, N. Manyala and M. Maaza, J. Alloys Compd., 2015, 637, 219 CrossRef CAS.
- Y. Han, L. Dong, J. M. Feng, D. J. Li, X. F. Li and S. X. Liu, Electrochim. Acta, 2015, 167, 246 CrossRef CAS.
- Z. Y. Fan, J. Liang, W. Yu, S. J. Ding, S. D. Cheng, G. Yang, Y. L. Wang, Y. X. Xi, K. Xi and R. V. Kumar, Nano Energy, 2015, 16, 152 CrossRef CAS.
- G. H. Qin, H. J. Zhang and C. Y. Wang, J. Power Sources, 2014, 272, 491 CrossRef CAS.
- T. R. Penki, S. Shivakumara, M. Minakshi and N. Munichandraiah, Electrochim. Acta, 2015, 167, 330 CrossRef CAS.
- X. N. Shang, X. W. Li, H. W. Yue, S. Xue, Z. J. Liu, X. Y. Hou and D. Y. He, Mater. Lett., 2015, 157, 7 CrossRef CAS.
- D. Lei, B. H. Qu, H. T. Lin and T. H. Wang, Ceram. Int., 2015, 41, 10308 CrossRef CAS.
- R. Guo, W. Yue, Y. An, Y. Ren and X. Yan, Electrochim. Acta, 2014, 135, 161 CrossRef CAS.
- X. N. Shang, X. W. Li, H. W. Yue, S. Xue, Z. J. Liu, X. Y. Hou and D. Y. He, Mater. Lett., 2015, 157, 7 CrossRef CAS.
- P. P. Lv, H. L. Zhao, Z. P. Zeng, C. H. Gao, X. Liu and T. H. Zhang, Appl. Surf. Sci., 2015, 329, 301 CrossRef CAS.
- H. Y. Yue, Q. X. Wang, Z. P. Shi, C. Ma, Y. M. Ding, N. N. Huo, J. Zhang and S. T. Yang, Electrochim. Acta, 2015, 180, 622 CrossRef CAS.
- R. Guo, W. B. Yue, Y. Ren and W. Z. Zhou, Mater. Res. Bull., 2016, 73, 102 CrossRef CAS.
- G. Y. Zhao, Z. M. Xu, L. Zhang and K. N. Sun, Electrochim. Acta, 2013, 114, 251 CrossRef CAS.
- Y. S. Yun and H.-J. Jin, Mater. Lett., 2013, 108, 311 CrossRef CAS.
- S. Yuan, D. L. Ma, S. Wang, Y. B. Liu, X. H. Yang and Z. Y. Cao, Mater. Lett., 2015, 145, 104 CrossRef CAS.
- Y. Cai, H. E. Wang, J. Jin, S. Z. Huang, Y. Yu, Y. Li, S. P. Feng and B. L. Su, Chem. Eng. J., 2015, 281, 844 CrossRef CAS.
- S. K. Park, H. J. Lee, M. H. Lee and H. S. Park, Chem. Eng. J., 2015, 281, 724 CrossRef CAS.
- S. X. Bao, N. Yan, X. H. Shi, R. Li and Q. W. Chen, Appl. Catal., A, 2014, 487, 189 CrossRef CAS.
- H. Y. Niu, S. L. Liu, Y. Q. Cai, F. C. Wu and X. L. Zhao, Microporous Mesoporous Mater., 2016, 219, 48 CrossRef CAS.
- Y. H. Song, L. Zuo, S. H. Chen, J. F. Wu, H. Q. Hou and L. Wang, Electrochim. Acta, 2015, 173, 588 CrossRef CAS.
- W. J. Meng, W. Chen, L. Zhao, Y. Huang, M. S. Zhu, Y. Huang, Y. Q. Fu, F. X. Geng, J. Yu, X. F. Chen and C. Y. Zhi, Nano Energy, 2014, 8, 133 CrossRef CAS.
- X. Yang, Y. B. Tang, X. Huang, H. T. Xue, W. P. Kang, W. Y. Li, T. W. Ng and C. S. Lee, J. Power Sources, 2015, 284, 109 CrossRef CAS.
- Z. Q. Li and L. W. Yin, Nanoscale, 2015, 7, 9597 RSC.
- W. Chen, Z. Zhang, W. Z. Bao, Y. Q. Lai, J. Li, Y. Q. Gan and J. J. Wang, Electrochim. Acta, 2014, 134, 293 CrossRef CAS.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464 CrossRef CAS.
- L. Zhao, W. Liu, S. Liu, J. F. Wang, H. L. Wang and J. X. Chen, J. Mater. Chem. A, 2015, 3, 14210 Search PubMed.
- Q. H. Tian, Y. Tian, Z. X. Zhang, L. Yang and S. I. Hirano, J. Power Sources, 2015, 291, 173 CrossRef CAS.
- W. W. Yuan, J. Zhang, D. Xie, Z. M. Dong, Q. M. Su and G. H. Du, Electrochim. Acta, 2013, 108, 506 CrossRef CAS.
- Z. L. Xiu, X. P. Hao, Y. Z. Wu, Q. F. Lu and S. W. Liu, J. Power Sources, 2015, 287, 334 CrossRef CAS.
- H. Luo, K. Huang, B. Sun and J. X. Zhong, Electrochim. Acta, 2014, 149, 11 CrossRef.
- L. Fan, Y. C. Zhu, J. J. Zhang, J. W. Liang, L. L. Wang, D. H. Wei, X. N. Li and Y. T. Qian, Electrochim. Acta, 2014, 121, 21 CrossRef CAS.
- J. Z. Wang, C. Zhong, D. Wexler, N. H. Idris, Z. X. Wang, L. Q. Chen and H. K. Liu, Chem.–Eur. J., 2011, 17, 661 CrossRef CAS PubMed.
- L. Wang, Y. Yu, P. C. Chen, D. W. Zhang and C. H. Chen, J. Power Sources, 2008, 83, 717 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra24166a |
| This journal is © The Royal Society of Chemistry 2015 |