DOI:
10.1039/C5RA23585E
(Paper)
RSC Adv., 2015,
5, 103713-103721
Synthesis and properties of novel poly(ethylene succinate-co-decamethylene succinate) copolymers
Received
9th November 2015
, Accepted 26th November 2015
First published on 30th November 2015
Abstract
In this work, a series of novel poly(ethylene succinate-co-decamethylene succinate) (PEDS) copolymers with different decamethylene succinate (DS) contents and their homopolymer poly(ethylene succinate) (PES) were synthesized by a two-stage melt polycondensation method. The thermal stability, crystal structure, crystallization behavior, mechanical properties, and hydrolytic degradation of PEDS were investigated with various techniques and compared with those of PES. The PEDS copolyesters have the similar thermal stability and crystal structure as PES, but the crystallinity values of PEDS are smaller than that of PES. The values of the glass transition temperature, melting temperature, and melt crystallization peak temperature of PEDS decrease apparently, relative to PES. The elongation at break values of PEDS were improved significantly with the presence of the DS units, while the yield strength and Young's modulus values become smaller compared with those of PES. The hydrolytic degradation rates of PEDS are slower than that of PES, regardless of the DS contents. When the DS content increases to 18 mol%, the sample has a faster hydrolytic degradation rate than other copolyesters.
Introduction
Recently, more and more researchers have focused on the fundamental study and practical application of some aliphatic polyesters, such as poly(butylene succinate) (PBS), poly(ethylene adipate) (PEA), poly(butylene adipate) (PBA), poly(ethylene succinate) (PES), and so on.1–15 Among these aliphatic polyesters, PES is regarded as one of the most promising polyesters due to its excellent biocompatibility, favorable biodegradability, thermal stability, relatively high melting temperature, and environment-friendly properties. The synthesis and physical properties of PES have been extensively investigated.16–27 However, compared with conventional plastics such as polypropylene (PP) and polyethylene (PE), PES has relatively poor mechanical properties, which may restrict its development and applications in films for packaging and agriculture.
Blending and copolymerization are widely used to modify the properties of polymers. Blending is a simple but effective way to obtain the materials with desired performance. Up to now, many polymers have been blended with PES, including poly(ethylene oxide), PBS, poly(4-vinyl phenol), poly(L-lactide), and poly(3-hydroxybutyrate-co-hydroxyvalerate).28–36 However, most polymers are immiscible with PES, leading to phase separation which may influence the physical properties of the blends.32,35
The properties of copolymers have been improved significantly compared with those of their homopolymers, including mechanical properties, degradation rate, and processability.37–39 Therefore, copolymerization is used widely to modify the properties of polymers, which could be adjusted by the kind and content of the chemical units. Many PES based copolymers have been synthesized, such as poly(ethylene succinate-co-ethylene terephthalate), poly(ethylene succinate-co-ethylene glycol), poly(ethylene succinate-co-ethylene adipate), poly(ethylene succinate-co-butylene succinate), etc.40–45 Kondratowicz et al. prepared a series of poly(ethylene succinate-co-ethylene terephthalate) copolymers by melt polycondensation of ethylene glycol, succinic acid, and bis-(β-hydroxyethylene terephthalate).40 The copolymers with 20–40 mol% ethylene glycol were amorphous and were unfit for packaging application due to their poor thermal and mechanical stability. When the content of ethylene glycol increased to 50–60 mol%, the copolyesters were able to crystallize. The hydrolytic degradation of the copolyesters containing 40–60 mol% ethylene glycol was investigated under different conditions. The fastest hydrolytic degradation rate was observed for pH = 4 and for compost; moreover, the copolyester with 40 mol% ethylene glycol was chosen for industrial synthesis due to its good processing and compatibility properties. Wang et al. synthesized a novel poly(ester ether urethane) (PEEU) through chain-extension reaction of PES and poly(ethylene glycol) (PEG) with 1,6-hexamethylene diisocyanate as a chain-extender.41 The glass transition temperature and crystallinity of PEEU decreased with the increment of PEG content, while the hydrophilicity and biocompatibility of the copolyesters increased with increasing the PEG content compared with PES. The tension strength and modulus of PEEU decreased while elongation at break increased with PEG content. Chen et al. studied the crystallization kinetics and melting behavior of poly(ethylene succinate-co-5 mol% trimethylene succinate).42 The random parameter and single glass transition temperature suggested that this copolymer was a random copolyester. The Hoffman–Lauritzen nucleation theory was used to analyze the growth rate data, and the regime II to III transition temperature was around 65 °C. The crystal structure remained unchanged and triple melting peaks were found after crystallizing between 30 and 80 °C. In previous works, we reported the crystallization behavior, crystal structure, and spherulitic morphology of poly(ethylene succinate-co-ethylene adipate) and poly(ethylene succinate-co-butylene succinate), and the effects of the second composition on the properties of PES were analyzed in detail.43,44 The crystallization of the copolyesters was weaker than that of PES, while the crystal structures were not modified in the presence of the second composition. In this work, we intended to synthesize a series of high molecular weight of PES based linear aliphatic polyesters through a two-stage melt polycondensation method without aromatic groups and chain-extender, because aromatic groups may influence the degradation properties of the polymers and most of the chain-extenders have a certain extent of toxicity. However, most of the above linear aliphatic polyesters did not possess excellent mechanical properties due to their low molecular weights,43,44 which may influence their further application as films in packaging and agriculture fields.
In addition to the above PES based copolyesters, it is of interest and importance to develop novel PES based copolyesters to improve the physical properties and extend the practical application. To the best of our knowledge, the synthesis and properties of poly(ethylene succinate-co-decamethylene succinate) (PEDS) copolyesters have not been reported in literature until now. The chemical structure of PEDS is similar to that of poly(ethylene succinate-co-diethylene glycol succinate) (P(ES-co-DEGS)), which was synthesized in our recent work.45 The crystallization rates of P(ES-co-DEGS) were slower than that of PES, while the crystal structure of the copolymers remained unchanged. The crystallinity of P(ES-co-DEGS) was slightly reduced with the incorporation of diethylene glycol succinate (DEGS) units. The differences between PEDS and P(ES-co-DEGS) are as follows. First, the monomers are different in these copolyesters. Diethylene glycol and decamethylene glycol were used to synthesize P(ES-co-DEGS) and PEDS, respectively. Second, the crystallization ability of the second composition in P(ES-co-DEGS) and PEDS is different. Normally, poly(diethylene glycol succinate) (PDEGS) is amorphous, and poly(decamethylene succinate) (PDS) is able to crystallize.46,47 Therefore, in this work, we synthesized a series of high molecular weight novel PEDS copolymers with different decamethylene succinate (DS) contents and their homopolymer PES; moreover, the effect of different DS compositions on the crystal structure, thermal stability, crystallization behavior, mechanical properties, and hydrolytic degradation of PEDS were investigated with various techniques. The aims of this work are not only to synthesize and characterize some novel PES based copolyesters but also to better understand the structure and properties relationship of aliphatic copolyesters. The experimental results indicated that the thermal and mechanical properties of PEDS may be regulated depending on the DS contents. The results reported herein may be of interest and importance from the viewpoint of the structure and properties relationship of biodegradable polyesters.
Experimental
Materials
Succinic acid (SA) was bought from Tianjin Fuchen Chemical Solvent Factory. Ethylene glycol (EG) and decamethylene glycol (DG) were purchased from Beijing Chemical Works and Xiya Reagent Research Center, respectively. Tetrabutyl titanate (TBT), used as a catalyst, was purchased from Beijing Chang Ping Jing Xiang Chemical Factory. All the regents were used without purification.
Preparation of PEDS and PES
PEDS copolyesters with different DS contents were prepared by a two-stage melt polycondensation method (esterification and polycondensation). During the esterification process, appropriate amounts of SA, EG, and DG were added into a three-necked flask under a nitrogen atmosphere. The reaction mixtures were heated to 150 °C for a period of time until all the reagents were melted completely, then further heated to 190 °C slowly and remained for 2.5 h. During the polycondensation process, a small quantity of TBT (0.3 molar% of SA) was injected into the flask. A vacuum of 350 Pa was applied, and the temperature was increased to 230 °C. The reaction continued for 5–6 h. Finally, the resulting product was cooled down to room temperature, dissolved into chloroform, precipitated and washed by methanol, volatilized at room temperature for one night, and dried at 50 °C under vacuum for 5 days to remove the residual solvent. Through a similar process, PES was also synthesized.
Characterizations
Gel permeation chromatography (GPC). The average molecular weight (Mw) and polydispersity index (PDI) of PEDS and PES were obtained from the GPC analysis (Agilent 2006 Series) using dimethylformamide (DMF) as the solvent.
Nuclear magnetic resonance spectroscopy (1H NMR). 1H NMR spectra of PEDS and PES were obtained by Bruker AV 600 at a frequency of 600 MHz. Deuterated chloroform (CDCl3) was used as the solvent.
Thermogravimetric analysis (TGA). The thermal stabilities of PEDS and PES were investigated by TGA Q50 under a nitrogen atmosphere. The samples were heated from room temperature to 580 °C at 20 °C min−1.
Wide-angle X-ray diffraction (WAXD). WAXD experiments were performed on an X-ray diffractometer (Rigaku D/Max 2500 VB2+/PC) from 5° to 45° with a scan rate of 5° min−1 at room temperature. The samples were first pressed into films on a hot stage at 130 °C for 5 min to erase the thermal history and then transferred into an oven at 50 °C for 1 day.
Differential scanning calorimeter (DSC). The thermal properties of PEDS and PES were characterized by DSC Q100 under a nitrogen atmosphere, and each sample was around 5 mg. The samples were heated to 130 °C, kept for 3 min to erase the thermal history, cooled to −40 °C at a constant cooling rate (60 or 2 °C min−1), and reheated to 130 °C at 10 °C min−1.
Mechanical properties. Tensile tests were performed with an Instron 1185 Universal Testing Machine at ambient temperature with a speed of 50 mm min−1. PEDS and PES were first molded into films through a melting and pressing method at 130 °C for 10 min and then quenched into ice water. The films were cut into dumbbell-shaped specimens with a dimension of 40 mm × 4 mm × 0.5 mm. The results were obtained by an average of three specimens at least.
Hydrolytic degradation. The PEDS and PES films used for the hydrolytic degradation experiments were prepared by a melting and pressing method and then cooled in the ice water. The films were cut into a size of 10 mm × 6 mm × 1 mm before placed into a NaOH solution of pH = 14 at 37 °C. After a specific time of degradation, the films were washed with deionized water several times and dried at room temperature until the weights were constant. The weight loss (Wloss) was calculated by the following equation:| |
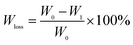 | (1) |
where W0 is the original mass and W1 is the residual mass.
Scanning electron microscopy (SEM). The surfaces of PEDS and PES films before and after hydrolytic degradation were observed by a Hitachi S-4700 SEM. The films were coated with gold before observation.
Results and discussion
Composition and crystal structure of PEDS
PEDS and PES were synthesized through a two-step melt polycondensation method. The chemical structure of PEDS is shown in Fig. 1a. It is well known that the composition of copolymers plays an important role on their properties. Therefore, 1H NMR spectra were used to determine the exact compositions of PEDS. Fig. 1b displays the 1H NMR spectra of PEDS82 as an example. The characteristic resonance signal of methylene protons of SA appears at 2.66 ppm, and that of EG appears at 4.30 ppm. Those of methylene protons of DG occur at 4.08, 1.62, and 1.28 ppm for H3, H4, and H5 + H6 + H7. The composition of PEDS was calculated from the area ratio of the peaks of H1 from ES units and H3 or H4 from DS units. The results are listed in Table 1. The difference between the feed composition and the copolymer composition for PEDS is rather small, indicating that a series of expected products were obtained. The PEDS copolyesters with different DS contents are named as PEDS94, PEDS87, and PEDS82, respectively, where the number refers to the molar ratio of ethylene succinate (ES) units in the copolyesters according to the calculation of the 1H NMR spectra. The Mw values of PEDS and PES were measured and are also summarized in Table 1. From Table 1, all the samples show high Mw values; therefore, they may possess good processability and excellent mechanical properties from a viewpoint of practical application. The Mw values of the PEDS copolyesters are higher than that of PES, which may be attributed to the longer molecular chain of DS units. Similar Mw values (around 1.7 × 105 g mol−1) were also found in PEEU, which was synthesized through a chain-extension reaction of PES and PEG, suggesting that the Mw value of PEDS could achieve the level of the polyesters prepared by a chain-extension reaction.41
 |
| | Fig. 1 (a) Chemical structure of PEDS and (b) 1H NMR spectrum for PEDS82. | |
Table 1 Summary of composition, molecular weight, and crystallinity values of PES and PEDS
| Samples |
fa |
Fb |
Mw (g mol−1) |
PDI |
Xc (%) |
| f: EG/DG molar ratio in feed. F: ES/DS molar ratio in copolymer based on the 1H NMR spectra. |
| PES |
100/0 |
100/0 |
9.05 × 104 |
1.7 |
35 |
| PEDS94 |
95/5 |
94/6 |
1.45 × 105 |
1.3 |
31 |
| PEDS87 |
90/10 |
87/13 |
1.74 × 105 |
1.3 |
29 |
| PEDS82 |
85/15 |
82/18 |
1.60 × 105 |
1.2 |
28 |
WAXD is an effective technique to characterize the crystal structure of polymers, and the WAXD patterns of PEDS and PES are shown in Fig. 2. For PES, three strong diffraction peaks located at 2θ = 20.1°, 22.7°, and 23.2° are attributed to (021), (121), and (200) planes, respectively.48 Similar diffraction peaks are observed for the three PEDS copolyesters with different DS contents, indicating that the crystal structures of PEDS copolyesters are not modified by the incorporation of DS units, and the DS units should be excluded from crystalline domains.49 The peak intensities of PEDS were reduced compared to those of PES. The crystallinity (Xc) of PES is about 35%, which decreases to around 30% for PEDS, regardless of the DS contents. The reduced Xc values of PEDS may be attributed to the reduced crystallization ability of PEDS with the presence of DS units. Similar results were found in other works.43,44 The Xc values are summarized in Table 1. In brief, the incorporation of different contents of DS units does not change the crystal structures but slightly reduces the crystallinity values of PEDS, with respect to PES.
 |
| | Fig. 2 WAXD patterns showing the crystalline structures of PES and PEDS after crystallizing at 50 °C for 1 day. | |
Thermal properties of PEDS and PES
The thermal stability of PEDS and PES has a direct influence on their processing conditions. Fig. 3 depicts the TGA curves of PEDS and PES. There is no apparent weight loss until 270 °C for PEDS and PES, suggesting that all the samples have a relative high thermal stability. PEDS and PES have a similar degradation behavior within the investigated temperature range, regardless of the DS contents. From Fig. 3, the degradation temperatures at 5% weight loss of PEDS and PES are around 330 °C, suggesting that the incorporation of DS units does not obviously influence the thermal stability of PES. Similar thermal stabilities were also found in the poly(ethylene succinate-co-ethylene adipate) copolyesters, whose degradation temperatures at 5% weight loss were around 320 °C.43 The residual weight of PES is 4.9% at 550 °C, which decreases to 4.1, 3.4, and 1.5% for PEDS94, PEDS87, and PEDS82 at the same temperature, respectively, suggesting that the residual weight of PEDS decreases with the increment of DS contents. Similar phenomena were also found in PES with different molecular weights and other polyesters.50–52
 |
| | Fig. 3 TGA curves of PES and PEDS at 20 °C min−1 under nitrogen atmosphere. | |
The glass transition temperature (Tg) values of PEDS and PES were obtained from the second heating curves on the melt-quenched samples, which are shown in Fig. 4. PES has a Tg value of −10 °C, and the Tg values of PEDS94, PEDS87, and PEDS82 are around −17, −22, and −25 °C, respectively, indicating that the flexibility of the PEDS chains has been apparently improved, relative to PES. Due to their weak crystallization ability, all the samples display a cold crystallization peak temperature (Tch). PES has a Tch of 43.1 °C with a cold crystallization enthalpy (ΔHch) of 53.3 J g−1. With increasing the DS contents to 6, 13, and 18 mol%, the Tch values of PEDS shift upward to 32.2, 46.7, and 47.9 °C, and the corresponding ΔHch values decrease to 50.3, 29.3, and 1.4 J g−1, respectively, indicating the cold crystallization of PEDS has been restricted with the increment of DS units. From Fig. 4, the melting temperature (Tm) and melting enthalpy (ΔHm) of PES were measured to be 102.5 °C and 67.1 J g−1, respectively. The Tm values of PEDS94, PEDS87, and PEDS82 decrease to 94.1, 86.2, and 81.7 °C, respectively, and the corresponding ΔHm values decrease to 61.1, 33.6, and 1.5 J g−1, respectively, suggesting that the crystallization ability of PEDS becomes weaker with increasing the DS composition. The data are listed in Table 2.
 |
| | Fig. 4 DSC heating curves of PES and PEDS at 10 °C min−1 after quenching from the melt at 60 °C min−1. | |
Table 2 Basic thermal properties of PES and PEDS for the melt-quenched samples
| Samples |
Tg (°C) |
Tch (°C) |
ΔHch (J g−1) |
Tm (°C) |
ΔHm (J g−1) |
| PES |
−10 |
43.1 |
53.5 |
102.5 |
67.1 |
| PEDS94 |
−17 |
32.2 |
50.3 |
94.1 |
61.1 |
| PEDS87 |
−22 |
46.7 |
29.3 |
86.2 |
33.6 |
| PEDS82 |
−25 |
47.9 |
1.4 |
81.7 |
1.5 |
The nonisothermal melt crystallization behavior of PEDS copolyesters with different DS contents and PES was also investigated with DSC. As the PEDS copolyesters with high DS contents could not crystallize at a fast cooling rate, all the samples were crystallized nonisothermally from the crystal-free melt at a very slow cooling rate of 2 °C min−1 in this work. Fig. 5 displays the nonisothermal melt crystallization exotherms at a cooling rate of 2 °C min−1 for PEDS and PES. As shown in Fig. 5, PES shows a nonisothermal melt crystallization peak temperature (Tcc) of 59.0 °C with a melt crystallization enthalpy (ΔHcc) of 55.9 J g−1. With increasing the DS content, the crystallization exotherms of PEDS94 and PEDS87 shift downward to low temperature range and become broader than PES. It should be noted that PEDS82 could not finish crystallization even at a cooling rate of 2 °C min−1. PEDS94 displays a Tcc of 38.6 °C and a ΔHcc of 47.9 J g−1, while PEDS87 presents a Tcc of 27.9 °C and a ΔHcc of 30.4 J g−1. The above results clearly indicate that the nonisothermal melt crystallization of PEDS become weaker with the increment of DS content.
 |
| | Fig. 5 DSC cooling traces of PES and PEDS during the nonisothermal melt crystallization at 2 °C min−1. | |
Mechanical properties of PEDS and PES
The mechanical properties of PEDS and PES play an important role in their application field. The stress–strain curves of PEDS and PES are shown in Fig. 6a. To see more clearly, Fig. 6b is an enlarged image of Fig. 6a within the strain range of 0–60%. It is clear that all the samples show obvious yield behavior with an elongation at yield (ε1) value of around 10%, regardless of the DS contents. PES has a yield strength (σ) value of 19.3 ± 0.3 MPa. With increasing the DS content, the σ values for PEDS94, PEDS87, and PEDS82 decrease to 19.0 ± 1.2, 15.4 ± 1.4, and 11.3 ± 0.6 MPa, respectively, the Young's modulus (E) value of PES is 371 ± 80 MPa, which decreases to 342 ± 23, 324 ± 21, and 281 ± 30 MPa for PEDS94, PEDS87, and PEDS82, respectively. The decreased values of σ and E for PEDS are attributed to their reduced crystallinity values compared with that of PES. PES has an elongation at break (ε2) value of 272 ± 48%. With the increasing DS content to 6, 13, and 18 mol%, the ε2 values of PEDS increase to 366 ± 32, 833 ± 167, and 1268 ± 22%, respectively, owing to the improved flexibility of chain segments and decreased crystallinity values of PEDS. The result may also have a relationship with the higher molecular weight of PEDS compared with that of PES. The relative data are summarized in Table 3. The ε2 values of PEDS increase by about 1.3 to 4.7 times, while the σ and E values only decrease by about 41 and 25% compared with those of PES. In a word, the mechanical properties of PEDS have been significantly improved and can be regulated with the content of the DS units.
 |
| | Fig. 6 (a) Stress–strain curves of PES and PEDS and (b) enlarged portion of the strain from 0–60%. | |
Table 3 Summary of the mechanical properties for PES and PEDS
| Samples |
ε1 (%) |
σ (MPa) |
ε2 (%) |
E (MPa) |
| PES |
9.6 ± 1.6 |
19.3 ± 0.3 |
272 ± 48 |
371 ± 80 |
| PEDS94 |
9.3 ± 0.3 |
19.0 ± 1.2 |
366 ± 32 |
342 ± 23 |
| PEDS87 |
10.3 ± 1.3 |
15.4 ± 1.4 |
833 ± 167 |
324 ± 21 |
| PEDS82 |
10.6 ± 0.9 |
11.3 ± 0.6 |
1268 ± 22 |
281 ± 30 |
Similar improved mechanical properties were also found in PEEU, which were synthesized through a chain-extension reaction of PES and PEG.41 Take the copolymer containing 18.9 mol% PEG as an example, the ε2, σ and E values were around 1000%, 10 and 280 MPa, respectively. In the present work, the ε2 value of PEDS82 is larger than that of PEEU, and the σ and E values are close to those of PEEU. Therefore, the mechanical properties of PEDS are comparable to those of PEEU when they have the similar second comonomer composition. The excellent tensile properties of the copolyesters may find their practical applications as films in packaging, agriculture, etc.
Hydrolytic degradation of PEDS and PES
The hydrolytic degradation behavior of PEDS and PES was further investigated. The plots of weight loss versus hydrolytic degradation time for PEDS and PES are displayed in Fig. 7. The weight loss displays almost a linear increase with increasing degradation time for PEDS and PES within the investigated time. The hydrolytic degradation rates were obtained from the slopes of the lines.53,54 For PES, the degradation rate is 3.32 wt% per day, which decreases to 2.92, 2.19, and 3.14 wt% per day for PEDS94, PEDS87, and PEDS82, respectively. It is interesting to find the hydrolytic degradation rate becomes slower with increasing the DS content up to 13 mol%, while when the content of DS units further increases to 18 mol%, the hydrolytic degradation rate of PEDS82 becomes faster. The results may be attributed to the following two factors. First, the density of ester bonds of PEDS is smaller than that of PES in the presence of the DS units; moreover, the hydrophilicity of the samples becomes weaker with a reduced density of ester bonds. Therefore, the PEDS copolymers may have a slower hydrolytic degradation rate than PES. Second, the crystallinity values of the samples have a close relationship with their hydrolytic degradation rates, because the amorphous region is easier to degrade than the crystallization region of a semicrystalline polymer.55,56 Before hydrolytic degradation, the crystallinity of PES, calculated from the XRD experiment, is about 40%. While those of PEDS94, PEDS87, and PEDS82 are about 33, 31, and 25%, respectively. After a hydrolytic degradation of 15 days, the crystallinity of PES increases to about 51%, and those of PEDS are about 39, 37, and 35% with the DS contents of 6, 13, and 18 mol%, respectively. The decreased crystallinity may accelerate the hydrolytic degradation process of PEDS, relative to PES; moreover, increasing the DS content seems to favor the hydrolytic degradation of PEDS. As a result, the hydrolytic degradation process of PEDS must be influenced by both the density of ester bonds and the crystallinity values. Regardless of the DS contents, the hydrolytic degradation rates of PEDS are slower than that of PES, indicating that the decreased density of ester bonds determines the hydrolytic degradation process. In the case of the PEDS copolyesters with different DS contents, the reduced density of ester bonds may decelerate while the decreased crystallinity values may accelerate the hydrolytic degradation process. Therefore, the hydrolytic degradation is a competitive result of the two factors. For PEDS94 and PEDS87, the reduced density of ester bonds controls the hydrolytic degradation process; therefore, PEDS87 degrades slowly than PEDS94 with increasing the DS content from 6 to 13 mol%. With further increasing the DS content up to 18 mol%, the reduced crystallinity is more important than the decreased density of ester bonds to affect the hydrolytic degradation process; therefore, PEDS82 degrades faster than PEDS87 and PEDS94.
 |
| | Fig. 7 Plots of weight loss versus hydrolytic degradation time for PES and PEDS at 37 °C and pH = 14. | |
The hydrolytic degradation behavior of PEDS and PES was further studied by SEM, and the morphology of the film surfaces for PEDS and PES before and after hydrolytic degradation was observed. As seen from Fig. 8, the surfaces are smooth for all the PEDS and PES films before hydrolytic degradation, and they become rough after a hydrolytic degradation of 15 days for all the samples. Moreover, some porous structures are observed on the film surfaces, resulting from the hydrolytic degradation of the samples. Furthermore, the order of the surface roughness of the films is clear: PES > PEDS82 > PEDS94 > PEDS87. The results are consistent with the results shown in Fig. 7.
 |
| | Fig. 8 SEM images displaying the morphology of film surface before and after a hydrolytic degradation of 15 days at 37 °C and pH = 14 for PES (a and b), PEDS94 (c and d), PEDS87 (e and f), and PEDS82 (g and h). | |
Conclusions
In this work, a series of novel PEDS copolymers with the DS contents ranging from 6 to 18 mol% and their homopolymer PES were successfully synthesized through a melt polycondensation method. The thermal stability, crystal structure, crystallization behavior, mechanical properties, and hydrolytic degradation of PEDS were studied in detail and compared with those of PES. The thermal stability and crystal structure of PEDS are similar to those of PES, but the crystallinity values are smaller than that of PES. With the increment of the DS content, the values of the glass transition temperature, melting temperature, and nonisothermal melt crystallization peak temperature of PEDS decrease apparently compared with those of PES. Moreover, the mechanical properties of PEDS were improved obviously with the incorporation of the DS units. The Young's modulus and yield strength values decrease while the elongation at break values for PEDS were improved obviously with increasing the DS content, relative to PES. In addition, the hydrolytic degradation behavior of PEDS was investigated. The hydrolytic degradation rates of PEDS are slower than that of PES, regardless of the DS contents, while PEDS82 degrades faster than PEDS87 and PEDS94.
Acknowledgements
This work was supported by the National Natural Science Foundation, China (51573016, 51373020 and 51221002).
Notes and references
- Z. Qiu, T. Ikehara and T. Nishi, Polymer, 2003, 44, 3095 CrossRef CAS.
- K. Cho, J. Lee and K. Kwon, J. Appl. Polym. Sci., 2001, 79, 1025 CrossRef CAS.
- T. Wang, H. Li, F. Wang, S. Yan and J. Schultz, J. Phys. Chem. B, 2011, 115, 7814 CrossRef CAS PubMed.
- E. M. Woo, P. L. Wu, M. C. Wu and K. C. Yan, Macromol. Chem. Phys., 2006, 207, 2232 CrossRef CAS.
- A. Meyer, K. C. Yen, S. H. Li, S. Förster and E. M. Woo, Ind. Eng. Chem. Res., 2010, 49, 12084 CrossRef CAS.
- E. M. Woo, L. Y. Wang and S. Nurkhamidah, Macromolecules, 2012, 45, 1375 CrossRef CAS.
- Y. Bian, L. Han, C. Han, H. Lin, H. Zhang, J. Bian and L. Dong, CrystEngComm, 2014, 16, 2702 RSC.
- J. Yang, P. Pan, T. Dong and Y. Inoue, Polymer, 2010, 51, 807 CrossRef CAS.
- T. Wang, H. Wang, H. Li, Z. Gan and S. Yan, Phys. Chem. Chem. Phys., 2009, 11, 1619 RSC.
- L. Zhao, X. Wang, L. Li and Z. Gan, Polymer, 2007, 48, 6152 CrossRef CAS.
- E. M. Woo, K. C. Yen and M. C. Wu, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 892 CrossRef CAS.
- N. Yin, Z. X. Zeng and W. L. Xue, J. Appl. Polym. Sci., 2010, 117, 1883 CrossRef CAS.
- Y. Y. Song, H. M. Ye, J. Xu, K. Hou, Q. Zhou and G. W. Lu, Polymer, 2014, 55, 3054 CrossRef CAS.
- Z. Gan, K. Kuwabara, H. Abe, T. Iwata and Y. Doi, Biomacromolecules, 2004, 5, 371 CrossRef CAS PubMed.
- Z. Cui and Z. Qiu, Polymer, 2015, 67, 12 CrossRef CAS.
- Z. Qiu, M. Komura, T. Ikehara and T. Nishi, Polymer, 2003, 44, 7781 CrossRef CAS.
- K. Kuwabara, Z. Gan, T. Nakamura, H. Abe and Y. Doi, Polym. Degrad. Stab., 2004, 84, 105 CrossRef CAS.
- Z. Gan, H. Abe and Y. Doi, Biomacromolecules, 2000, 1, 704 CrossRef CAS PubMed.
- Z. Gan, H. Abe and Y. Doi, Biomacromolecules, 2000, 1, 713 CrossRef CAS PubMed.
- G. Liu, L. Zheng, X. Zhang, C. Li and D. Wang, Polymer, 2013, 54, 6860 CrossRef CAS.
- Y. Ichikawa, J. Washiyama, Y. Moteki, K. Noguchi and K. Okuyama, Polym. J., 1995, 27, 1264 CrossRef CAS.
- J. B. Zeng, F. Wu, C. L. Huang, Y. S. He and Y. Z. Wang, ACS Macro Lett., 2012, 1, 965 CrossRef CAS.
- Z. Qiu, T. Ikehara and T. Nishi, Polymer, 2003, 44, 5429 CrossRef CAS.
- Y. Ichikawa, K. Noguchi, K. Okuyama and J. Washiyama, Polymer, 2001, 42, 3703 CrossRef CAS.
- Z. Qiu, S. Fujinami, M. Komura, K. Nakajima, T. Ikehara and T. Nishi, Polym. J., 2004, 36, 642 CrossRef CAS.
- K. Chrissafis, K. M. Paraskevopoulos and D. N. Bikiaris, Thermochim. Acta, 2005, 435, 142 CrossRef CAS.
- G. Z. Papageorgiou and D. N. Bikiaris, Polymer, 2005, 46, 12081 CrossRef CAS.
- P. Pan, L. Zhao and Y. Inoue, Macromol. Mater. Eng., 2013, 298, 919 CrossRef CAS.
- Y. S. He, J. B. Zeng, S. L. Li and Y. Z. Wang, Thermochim. Acta, 2012, 529, 80 CrossRef CAS.
- Q. Li and J. Liu, Colloid Polym. Sci., 2013, 291, 2007 CAS.
- H. Ni'mah and E. M. Woo, Cryst. Growth Des., 2014, 14, 576 Search PubMed.
- J. Lu, Z. Qiu and W. Yang, Polymer, 2007, 48, 4196 CrossRef CAS.
- J. Lu, Z. Qiu and W. Yang, Macromolecules, 2008, 41, 141 CrossRef CAS.
- Z. Qiu, S. Fujinami, M. Komura, K. Nakajima, T. Ikehara and T. Nishi, Polymer, 2004, 45, 4515 CrossRef CAS.
- L. Miao, Z. Qiu, W. Yang and T. Ikehara, React. Funct. Polym., 2008, 68, 446 CrossRef CAS.
- Z. Qiu, T. Ikehara and T. Nishi, Macromolecules, 2002, 35, 8251 CrossRef CAS.
- L. Feng, X. Bian, Z. Chen, G. Li and X. Chen, Polym. Degrad. Stab., 2013, 98, 1591 CrossRef CAS.
- A. Cao, T. Okamura, K. Nakayama, Y. Inoue and T. Masuda, Polym. Degrad. Stab., 2002, 78, 107 CrossRef CAS.
- J. Zeng, C. Huang, L. Jiao, X. Lu, Y. Wang and X. Wang, Ind. Eng. Chem. Res., 2012, 51, 12258 CAS.
- F. Kondratowicz and R. Ukielski, Polym. Degrad. Stab., 2009, 94, 375 CrossRef CAS.
- C. Liu, J. B. Zeng, S. L. Li, Y. S. He and Y. Z. Wang, Polymer, 2012, 53, 481 CrossRef CAS.
- M. Chen, W. C. Chang, H. Y. Lu, C. H. Chen, J. S. Peng and C. J. Tsai, Polymer, 2007, 48, 5408 CrossRef CAS.
- H. Wu and Z. Qiu, CrystEngComm, 2012, 14, 3586 RSC.
- Y. Yang and Z. Qiu, J. Appl. Polym. Sci., 2011, 122, 105 CrossRef CAS.
- P. Xue and Z. Qiu, Thermochim. Acta, 2015, 606, 45 CrossRef CAS.
- M. Soccio, N. Lotti, L. Finelli, M. Gazzano and A. Munari, Eur. Polym. J., 2008, 44, 1722 CrossRef CAS.
- G. Zhu, Y. Wang, W. Zhu, K. Zhu, X. Xu and Z. Shen, J. Appl. Polym. Sci., 2013, 128, 2817 CrossRef CAS.
- A. S. Ueda, Y. Chatani and H. Tadokoro, Polym. J., 1971, 2, 387 CrossRef CAS.
- Y. Ichikawa, J. Washiyama, Y. Moteki, K. Noguchi and K. Okuyama, Polym. J., 1995, 27, 1264 CrossRef CAS.
- K. Chrissafis, K. M. Paraskevopoulos and D. N. Bikiaris, Thermochim. Acta, 2006, 440, 166 CrossRef CAS.
- H. Chen, X. Wang, J. Zeng, L. Li, F. Dong and Y. Wang, Ind. Eng. Chem. Res., 2011, 50, 2065 CrossRef CAS.
- T. Zorba, K. Chrissafis, K. M. Paraskevopoulos and D. N. Bikiaris, Polym. Degrad. Stab., 2007, 92, 222 CrossRef CAS.
- H. Y. Lu, S. F. Lu, M. Chen, C. S. Yang, C. H. Chen and C. J. Tsai, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2431 CrossRef CAS.
- M. Mochizuki, K. Mukai, K. Yamada, N. Ichise, S. Murase and Y. Iwaya, Macromolecules, 1997, 30, 7403 CrossRef CAS.
- H. Zhao, Y. Bian, Y. Li, Q. Dong, C. Han and L. Dong, J. Mater. Chem. A, 2014, 2, 8881 CAS.
- H. Pan and Z. Qiu, Macromolecules, 2010, 43, 1499 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 








