
Thermo-chemical modification to produce citric acid–yeast superabsorbent composites for ketoprofen delivery
Diejing Fenga,
Bo Bai*b,
Honglun Wangb and
Yourui Suob
aCollege of Environmental Science and Engineering, Chang’an University, Xi’an, 710054, P. R. China
bKey Laboratory of Tibetan Medicine Research, Northwest Plateau Institute of Biology, Chinese Academy of Sciences, Xining, 810001, P. R. China. E-mail: baibochina@163.com
First published on 25th November 2015
Abstract
Owing to their unique physicochemical/biological properties and natural abundance, native yeast microbes were used to prepare a novel eco-friendly superabsorbent composite through thermo-chemical modification of yeast with citric acid in semi-dry conditions. The structure and morphology of the as-produced citric acid–yeast superabsorbent composites were characterized by Fourier-transform infrared spectroscopy (FTIR) and scanning electron microscopy (SEM), respectively. The surface characteristics were studied systematically, including carboxyl content, degree of esterification, degree of substitution and zeta potential. The detailed formation mechanism was proposed. The evaluation of equilibrium welling showed that the species were able to absorb up to 38 times their weight in distilled water, and the water absorption capacity was affected by reaction temperature and external solution (the charge number, salt concentration and pH value). The results implied that citric acid–yeast superabsorbent composites exhibited enhanced water absorption, salt tolerance and pH sensitivity. The excellent swelling performance and special functional groups allowed the citric acid–yeast composites to target drug delivery. Ketoprofen, as a model drug, was utilized to monitor the loading and cumulative release efficiency to evaluate the suitability of citric acid–yeast composites as drug carriers. Its pH-sensitivity, biocompatibility and degradability make it a potential candidate for drug delivery.
1. Introduction
Superabsorbent hydrogels are cross-linked hydrophilic polymers with the ability of retaining large amounts of water. Lots of maze-like network structures within the superabsorbent hydrogels ensured that the hydrogel products can absorb water, swell in water and hold a large amount of water while still maintaining their physical dimensional traits.1,2 The traditional strategies for the synthesis of superabsorbent materials have mainly centered on the use of petroleum-based polymers, which are nonrenewable, difficult to degrade, and harmful to the environment. Recently, substitution of conventional petroleum-based polymers in the construction of composite superabsorbent materials using biomaterials have sparked another particular interest in various uses including drug delivery,3 immobilization of enzymes,4 tissue engineering,5 healthcare6 as well as agro-industries,7 due to the excellent nontoxic, hydrophilic, biocompatible and biodegradable of the bio-matrices. It has been verified that the so formed biomaterial-based superabsorbent composites with unique physical/chemical properties have significantly overcome some conspicuous defects of petroleum-based polymers, such as nonrenewable resources, and persistence and accumulation in the environment.8 In this regard, the design and development of superabsorbents using naturally available raw bio-materials have opened up a new window to obtain novel environmentally safe materials for promising applications. Typically, various bio-materials have been used for incorporation into superabsorbent matrices through chemical surface modification, grafting copolymerization and symmetry methods, etc. Hereinto, natural starch,9 chitosan,10 cellulose,11 tara gum,12 konjac glucomannan13 and guar gum,14 were especially notable because of their stable raw material supply and cost, renewable resource, nontoxicity, and biodegradable advantages.Yeast, one of the most abundant microorganisms, is extracted from the brewing industry, and used extensively in foodstuffs. The special physicochemical/biological properties of yeast have made it eligible as an ideal candidate for the fabrication of superabsorbent composite materials.15 Specifically, the cell wall of yeast is composed of ∼90% polysaccharides, mainly polymers of mannose (mannoproteins, ca. 40% of the cell wall dry mass), polymers of glucose (β-glucan, ca. 60% of the cell wall dry mass) and polymers of N-acetylglucosamine (chitin, ca. 2% of the cell wall dry mass), and the remainder consists of a small portion of proteins and lipids.16–18 Thus, the abundant functional groups, including hydroxyl, amidogen, carboxyl, acylamino and sulfonyl groups inherited from yeast cell walls, can not only provide the prerequisite hydrophilicity but also serve as the versatile platform for further chemical modification with other desired molecules to tune their chemical and functional properties. More importantly, the inherent semi-permeable property of the cell wall permits the passage of water molecules, especially water and carbon dioxide, with size exclusion estimated to be 30–60 kDa.19,20 The stable osmotic environment within the yeast cell walls can help yeast cells to retain water by preventing osmotic lysis.20 The unique hollow shape inside the yeast cell can act as a reservoir to store water. Apart from the above highlights, further chemical modification of yeast presents a means for preparing superabsorbent materials with unique properties that can dramatically increase the value and utility of these biopolymers.
Citric acid is a regenerative resource-based substance, mainly manufactured by fermentation of carbohydrate and produced in more than a million tons every year.21 Citric acid is an inexpensive natural preservative present in citrus fruits and has been widely used as an acidifier in food and drinks.22 In terms of biochemistry, citric acid is an important intermediate metabolic product of all aerobic organisms (Krebs or citric acid cycle).23,24 Citric acid is a naturally occurring organic acid with three carboxylic groups and one hydroxyl group in the chemical structure, and therefore citric acid shares similar properties with carboxylic acids. For example, the three carboxylic groups on citric acid could react with free hydroxyl groups on the biomaterials to form an ester linkage. In view of these characteristics, chemical modification, as a creative usage of citric acid, has been proposed to confer new chemical and physical functionalities for natural biomaterials. Typically, cellulose,25 starch26 and protein27 have been incorporated in to carboxylic acid groups into backbones through thermo-chemical modification with citric acid molecules. The obtained biomaterial-based superabsorbents with high carboxylic acid content have exerted superior hydrophilic properties useful in absorbent applications. For instance, Salam et al. have determined that starch citrate and hemicellulose citrate have significantly increased water affinity and saline absorption relative to native starch and hemicellulose, producing a water soluble polymer.28,29 Farjami et al. have investigated the influences of crosslinking of whey proteins with citric acid, and proved that the superior water-holding capacity and dense microstructure of the citric acid cross-linked whey protein microgels were attributed to the existence of a high number of carboxyl residues in the structure.30 However, the combination of citric acid and yeast to produce a yeast-based superabsorbent material through the incorporation of carboxyl groups into the yeast cell wall has not been put into practice yet.
Herein, in the present paper yeast cells were firstly thermo-chemically modified with citric acid to yield potentially biocompatible and biodegradable superabsorbent composites. The structure and morphology of obtained citric acid–yeast superabsorbent composites were characterized by Fourier transform infrared spectroscopy (FTIR) and scanning electron microscopy (SEM), respectively. The detailed formation mechanism is proposed. The surface characteristics were studied systematically, including carboxyl content, degree of esterification (DE), degree of substitution (DS) and point of zero charge (pHPZC), in order to further verify the synthesis procedure. The effect of reaction temperature and external solution (the charge number, salt concentration and pH value) on water absorption capacity was also investigated. Moreover, by virtue of the excellent swelling performances and special functional groups we chose ketoprofen as a model drug to evaluate the loading and cumulative release efficiency of the citric acid–yeast composites as a drug carrier. Obviously, as compared to the conventional petroleum-based superabsorbent, the present route of thermo-chemical linkage in semi-dry conditions was more eco-friendly and simpler, and the citric acid–yeast superabsorbent composite materials were more eco-friendly, biocompatible and biodegradable. The present research has provided basic techniques for further preparation and application of yeast-based products.
2. Experimental
2.1 Materials
Yeast powder was purchased from the Angel Yeast (Wuhan, China). Citric acid, sodium hydroxide and ethyl alcohol were supplied by Tianjin Chemical Reagent Factory (Tianjin, China). Ketoprofen was furnished by Zhejiang Chetou Pharmaceutical Corp. Phenolphthalein, hydrochloric acid, potassium chloride, calcium chloride and ferric chloride were provided by Xi’an Chemical Reagent Factory (Shaanxi, China). All reagents were of analytical grade and used without further purification. Deionized water was used throughout the work, and all solutions were prepared with deionized water.2.2 Preparation of citric acid–yeast superabsorbent composites
The yeast cells were thermo-chemically modified by the following procedures.31 Citric acid (5 g) was dissolved in a minimal amount of deionized water (about 10 mL) in a beaker. A portion of 2.5 g of air-dried yeast powder was dispersed in the above citric acid solution and mixed vigorously. The mixture was transferred to Pyrex dishes and placed in a forced air oven to dehydrate at 55 °C for 4 days. At this point all surface moisture was removed and yeast cells were coated with CA. Then the oven temperature was adjusted to the desired level (100–140 °C) and the mixtures were allowed to react for 3 h. After that, the reaction products (CAY100–140) were suspended in excess distilled water for 30 min, adjusted to pH 2 using acetic acid and washed with distilled water 3 times. Finally, the produced citric acid–yeast superabsorbent composites were air dried overnight.2.3 Characterizations
FTIR spectra were recorded on a Nicolet FT-IR spectrometer using a KBr disc containing 10% finely ground samples. All the spectra were obtained by the accumulation of 256 scans, with a resolution of 400–4000 cm−1. The morphological observation was performed on images acquired using a scanning electron microscope (SEM, Hitachi S-4800), and the samples were coated with platinum of 10 nm thickness to make them conductive.2.4 Determination of carboxyl content
Carboxyl content in the citric acid–yeast superabsorbent composites was estimated by using an acid–base titration according to the method of Salam.29 1.0 g of synthesized products was firstly dissolved in excess 0.1 M sodium hydroxide solution (pH 12.5) and the mixture was allowed to react for 1 h. The remaining excess amount of sodium hydroxide was determined by titration with 0.1 M hydrochloride acid solution with phenolphthalein as an indicator.31 The carboxyl content (meq per 100 g) in milliequivalents of acidity per 100 g calculated as:
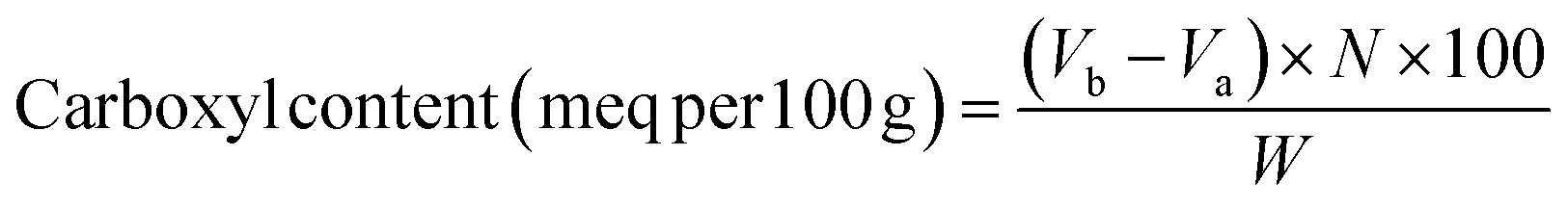 | (1) |
2.5 Determination of degree of esterification and degree of substitution
Degree of esterification (DE) and degree of substitution (DS) was determined by titration involving complete basic hydrolysis of the ester linkages and potentiometric titration of the excess alkaline according to the method of Wurzburg.32 Specifically, 1 g of ground citric acid–yeast superabsorbent composites accurately weighed was added to 50 mL of 75% ethanol solution. The slurry was kept in the water bath (50 °C) for 30 min with continuous agitation. After the slurry was cooled, 30 mL of aqueous solution of 0.5 M potassium hydroxide was added to saponify the ester for 72 h with stirring at room temperature. The excess alkali was back titrated with 0.5 M hydrochloride acid using phenolphthalein as indicator. Reference and duplicate samples were treated in a similar way. The degree of esterification and the degree of substitution followed the formula below.33,34
 | (2) |
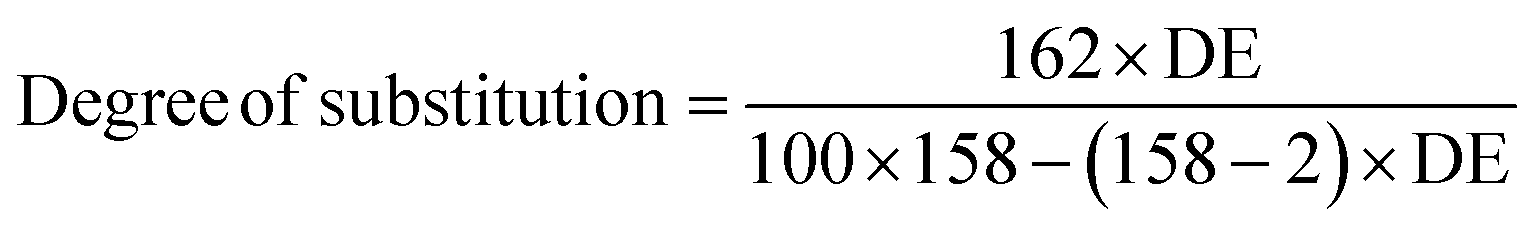 | (3) |
2.6 Zeta potential
The point of zero charge (pHPZC) of citric acid–yeast superabsorbent composites was determined according to the literature.35 All the samples were dispersed in distilled water and then adjusted to predesigned pH values from 2–9 by 0.1 M NaOH and HCl solutions. The zeta potential was indicated by conducting electrophoretic mobility measurements using Malvern Zetasizer Nano ZS90. The zeta potential ζ was determined from electrophoretic mobility measurement of charged samples using the Smoluchowski approximation:
 | (4) |
2.7 Equilibrium swelling method
The equilibrium swelling (qe) was determined by a gravimetric method. Citric acid–yeast composites (1 g) were immersed into deionized water in a Petri dish at room temperature for 2 h, and then poured into a pre-weighted tea bag. The excess water was allowed to drip off the sample due to gravity. The weight of wet tea bag and wet sample was then measured (Xa), and the equilibrium swelling (qe) was calculated according to eqn (5):6
 | (5) |
2.8 Evaluation of pH sensitivity
The solutions with various pH values were adjusted by the dilutions of HCl (pH 1.0) and NaOH (pH 13.0) solutions, and the pH values of the solutions were determined by a pH meter. The pH sensitivity of the samples was investigated in terms of the swelling and deswelling in two buffer solutions with pH 2 and 7, respectively.36 The consecutive time interval for each swelling and deswelling cycle was 120 min.2.9 In vitro ketoprofen loading and controlled release
The drug-delivery system was prepared according to the method described previously with some modifications.37,38 Briefly, ketoprofen was firstly dissolved in ethanol to a concentration of 200 mg mL−1 while protected from light. An appropriate amount of the citric acid–yeast composites was added to form a drug–carrier ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This mixture was shaken in an incubator shaker (sealed with a cover to avoid the volatilization of alcohol) for 6 h to reach the equilibrium state. To monitor the process of drug loading, 5 mL of the solution was withdrawn at predetermined time intervals. The particles were separated out from the mixture by centrifugation and then 1 mL of the supernatant was collected for measurement with a UV-vis spectrophotometer at a wavelength of 260 nm to determine the adsorbed amount of ketoprofen. The drug-loaded products were washed with ethanol and distilled water three times each to remove any residual solutions.
:1. This mixture was shaken in an incubator shaker (sealed with a cover to avoid the volatilization of alcohol) for 6 h to reach the equilibrium state. To monitor the process of drug loading, 5 mL of the solution was withdrawn at predetermined time intervals. The particles were separated out from the mixture by centrifugation and then 1 mL of the supernatant was collected for measurement with a UV-vis spectrophotometer at a wavelength of 260 nm to determine the adsorbed amount of ketoprofen. The drug-loaded products were washed with ethanol and distilled water three times each to remove any residual solutions.
In vitro ketoprofen release from drug-loaded citric acid–yeast composites was carried out by a dissolution study in hydrochloric acid solution (HCl, pH 1.2) and phosphate buffer solution (PBS, pH 6.8).39 Specifically, 20 mg of ketoprofen-loaded samples in a dried state were put into a 500 mL HCl solution or phosphate buffer solution and kept in a water bath maintaining a constant temperature of 37 °C. Aliquots of 5 mL were withdrawn at designated time intervals and analyzed spectrophotometrically at 259 nm for HCl and at 260 nm for PBS. After each measurement, the samples used were returned into the original medium solution to maintain a constant volume of surrounding release medium. Each experiment was conducted in triplicate and the average values were recorded to plot the profiles. The drug loading efficiency and cumulative in vitro release efficiency were calculated according to the following equations:
 | (6) |
 | (7) |
3. Results and discussion
3.1 Preparation and characterization of citric acid–yeast superabsorbent composites
A proposed mechanism for the formation of citric acid–yeast superabsorbent composites by thermo-chemical modification is shown in Scheme 1. | ||
| Scheme 1 The formation mechanism of citric acid–yeast superabsorbent composites via thermo-chemical reaction between citric acid and yeast. | ||
According to a previous report, citric acid is a triprotic α-hydroxy acid with three carboxylic acid functional groups instead of one, which structurally differs from glycolic and lactic acid.40 And consequently, the two carboxylic groups on citric acid could react with all three free hydroxyl groups in the anhydroglucose of biomaterials to form two ester linkages either inter-molecularly between two polymer molecules or intramolecularly within the same polymer molecule.41 In the current research, the complicated components within the yeast microorganism have endowed the cell wall with a number of hydrophilic groups like hydroxyl (–OH), carboxyl (–COOH), amidogen (–NH2), phosphate (–OPO3H2), acylamino (–CONH2) and sulfonyl (–SO3H), which contribute to capturing citric acid firstly by hydrogen bond linkage. When the temperature was heated to boil the water away, the surface of yeast could thereby be coated with lots of citric acid molecules in the semi-dry conditions. As the temperature was heated above 100 °C, the two adjacent carboxylic acid groups on citric acid dehydrated to yield a cyclic anhydride, resulting in the loss of one water molecule. The reactive anhydride subsequently reacted with another hydroxyl group on the polysaccharide or protein of the yeast cell wall to form another ester linkage. At the same time, some other citric anhydrides were formed between unreacted carboxylic acid groups and the neighboring restorable one on the attached citric acid through anhydridization. In these ways, the lots of maze-like crosslinking network structures came into being when these citric anhydrides reacted with the hydroxyl groups from a different polysaccharide or protein molecule of a yeast cell wall, giving rise to the formation of other ester bonds. Correspondingly, the overall surface of yeast cell walls were eventually modified with citric acid by introducing carboxylic acid groups on it, and the independent and decentralized yeast cells were gradually cross-linked by citric acid in the formation of a cross-linked network structure of citric acid–yeast superabsorbent material. Such formation mechanisms are very similar with the formation procedure of Socrates of starch,24 cellulose,42 cyclodextrin,43 and soy protein fiber.44
To confirm the interaction between citric acid and yeast during the formation of citric acid–yeast superabsorbent composites by thermo-chemical modification, FTIR spectroscopy was employed to monitor the chemical bond transformation of parallel native yeast and citric acid–yeast superabsorbent materials synthesized at various temperatures (100–140 °C). Fig. 1 depicts the FTIR spectra of bare yeast and citric acid–yeast superabsorbent composites at different reaction temperatures from (100–140 °C). In Fig. 1(a), the strong and broad band at 3306 cm−1 (O–H and N–H stretching vibrations), 1649 cm−1 (N–H bending vibration and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in amide I), 1547 cm−1 (N–H in amide II), 1406 cm−1 (C–N in amide III), 1240 cm−1 (C–O stretching vibration) and the bands ranged from 1200 to 800 cm−1 (C–O–H and C–O–C stretching vibrations of glucosidic bonds) were the characteristic absorption peaks of the native yeast structure.45,46 After thermo-chemical reaction, the peak at 3306 cm−1 was shifted to 3446 cm−1 (integrated O–H stretching vibrations of –OH and –COOH groups), 1649 cm−1 and 1547 cm−1 were weakened, 1406 cm−1 was strengthened, and 1240 cm−1 was shifted to 1174 cm−1. These changes in Fig. 1(b–f) indicate the participation of –OH groups of the yeast cell wall in graft-polymerization with citric acid through esterification reactions. In addition, other evidence can also verify the reaction, i.e., an intensive absorption band appeared at 1737 cm−1 (CO stretching vibration of ester moiety) and 1365 cm−1 (COO− symmetric stretching),8,47 which was indeed not present in the spectra of native yeast. Previous studies on corn gluten meal and distiller’s dried grain showed similar peaks after reaction with citric acid.48 Overall, all the above indicate that citric acid had reacted with hydroxyl groups on yeast cell walls to form ester linkages, and the carboxylic acid groups have been successfully introduced onto the yeast cell wall.
O in amide I), 1547 cm−1 (N–H in amide II), 1406 cm−1 (C–N in amide III), 1240 cm−1 (C–O stretching vibration) and the bands ranged from 1200 to 800 cm−1 (C–O–H and C–O–C stretching vibrations of glucosidic bonds) were the characteristic absorption peaks of the native yeast structure.45,46 After thermo-chemical reaction, the peak at 3306 cm−1 was shifted to 3446 cm−1 (integrated O–H stretching vibrations of –OH and –COOH groups), 1649 cm−1 and 1547 cm−1 were weakened, 1406 cm−1 was strengthened, and 1240 cm−1 was shifted to 1174 cm−1. These changes in Fig. 1(b–f) indicate the participation of –OH groups of the yeast cell wall in graft-polymerization with citric acid through esterification reactions. In addition, other evidence can also verify the reaction, i.e., an intensive absorption band appeared at 1737 cm−1 (CO stretching vibration of ester moiety) and 1365 cm−1 (COO− symmetric stretching),8,47 which was indeed not present in the spectra of native yeast. Previous studies on corn gluten meal and distiller’s dried grain showed similar peaks after reaction with citric acid.48 Overall, all the above indicate that citric acid had reacted with hydroxyl groups on yeast cell walls to form ester linkages, and the carboxylic acid groups have been successfully introduced onto the yeast cell wall.
 | ||
| Fig. 1 FTIR spectra of yeast (a) and citric acid–yeast superabsorbent composites (CAY100–140) at different temperatures varying from 100 °C to 140 °C (b–f). | ||
Clearly, the formation of citric acid–yeast superabsorbent composites was largely dependent on the reaction temperature, which should result in different modification degrees of yeast with citric acid. Thus, the surface characteristics of citric acid–yeast superabsorbent composites prepared at different reaction temperatures (100–140 °C) have been determined by detecting the carboxyl content, degree of esterification, degree of substitution and point of zero charge. The experimental results are shown in Table 1.
| Samples | Carboxyl content (meq per 100 g) | Degree of esterification (%) | Degree of substitution | Point of zero charge (pHPZC) |
|---|---|---|---|---|
| a CAY100–140 is the citric acid–yeast superabsorbent composites prepared at 100–140 °C, respectively. | ||||
| Yeast | 126 | — | — | 4.5 |
| CAY100 | 328 | 26 | 0.29 | 3.3 |
| CAY110 | 425 | 36 | 0.47 | 3.2 |
| CAY120 | 483 | 45 | 0.69 | 3.1 |
| CAY130 | 389 | 49 | 0.81 | 3.4 |
| CAY140 | 304 | 52 | 0.91 | 3.6 |
As presented in Table 1, the carboxyl content of citric acid–yeast superabsorbent composites was distinctly increased in comparison to that of native yeast. Specifically, the carboxyl content increased from 328 meq per 100 g to 483 meq per 100 g with increasing temperature from 100 °C to 120 °C. Above the temperature range, however, the carboxyl content was reduced. It can be attributed to the fact that additional crosslinking was taking place drastically between the two free carboxyl groups nearby on the modified citric acid–yeast composite samples at a higher temperature, and slower crosslinking was allowed to yield the citric acid–yeast superabsorbent products with higher carboxyl content at a lower temperature. Previous study of citric acid with starch had shown similar phenomenon that a growth in reaction temperature from 110 °C to 130 °C led to attachment of more carboxylic acid groups onto starch, and crosslinking occurred at 140 °C resulting in lower carboxyl content.31 Sessa et al. also found that an enhancement in reaction temperature of corn gluten meal and soy protein isolate with citric acid from 110 °C to 120 °C had caused an increase of carboxyl content.48
The degree of esterification (DE) between the hydroxyl group of yeast and carboxyl groups of citric acid molecules and the degree of substitution (DS) were ascertained by a titration method in order to further investigate the response of reaction efficiency to temperature. DE was the percentage of the reacted hydroxyl groups relative to the total initial hydroxyl groups of the yeast cell wall, and DS was the average number of moles of substituent per anhydroglucose unit.29 The degree of esterification and degree of substitution were increased with the temperature varying from 100 °C to 140 °C. The cause lies in that a high temperature provided a vital driving force to the thermo-chemical modification and crosslinking of yeast with citric acid. As the temperature gradually rose, the anhydride functionality from the carboxyl groups of citric acid was much more reactive towards the hydroxyl group of yeast cell wall, resulting in a higher degree of esterification and degree of substitution.
The successful fixation of citric acid molecular onto the yeast cell wall also can be revealed by its own surface charge. In order to obtain the reliability of surface traits of citric acid–yeast superabsorbent materials and yeast, the zeta potential of bare yeast and citric acid–yeast superabsorbent samples were confirmed to seek the surface charge distribution. The values of the pH necessary to affect a net zero charge on a solid surface in the absence of specific biosorption is called the point of zero charge (pHPZC). This is a convenient index of a surface when it becomes either positively or negatively charged as a function of pH.49 As can be seen from Fig. 2, native yeast exhibited negative zeta potential values (pHPZC 4.5), equal to the value of 4.5 reported by Tian’s group,50 which was due to its negative surface charge both in acidic and alkaline environment. Obviously, the pHPZC of CAY100–140 samples were determined to be 3.3, 3.2, 3.1, 3.4 and 3.6, respectively, with a decreasing order as follows: CAY140 > CAY130 > CAY100 > CAY110 > CAY120. All these values were lower than that of bare yeast, corroborating that the surface of CAY100–140 samples was acidic. It can be noted that the thermo-chemical modification process resulted in more negative values of zeta potential, indicating the increase in acidic sites of pristine yeast. These acidic sites were increased with the increase of reaction temperature from 100 °C to 120 °C, and then decreased with further increasing temperature to 140 °C. The results were very in conformity to that of carboxyl content, which can be accounted to the crosslinking of –COOH and –OH groups in various reaction temperatures.
 | ||
| Fig. 2 Zeta potential of native yeast and citric acid–yeast superabsorbent composites prepared at different temperatures (100–140 °C). | ||
SEM micrographs of native yeast cells before and after thermo-chemical treatment with citric acid (as illustrated in Fig. 3) give further insight into yeast morphology and its modification during the treatment. In Fig. 3(a), the primitive yeast cells displayed regular and orderly ellipsoid shapes with a tight and smooth surface and a mean diameter of about 3.92 ± 0.2 μm in length and 3.38 ± 0.2 μm in width. However, in Fig. 3(b), the structure of the granule surface was completely changed after thermo-chemical modification with citric acid at 120 °C in comparison to the unmodified yeast cells. The citric acid–yeast superabsorbent products had various diameters larger than individual primitive yeast cell, ascribing to the dramatic crosslinking of yeast cells by citric acid. Moreover, the initial smooth appearance was turned to a rough surface with large quantities of pleats and holes, as can be seen in Fig. 3(c). Such surface morphology with a high surface area and abundant –COOH hydrophilic groups endowed the citric acid–yeast superabsorbent material with much more water-absorbing sites than that of bare yeast cells, and was apt for water molecules to penetrating in the internal cross-kinked structure. In Fig. 3(d), the citric acid–yeast superabsorbent sample prepare at 140 °C exhibited a relatively rough surface, instead of distinct holes and cracks. It could be a consequence of excessive esterification and crosslinking of yeast cells by citric acid during the treatment process. It can be concluded from the results that the modification of yeasts with citric acid had greatly changed the surface morphology and structure.
 | ||
| Fig. 3 Morphologies of primitive yeasts (a) and citric acid–yeast superabsorbent composites prepared at 120 °C (b and c) and 140 °C (d). | ||
3.2 Swelling capacity of citric acid–yeast superabsorbent composites
Due to the diverse surface characteristics of citric acid–yeast superabsorbent composites prepared at various temperatures, the citric acid–yeast superabsorbent samples behaved differently in swelling capacity. The absorption curves of yeast and citric acid–yeast superabsorbent species in distilled water are presented in Fig. 4.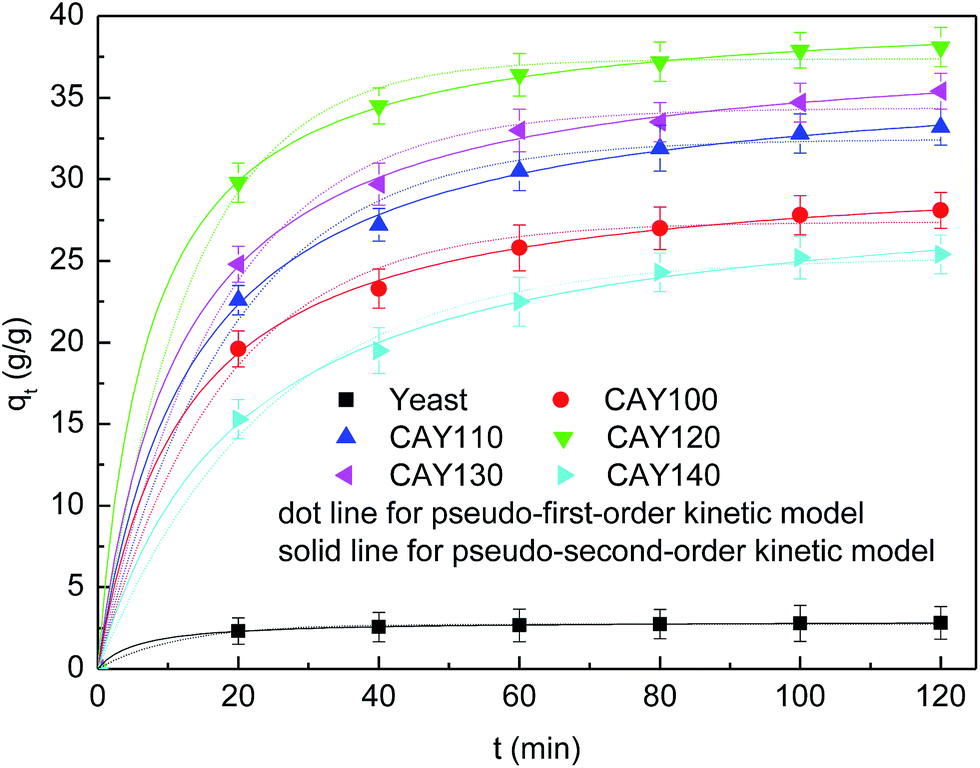 | ||
| Fig. 4 Kinetic models for water absorption of both yeast and citric acid–yeast superabsorbent composites. | ||
As can be perceived, the initial rate of water absorption abruptly rose and then began to level off. The swelling equilibrium could be achieved approximately within 90 min. Similar trends of water absorbency have been noted in the previous literature.51,52 In contrast, the bare yeast had a water uptake value of 2.82, while the water absorption of citric acid–yeast superabsorbent composites was largely higher that of native yeasts. This enhanced water absorption capacity of citric acid–yeast superabsorbent composites, on the one hand, profited from the abundantly hydrophilic carboxyl groups from modification with citric acid. When citric acid–yeast superabsorbent materials were put into water, the carboxylic acid groups lost a proton to become carboxylate groups. These negatively-charged carboxylate groups repelled each other within the superabsorbent matrix and began to swell. At the same time, the enhanced the electrostatic repulsion in the network strengthened the osmotic pressure difference. In theory, the larger the osmotic pressure difference that existed, the faster the water molecules permeated into the absorbents. On the other hand, it may also be originated from porosity of citric acid–yeast superabsorbent particles obtained from the drying process. The fast water extraction during drying procedure induced the high and connected pores in the product, which result in large specific surface area to absorb lots of water in a short time. So, the synthesized citric acid–yeast superabsorbent composites showed a higher velocity in premier swelling progress. As the swelling continued, more water molecular diffused into the network and gradually weakened the osmotic pressure difference. As a result of continuously overcoming the osmotic pressure inside the superabsorbent, the swelling rate became smaller, and the swelling ability finally reached equilibrium due to restriction in movement of cross-links of citric acid–yeast superabsorbent materials. Furthermore, there seemed to be an optimal reaction temperature that resulted in the highest water uptake. In this study, a reaction temperature of 120 °C contributed to the maximum water absorption capacity of citric acid–yeast superabsorbent sample (CAY120) up to 38.1 g H2O per g, and a higher or lower reaction temperature would led to a lower water absorption capacity. Such relevance of water absorption capacity to reaction temperature can be assigned to the facts that the maximum carboxyl content of citric acid–yeast superabsorbent composites may be yield at the optimal reaction temperature of 120 °C. By comparison, citric acid-modified gluten samples under the same reaction condition were able to absorb up to 78 times their weight in deionized water.8 When the temperature was further increased, the carboxyl groups began to dehydrate and changed into carboxylic acid anhydride. Hence, the carboxyl content was reduced, crosslink density was increased, and the water absorption capacity was decreased.
The swelling kinetics is beneficial to make clear mechanisms of swelling process and to evaluate the water absorption efficiency of the samples. To evaluate the swelling ability of citric acid–yeast superabsorbent composites and native yeasts, the experimental data were further fitted by applying pseudo-first-order and pseudo-second-order kinetic model as expressed by the following formulas.53,54
|
ln(qe − qt) = lnqe − k1t
| (8) |
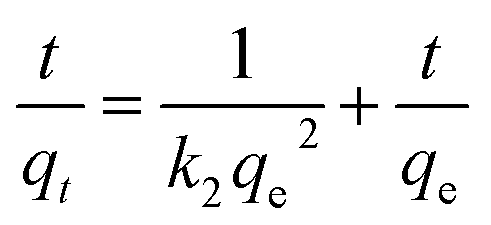 | (9) |
| Samples | qe,exp (g g−1) | Pseudo-first-order kinetic model | Pseudo-second-order kinetic model | ||||||
|---|---|---|---|---|---|---|---|---|---|
| k1 (min−1) | qe,cal (g g−1) | R2 | χ2 | k2 (g g−1 min−1) | qe,cal (g g−1) | R2 | χ2 | ||
| Yeast | 2.82 | 0.0883 | 2.7416 | 0.9953 | 0.0048 | 0.0619 | 2.9321 | 0.9998 | 2.37 × 10−4 |
| CAY100 | 28.1 | 0.0568 | 27.3946 | 0.9922 | 0.7780 | 0.0027 | 30.9469 | 0.9993 | 0.0719 |
| CAY110 | 33.2 | 0.0538 | 32.4583 | 0.9929 | 1.0002 | 0.0021 | 36.9456 | 0.9992 | 0.1117 |
| CAY120 | 38.1 | 0.0763 | 37.3699 | 0.9971 | 0.5431 | 0.0035 | 40.5503 | 0.9999 | 0.0149 |
| CAY130 | 35.4 | 0.0590 | 34.3625 | 0.9540 | 0.9939 | 0.0023 | 38.6179 | 0.9993 | 0.1125 |
| CAY140 | 25.4 | 0.0416 | 25.2118 | 0.9935 | 0.5357 | 0.0017 | 29.9017 | 0.9986 | 0.1128 |
The swelling performance of superabsorbent material is often affected drastically by the characteristics of the external solution, such as the charge number and salt concentration. Interestingly, as depicted in Fig. 5(a), the absorption capacity of citric acid–yeast superabsorbent sample (CAY120) in 0.01 M KCl, CaCl2 and FeCl3 solutions were significantly higher than that in deionized water. This trend was unlike the traditional absorbents, which had smaller volume expansion in saline solutions than pure water.6 A possible explanation was that citric acid–yeast superabsorbent composites acted as a polyelectrolyte with salt tolerance in saline solutions due to the chelating property, i.e., the free carboxylic acid groups of citric acid–yeast superabsorbent materials could chelate metal ions to form metal chelate compounds.55 Hence, the concentrations of K+, Ca2+ and Fe3+ cations were higher inside the structure than outside. As a result of the osmotic pressure difference, the absorption was enhanced consequently. When compared to K+ and Ca2+, Fe3+ has a greater chelating ability with –COO−. Therefore, the density of crosslinking structure increased, the swelling capability decreased and accordingly the absorption was reduced. It can be seen from Fig. 5(a), the swelling capacity of citric acid–yeast superabsorbent sample (CAY120) in different solutions was in the order: KCl > CaCl2 > FeCl3.
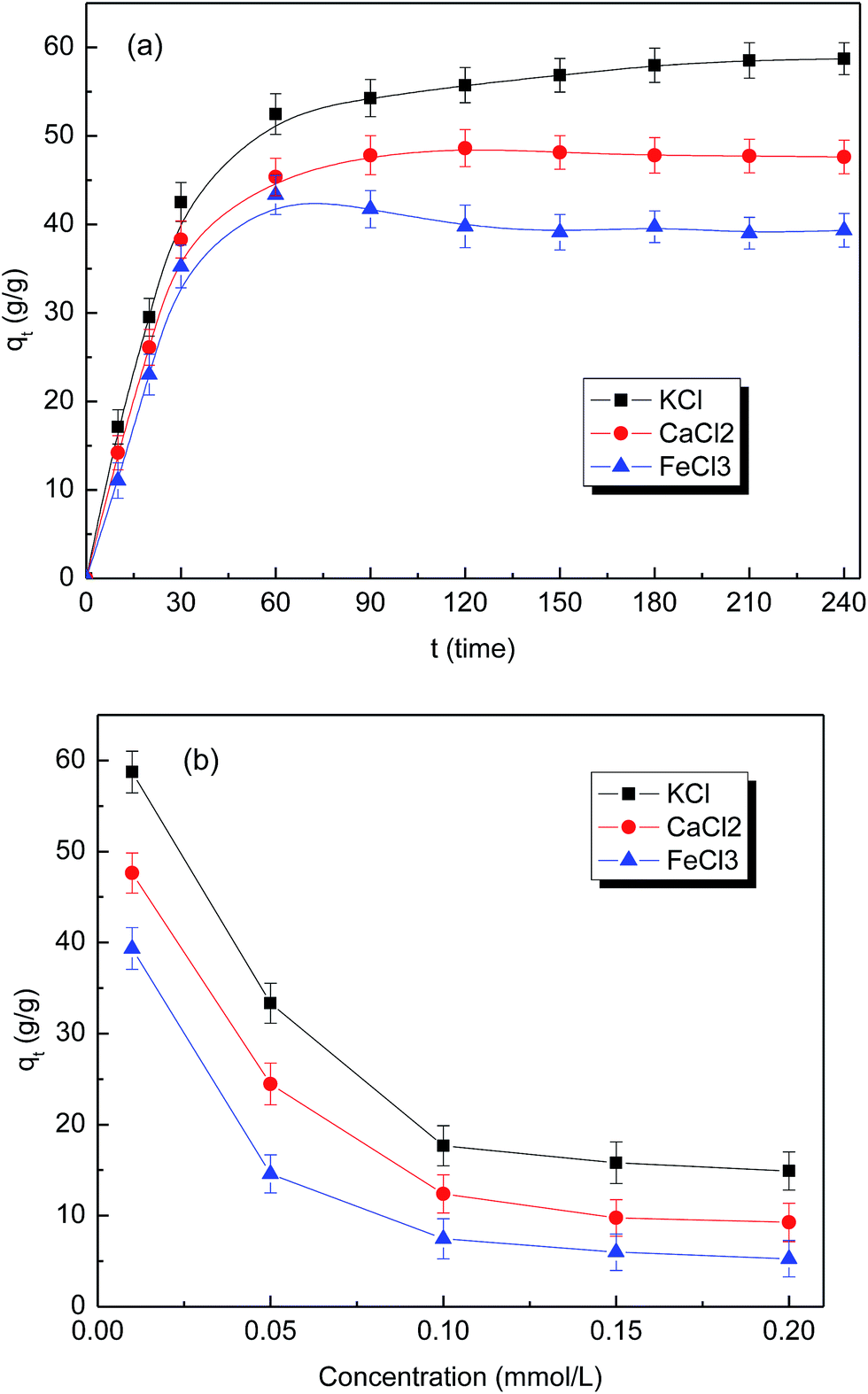 | ||
| Fig. 5 (a) Absorption curves of citric acid–yeast superabsorbent composite (CAY120) in different saline solutions (KCl, CaCl2 and FeCl3); (b) the effect of various salt concentrations on the equilibrium swelling ratio of citric acid–yeast superabsorbent composite (CAY120). | ||
Fig. 5(b) shows the effect of various salt concentrations on the equilibrium swelling ratio of citric acid–yeast superabsorbent materials (CAY120). It is obvious that the water absorbency decreased with increasing the concentration of external salt solutions from 0.01 M to 0.2 M, and the equilibrium swelling ratio in CaCl2 and FeCl3 solutions was lower than that in KCl solution, even at a higher concentration. As we know, the sensitivity of citric acid–yeast superabsorbent composites to monovalent saline was mainly attributed to the reduction of osmotic pressure difference between the interior of superabsorbent and the external solution. The charge screening effect of cations was another factor that influenced the equilibrium swelling ratio in saline solutions. In multivalent saline solution, Ca2+ and Fe3+ cations could form inter and intramolecular complexes with –COO− groups in citric acid–yeast superabsorbent sample (CAY120), which reduced the valid hydrophilic sites in polymeric chains and produced a denser crosslinked network. As a result, the rigid citric acid–yeast superabsorbent materials were restricted to expanding the crosslinked network and the water-holding capability was remarkably decreased. Therefore, the combination of osmotic pressure difference and the charge screening effect determined the swelling behaviors of citric acid–yeast superabsorbent composite (CAY120) in the given saline solution of various concentrations.
In a real-world context, the degree of acidity or alkalinity of swelling medium could impact on the water absorbency properties of the superabsorbent. Fig. 6(a) shows the swelling behavior of citric acid–yeast superabsorbent composite (CAY120) in different solutions of various pH values (2–12) adjusted by diluting HCl (pH 1.0) or NaOH (pH 10.0) solutions. As can be seen, the water absorptions of citric acid–yeast superabsorbent sample (CAY120) are low at strong acidic conditions (2 < pH < 5), but sharply increase and almost reach an equilibrium in the range of pH from 6 to 8. The further increase of pH from 9 to 12 caused a decrease of water absorbency. This phenomenon can be attributed to the abundant carboxylate groups on the surface of citric acid–yeast superabsorbent composite (CAY120). In theory, most carboxylate groups (–COO−) fixed on the structure of citric acid–yeast superabsorbent materials could combine with hydrogen ions (H+) in strong acidic media (2 < pH < 5) to form carboxyl groups (–COOH), decreasing the negative surface charge of the samples. With the reduction of –COO− groups, the electrostatic repulsive force among anionic groups would be weakened, hindering the superabsorbent matrix from swelling even further. Meanwhile, hydrogen ions (H+) would experience interactions with –COOH groups, forming intramolecular and intermolecular hydrogen bonds.56 Thereby, the swelling of the crosslinking structure was restrained and the water absorption was limited. When the pH was higher (6 < pH < 9), more –COOH were converted to free –COO− groups due to ionization, and the number of negatively charged –COO− groups increased, which strengthened the repulsive force, promoting the incorporation of more water into the structure. As a result, the citric acid–yeast superabsorbent composite (CAY120) was inclined to expand and swell more in this pH range. As the pH value reached higher than 10, the charge-screening effect was caused by interactions between Na+ ions and –COO− groups, and also the ester groups were hydrolyzed seriously, which reduced the amount of –COO− groups, destroyed the etherification structure and accordingly weakened the water absorption as well.
 | ||
| Fig. 6 (a) Swelling behavior of citric acid–yeast superabsorbent composite (CAY120) in different pH value solutions; (b) the on–off switching behaviors of citric acid–yeast superabsorbent composite (CAY120). | ||
Since the citric acid–yeast superabsorbent sample (CAY120) exhibited diverse swelling behaviors at different pH values, we tried to switch the pH sensitivity in swelling–deswelling cycles between the buffer solutions of pH 7.0 and 2.0 alternatively. Fig. 6(b) represented the dynamic swelling response of the superabsorbent at pH 7.0 and 2.0 at room temperature. In pH 7 medium, the sample achieved higher swelling, due to the anion–anion repulsive electrostatic forces among carboxylate groups after deprotonation of –COOH groups. In pH 2 solution, the swollen samples rapidly desired, because of the protonation of –COO− groups in acidic medium. In a pH oscillating solution, citric acid–yeast superabsorbent composites exhibited reversible on–off switchable swelling behavior and pH sensitivity. After four on–off cycles, the composites still had better responsivity, indicating that citric acid–yeast superabsorbent sample (CAY120) was promising for potential applications in drug delivery systems.57
3.3 Ketoprofen delivery study of citric acid–yeast superabsorbent composites
Ketoprofen, (RS)-2-(3-benzoylphenyl)-propionic acid, is a potent nonsteroidal anti-inflammatory drug (NSAID) with analgesic, antipyretic and anti-inflammatory effects through inhibiting prostaglandin and leukotriene synthesis.58 It is equally or much stronger than other NSAIDs (such as ibuprofen, phenylbutazone and aspirin) with respect to anti-inflammatory and analgesic activity.59 Ketoprofen has been commonly used for the treatment of inflammatory syndromes such as rheumatoid arthritis, osteoarthritis and acute gouty arthritis as well as for symptoms of trauma.60–62 Ketoprofen can be easily absorbed from the gastrointestinal tract with a plasma half-life of approximately 0.5–4 h, and thus the drug must be frequently administered orally.63 However, like all other NSAIDs, ketoprofen causes undesirable gastrointestinal side effects after being orally administered, like rash, ringing in the ears, headache, dizziness, drowsiness, abdominal pain, nausea, diarrhoea, constipation, heartburn, gastric irritation, ulceration, haemorrhage, etc.64,65 The short biological half-life and associated adverse side effects as well as low single administration dose make ketoprofen a suitable candidate for controlled release formulation.66 In particular, ketoprofen, being a weak acid (pKa 4.6), can be solubilized by adjusting the pH to a higher value as solubility increases with pH above its pKa.67 Therefore, the side effects of perorally administered ketoprofen on the stomach can be minimized by an oral controlled release drug delivery system, i.e., inhibiting the drug release in the gastric region (pH 1.2) and accelerating the drug absorption from the upper portion of the duodenum (pH 6.8) to avoid direct contact with mucosa.68–70 Previous studies have been conducted to ascertain that ketoprofen could be loaded onto the network of pH-sensitive hydrogels through hydrogen bonding and released in simulated gastrointestinal fluid.71,72 These modified gastro-resistant release dosage forms with multiple units, such as micro-particles, beads and pellets, have gained extensive attention due to their advantages over a single unit system, viz., reduced dosing frequency and lessened total dose to avoid high local drug concentration, free dispersion in the gastrointestinal tract to maximize drug absorption and reduce peak plasma fluctuations and side effects, and reduced inter-subject variability in drug absorption and the probability of dose dumping.73 Polymer-based drug delivery systems are used to optimize the therapeutic properties of drugs and render them safer and more efficient and reliable.74 Owing to these benefits, citric acid–yeast superabsorbent composites might be a promising vehicle for drug delivery of ketoprofen. In addition, citric acid–yeast composites are fabricated by modification of pristine yeasts with citric acid, which are natural raw materials and can be digested and degraded in vivo. These degradation products are biocompatible, metabolisable and removable from the human body. More importantly, the manufacturing processes of citric acid–yeast superabsorbent composites are handy, convenient and inexpensive.For medical applications, the drug loading efficiency of citric acid–yeast superabsorbent composite (CAY120) was firstly conducted as a function of time at room temperature and pH = 7, and the results are shown in Fig. 7(a). As can be noted, the loading curve exhibited a typical sustained swelling mode, which closely relied on the swelling properties of citric acid–yeast superabsorbent sample. During the initial three hours, the ketoprofen loading efficiency was promptly reached (62.7%), on account of the rapid swelling behavior of citric acid–yeast superabsorbent composites in aqueous medium that facilitated drug loading. Definitely, carboxylic acid groups on the matrix transformed into negatively-charged carboxylate groups as the samples were immersed in pH = 7 medium with loss of a proton. The formed carboxylate groups repelled each other within the matrix and the cross-linked network began to expand, conducive to a fast rate of drug permeation. As time went by, the drug adsorption capacity began to level off until the loading efficiency ascended to 95.6%, suggesting that the loading rate of ketoprofen was reduced gradually due to the swelling equilibrium of the system. As the illustration of Fig. 7(a) shows, it was the hydrogen bond that makes the drug tightly load onto the particular network of citric acid–yeast composites. These hydrogen bonds were the main junction of polar functional groups derived from citric acid–yeast composites and the chemical structure of ketoprofen, respectively.75 Also, the intermolecular π–π stacking interactions played a small role in drug loading, which was the noncovalent interaction between aromatic rings.
 | ||
| Fig. 7 (a) Drug loading curve of pure ketoprofen on citric acid–yeast composite (CAY120) at room temperature and pH = 7 and the proposed adsorption mechanism; (b) cumulative release profiles of citric acid–yeast composite (CAY120) in HCl solution (pH = 1.2) and phosphate buffer solution (pH = 6.8). | ||
To test the suitability of citric acid–yeast superabsorbent composite (CAY120) to deliver the entrapped ketoprofen further, in vitro cumulative release experiments were performed in simulated gastric fluid (pH 1.2) and simulated intestinal juice (pH 6.8) 37 °C, and the profiles are depicted in Fig. 7(b). As can be seen in Fig. 7, the release pattern of ketoprofen from critic acid–yeast composites was pH-dependent:76 the cumulative release efficiency of ketoprofen in pH 6.8 PBS was notably greater in comparison with that in HCl solution (pH 1.2). Specifically, at pH 6.8, the release pattern of ketoprofen from drug-loaded citric acid–yeast composites was characterized by a burst release during the initial 2 h period, followed by a more sustained release up to reaching a plateau during the remaining time period, and showed the ability to release more than 60% of impregnated ketoprofen in the first 2 h. Whereas at pH 1.2, there was not any distinct burst release revealed, and less than 30% of ketoprofen was released in the same time. As explained in detail previously, three important factors (i.e., diffusion, swelling, and matrix erosion) can be involved in the drug delivery system composed of biodegradable and water-soluble organic materials.77 During the first two hours, the beginning burst drug release at pH 6.8 occurred as soon as the ketoprofen-loaded citric acid–yeast composites transferred to phosphate buffer solution, possibly stemming from the rapid dissolution and diffusion of drug attached to the surface of citric acid–yeast composite matrix, as well as deprotonation of carboxylic groups in a medium of pH value higher than its pKa. Such trends have been reported earlier.78,79 The release process could be explained as follows: the phosphate buffer solution diffused into the citric acid–yeast composites; carboxylic groups deprotonated into carboxylate ions and the cross-linked network began to swell due to the anion–anion repulsion; H2PO4−, PO43− and a little HPO42− in the receiver medium of phosphate buffer solution exchanged with ketoprofen anions; drug ions diffused through the composite matrix and migrated into the receiver medium. Nevertheless, slow and inefficient drug release at pH 1.2 might be attributed to the poor solubility of ketoprofen in acidic medium (pH 1.2) lower than its pKa at about 4.6.80 In addition, protons were fiercely suppressed by the ionization of carboxylic acid groups of citric acid–yeast composites, and consequently the strong hydrogen bonds within the matrix compressed the cross-linked network, leading to a slow movement of solvent into the network, incomplete swelling of the network and frustrated release of interior drug diffusing into receiver medium. Also, the firm connection between drug and matrix restricted the dissolution of ketoprofen into HCl solution. Meanwhile, the periphery of the citric acid–yeast composites would be collapsed via hydrolysis in the face of acid erosion, and its tendency to uptake water was significantly hindered, which decreased the drug diffusion. Hence, there was a limited swelling of samples at pH 1.2 which inhibited the diffusion of drugs at a slower rate. After drug release for 8 h, the incomplete release observed (about 85%) at pH 6.8 could be explained by the combination of a low specific surface area, nonuniform particle sizes and residual drug–matrix chemical interactions. In the meantime, the release efficiency at pH 1.2 was finally reaching 93%, exceeding that (about 85%) at pH 6.8. This phenomenon was in agreement with previous work.77 This finding could be well explained by the citric acid–yeast composites inevitably being broken down into smaller particles by acid erosion gradually, or even the citric acid–yeast superabsorbent composite matrix finally degrading to fully release the integrated drug. Citric acid–yeast superabsorbent composite (CAY120) is a strong potential candidate for drug delivery since its components within the composites have complete degradability. Accordingly, it is an ideal site to improve the bioavailability of poorly absorbable drugs, proteins and vaccines.
4. Conclusion
In summary, the present study was focused on developing a simple and eco-friendly route for the preparation of a novel citric acid–yeast superabsorbent composite material via thermo-chemical modification of yeast with citric acid in semi-dry conditions. The abundant carboxylic acid groups derived from citric acid and special chemical/physical properties inherited from yeast have endowed the citric acid–yeast superabsorbent composites with enhanced water absorption, salt tolerance and pH sensitivity. More specifically, the citric acid–yeast superabsorbent composites were able to absorb up to 38 times their weight in distilled water, and the absorption curves followed a pseudo-second-order kinetic model. The absorption in different saline solutions with various valences were significantly higher than that in deionized water. Moreover, the swelling capacity of citric acid–yeast superabsorbent composites at pH 6 to 9 was stronger than that at other pH values, and exerted pulsation swelling behaviors by modulating the pH values. Such excellent complex characters have awarded an appropriate drug delivery application for the citric acid–yeast superabsorbent composites, i.e. they are very suitable for loading ketoprofen principally by means of hydrogen bonds since the release of the drug could be facilely triggered by adjusting pH with similar pH values of gastrointestinal in human body. Of particular interest regarding this technology that deserves to be mentioned is that it can be extended to the simple synthesis of other naturally biological resources/organic hybrid materials with similar structure or even to the design of more novel and effective on-command drug delivery systems due to their eco-friendly, biocompatible and biodegradable traits.Conflict of interest
The authors declare no competing financial interest.Nomenclature
| k1 | Rate constant for the pseudo-first-order kinetic model (min−1) |
| k2 | Rate constant for the pseudo-second-order kinetic model (g g−1 min−1) |
| Ma | Mass of drug adsorbed in the absorbent at time t (g) |
| Me | Mass of drug entrapped in the absorbent at equilibrium state (g) |
| Mr | Mass of drug released from the drug-loaded absorbent at time t (g) |
| N | Normality of used HCl |
| qe | Absorption capacity at equilibrium state (g g−1) |
| qt | Absorption capacity at time t (g g−1) |
| Va | Volume of HCl used to titrate in the presence of sample (mL) |
| Vb | Volume of HCl used to titrate in the absence of sample (mL) |
| V0 | Volume of HCl used to titrate blank sample (mL) |
| Vn | Volume of HCl used to titrate sample (mL) |
| W | Weight of dry sample (g) |
| UE | Electrophoretic mobility |
| Xa | Weight of wet tea bag and wet sample (g) |
| Yb | Weight of the wet tea bag (g) |
| Zp | Weight of the initial dry sample (g) |
Greek letter
| ε | Dielectric constant |
| ζ | Zeta potential (mV) |
| η | Viscosity (Pa s) |
Acknowledgements
This work was supported by Shaanxi Provincial Natural Science Foundation of China (No. 2015JM2071), National Natural Science Foundation of China (No. 21176031) and Fundamental Research Funds for the Central Universities (No. 2014G3292007).References
- O. S. Lawal, J. Storz, H. Storz, D. Lohmann, D. Lechner and W. M. Kulicke, Eur. Polym. J., 2009, 45, 3399–3408 CrossRef CAS.
- M. Kaur, D. P. S. Oberoi, D. S. Sogi and B. S. Gill, J. Food Sci. Technol., 2011, 48, 460–465 CrossRef CAS PubMed.
- B. Qiu, S. Stefanos, J. L. Ma, A. Lalloo, B. A. Perry, M. J. Leibowitz, P. J. Sinko and S. Stein, Biomaterials, 2003, 24, 11–18 CrossRef CAS PubMed.
- Q. Liu, A. M. Rauth and Y. W. Xiao, Int. J. Pharm., 2007, 339, 148–156 CrossRef CAS PubMed.
- C. T. Chiu, J. S. Lee, C. S. Chu, Y. P. Chang and Y. J. Wang, J. Mater. Sci.: Mater. Med., 2008, 19, 2503–2513 CrossRef CAS PubMed.
- A. Salam, R. A. Venditti, J. J. Pawlak and K. El-Tahlawy, Carbohydr. Polym., 2011, 84, 1221–1229 CrossRef CAS.
- C. Demitri, F. Scalera, M. Madaghiele, A. Sannino and A. Maffezzoli, Int. J. Polym. Sci., 2013, 21, 613–618 Search PubMed.
- B. S. Chiou, H. Jafri, T. Cao, G. H. Robertson, K. S. Gregorski, S. H. Imam, G. M. Glenn and W. J. Imam, J. Appl. Polym. Sci., 2013, 129, 3192–3197 CrossRef CAS.
- W. Zou, L. Yu, X. X. Liu, L. Chen, X. Q. Zhang, D. L. Qiao and R. Z. Zhang, Carbohydr. Polym., 2012, 87, 1583–1588 CrossRef CAS.
- S. Saber-Samandari, M. Gazi and E. Yilmaz, Polym. Bull., 2012, 68, 1623–1639 CrossRef CAS.
- W. B. Wang, Q. Wang and A. Q. Wang, Macromol. Res., 2011, 19, 57–65 CrossRef CAS.
- S. G. A. Alla, M. Sen and A. W. M. El-Naggar, Carbohydr. Polym., 2012, 89, 478–485 CrossRef PubMed.
- J. Li, J. Ji, J. Xia and B. Li, Carbohydr. Polym., 2012, 87, 757–763 CrossRef CAS.
- W. B. Wang and A. Q. Wang, Carbohydr. Polym., 2009, 77, 891–897 CrossRef CAS.
- F. M. Klis, Yeast, 1994, 10, 851–869 CrossRef CAS PubMed.
- T. H. Nguyen, G. H. Fleet and P. L. Rogers, Appl. Microbiol. Biotechnol., 1998, 50, 206–212 CrossRef CAS PubMed.
- P. Magnelli, J. F. Cipollo and C. Abeijon, Anal. Biochem., 2002, 301, 136–150 CrossRef CAS PubMed.
- B. Aguilar-Uscanga and J. M. François, Lett. Appl. Microbiol., 2003, 37, 268–274 CrossRef CAS PubMed.
- E. P. Feofilova, Microbiology, 2010, 79, 711–720 CAS.
- C. M. Hogan, Abiotic factor, in Encyclopedia of Earth, ed. E. Monosson and C. Cleveland, National Council for Science and the Environment, Washington, DC, 2010 Search PubMed.
- M. Berovic and M. Legisa, Biotechnol. Annu. Rev., 2007, 13, 303–343 CAS.
- K. L. Penniston, S. Y. Nakada, R. P. Holmes and D. G. Assimos, J. Endourol., 2008, 22, 567–570 CrossRef PubMed.
- D. S. Franklin and S. Guhanathan, Polym. Bull., 2014, 71, 93–110 CrossRef CAS.
- N. Reddy and Y. Yang, Food Chem., 2010, 118, 702–711 CrossRef CAS.
- D. Alonso, M. Gimeno, R. Olayo, H. Vázquez-Torres, J. D. Sepúlveda-Sánchez and K. Shirai, Carbohydr. Polym., 2009, 77, 536–543 CrossRef CAS.
- X. F. Ma, P. R. Chang, J. G. Yu and M. Stumborg, Carbohydr. Polym., 2009, 75, 1–8 CrossRef CAS.
- D. J. Sessa and R. E. Wing, Nahrung, 1998, 42, 266–268 CAS.
- S. Abdus, J. J. Pawlak, R. A. Venditti and E. T. Khaled, Biomacromolecules, 2010, 11, 1453–1459 CrossRef.
- A. Salam, J. J. Pawlak, R. A. Venditti and K. El-Tahlawy, Cellulose, 2011, 18, 1033–1041 CrossRef CAS.
- T. Farjami, A. Madadlou and M. Labbafi, Food Hydrocolloids, 2015, 50, 159–165 CrossRef CAS.
- R. E. Wing, Starch/Staerke, 1996, 48, 275–278 CrossRef CAS.
- O. B. Wurzburg, Cross-linked starches, in Modified starches: properties and uses, ed. O. B. Wurzburg, CRC Press, Boca Raton, Florida, 1986, pp. 41–53 Search PubMed.
- F. E. Soetaredjo, S. Ismadji, L. H. Huynh, N. S. Kasim, N. Y. Tran-Thi, A. Ayucitra and Y. S. Ju, Starch/Staerke, 2012, 64, 590–597 CrossRef CAS.
- M. Elomaa, T. Asplund, P. Soininen, R. Laatikainen, S. Peltonen, S. Hyvärinen and A. Urtti, Carbohydr. Polym., 2004, 57, 261–267 CrossRef CAS.
- S. Pitsari, E. Tsoufakis and M. Loizidou, Chem. Eng. J., 2013, 223, 18–30 CrossRef CAS.
- Y. Z. Wang, W. B. Wang, X. N. Shi and A. Q. Wang, J. Appl. Polym. Sci., 2013, 130, 161–167 CrossRef CAS.
- A. Abd-Elbary, M. A. E. Nabarawi, D. H. Hassen and A. A. Taha, Int. J. Pharm. Pharm. Sci., 2014, 6, 183–191 CAS.
- L. Z. Huang, H. X. Wang, B. Li, E. Li, Y. H. Zhou, Y. G. Yang, C. Dong and S. M. Shuang, J. Inclusion Phenom. Macrocyclic Chem., 2014, 80, 209–215 CrossRef CAS.
- L. Segale, P. Mannina, L. Giovannelli, H. Danan, P. Esposito, L. Galli and F. Pattarino, Eur. J. Pharm. Biopharm., 2012, 82, 491–497 CrossRef CAS PubMed.
- M. M. Fiume, B. A. Heldreth, W. F. Bergfeld, D. V. Belsito, R. A. Hill, C. D. Klaassen, D. C. Liebler, J. G. J. Marks, R. C. Shank, T. J. Slaga, P. W. Snyder and F. A. Andersen, Int. J. Toxicol., 2014, 33, 16S–46S CrossRef PubMed.
- C. Menzel, E. Olsson, T. S. Plivelic, R. Andersson, C. Johansson, R. Kuktaite, L. Jarnstrom and K. Koch, Carbohydr. Polym., 2013, 96, 270–276 CrossRef CAS PubMed.
- V. Coma, I. Sebti, P. Pardon, F. H. Pichavant and A. Deschamps, Carbohydr. Polym., 2003, 51, 265–271 CrossRef CAS.
- R. Anand, M. Malanga, I. Manet, F. Manoli, K. Tuza, A. Aykaç, C. Ladavière, E. Fenyvesi, A. Varqas-Berenquel, R. Gref and S. Monti, Photochem. Photobiol. Sci., 2013, 12, 1841–1854 CAS.
- N. Reddy and Y. Yang, Biotechnol.Prog., 2009, 25, 139–146 CrossRef CAS PubMed.
- B. Bo, N. Quici, Z. Li and G. L. Puma, Chem. Eng. J., 2011, 170, 451–456 CrossRef.
- D. J. Feng, B. Bai, C. X. Ding, H. L. Wang and Y. R. Suo, Ind. Eng. Chem. Res., 2014, 53, 12760–12769 CrossRef CAS.
- C. Demitri, R. D. Sole, F. Scalera, A. Sannino, G. Vasapollo, A. Maffezzoli, L. Ambrosio and L. Nicolais, J. Appl. Polym. Sci., 2008, 110, 2453–2460 CrossRef CAS.
- D. J. Sessa and R. E. Wing, Ind. Crops Prod., 1999, 10, 55–63 CrossRef CAS.
- A. E. Ofomaja and H. Yuh-Shan, J. Hazard. Mater., 2007, 139, 356–362 CrossRef CAS PubMed.
- Y. Tian, C. Y. Ji, M. J. Zhao, M. Xu, Y. S. Zhang and R. G. Wang, Chem. Eng. J., 2010, 165, 474–481 CrossRef CAS.
- W. B. Wang and A. Q. Wang, Carbohydr. Polym., 2010, 82, 83–91 CrossRef CAS.
- Y. Zhao, H. J. Su, L. Fang and T. W. Tan, Polymer, 2005, 46, 5368–5376 CrossRef CAS.
- S. Langergren and B. K. Svenska, Veternskapsakad Handlingar, 1898, 24, 1–39 Search PubMed.
- Y. S. Ho and G. McKay, Process Biochem., 1999, 34, 451–465 CrossRef CAS.
- P. Bernabé, C. Peniche and W. Argüelles-Monal, Polym. Bull., 2005, 55, 367–375 CrossRef.
- X. H. Xu, B. Bai, C. X. Ding, H. L. Wang and Y. R. Suo, Ind. Eng. Chem. Res., 2015, 54, 3268–3278 CrossRef CAS.
- M. Sadeghi and M. Yarahmadi, Afr. J. Biotechnol., 2011, 10, 12265–12275 CAS.
- G. C. Ceschel, P. Maffei and B. S. Lombardi, Drug Delivery, 2002, 9, 39–45 CrossRef CAS PubMed.
- T. G. Kantor, Pharmacotherapy, 1986, 6, 93–103 CrossRef CAS PubMed.
- I. E. Shohin, J. I. Kulinich, G. V. Ramenskaya, B. Abrahamsson, S. Kopp, P. Langguth, J. E. Polli, V. P. Shah, D. W. Groot, D. M. Barends and J. B. Dressman, J. Pharm. Sci., 2012, 101, 3593–3603 CrossRef CAS PubMed.
- D. Kovala-Demertzi, J. Organomet. Chem., 2006, 691, 1767–1774 CrossRef CAS.
- J. Suksaeree, L. Charoenchai, C. Monton, T. Chusut, A. Sakunpak, W. Pichayakorn and P. Boonme, Ind. Eng. Chem. Res., 2013, 52, 15847–15854 CrossRef CAS.
- D. B. Mazières, Topical Ketoprofen Patch, Drugs R&D, 2005, 6, 337–344 Search PubMed.
- R. L. Savage, P. W. Moller, C. L. Ballantyne and J. E. Wells, Arthritis Rheum., 1993, 36, 84–90 CrossRef CAS PubMed.
- G. C. Ceschel, P. Maffei and B. S. Lombardi, Drug Delivery, 2002, 9, 39–45 CrossRef CAS PubMed.
- G. F. Palmieri, G. Bonacucina, M. P. Di and S. Martelli, J. Microencapsulation, 2002, 19, 111–119 CrossRef CAS PubMed.
- S. Sridevi and P. V. R. Diwan, Eur. J. Pharm. Biopharm., 2002, 54, 151–154 CrossRef CAS PubMed.
- H. Cao, X. Y. Ma, S. H. Sun, H. J. Su and T. W. Tan, Polym. Bull., 2010, 64, 623–632 CrossRef CAS.
- R. Ganjoo, S. Soni and V. Ram, Inventi Impact: NDDS, 2013, 4, 283–288 Search PubMed.
- A. Roda, L. Sabatini, M. Mirasoli, M. Baraldini and E. Roda, Int. J. Pharm., 2002, 241, 165–172 CrossRef CAS PubMed.
- H. M. N. El-Din, A. W. M. El-Naggar and F. I. A. E. Fadle, Int. J. Polym. Mater., 2013, 62, 711–718 CrossRef CAS.
- A. I. Raafat, J. Appl. Polym. Sci., 2010, 118, 2642–2649 CrossRef CAS.
- L. Segale, P. Mannina, L. Giovannelli, H. Danan, P. Esposito, L. Galli and F. Pattarino, Eur. J. Pharm. Biopharm., 2012, 82, 491–497 CrossRef CAS PubMed.
- M. Nishikawa, Y. Takakura and M. Hashida, Adv. Drug Delivery Rev., 1996, 21, 135–155 CrossRef CAS.
- M. Prabaharan and J. F. Mano, e-Polym., 2013, 7, 503–516 Search PubMed.
- J. J. Sheng, N. A. Kasim, R. Chandrasekharan and G. L. Amidon, Eur. J. Pharm. Sci., 2006, 29, 306–314 CrossRef CAS PubMed.
- C. A. García-González, M. Jin, J. Gerth, C. Alvarez-Lorenzo and I. Smirnova, Carbohydr. Polym., 2015, 117, 797–806 CrossRef PubMed.
- A. Verma and J. K. Pandit, Trop. J. Pharm. Res., 2011, 10, 61–67 CAS.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS PubMed.
- R. V. Kulkarni and B. Sa, J. Drug Targeting, 2008, 16, 167–177 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |