
Adsorption of perrhenate ion by bio-char produced from Acidosasa edulis shoot shell in aqueous solution†
Abstract
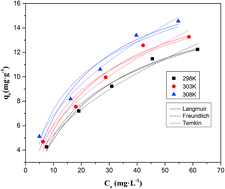
The adsorption of perrhenate ions by the bio-char prepared from Acidosasa edulis shoot shell at 773 K is investigated under acidic conditions. The effects of some important parameters including initial pH (1.0–6.0), adsorbent dose (0.8–8.0 g L−1), contact time (2–480 min) and initial perrhenate ion concentration (10–100 mg L−1), on the recovery of perrhenate ions from aqueous solution in batch experiments are tested. The adsorbent was characterized by scanning electron microscopy equipped with an energy-dispersive X-ray spectroscopy (SEM-EDX), attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) and specific surface area analysis. The adsorption data are well described by Freundlich isotherm and maximum perrhenate ions adsorption capacities of 14.6 mg g−1 for Acidosasa edulis shoot shell bio-char under the optimum conditions. Kinetics of adsorption are found to follow the pseudo-second-order rate equation. Thermodynamic analysis suggested that the adsorption is an endothermic process and occurs spontaneously. FTIR analysis confirmed the major involvement of hydroxyl and carboxyl groups during perrhenate ion adsorption. Further, more than 94% of total rhenium adsorbed could be recovered using 0.1 mol L−1 KOH as a desorption medium. The mechanism analysis indicates that the outer-sphere complexes and electronic attraction mechanism were involved in the adsorption of perrhenate ions. The results indicate that Acidosasa edulis shoot shell waste derived bio-char can act as an effective adsorbent material for perrhenate ions recovery from copper smelting acidic wastewater.
 Please wait while we load your content...
Please wait while we load your content...

