
Copper promoted synthesis of substituted quinolines from benzylic azides and alkynes†
Ching-Zong Luo,
Parthasarathy Gandeepan,
Yun-Ching Wu,
Wei-Chen Chen and
Chien-Hong Cheng*
Department of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: chcheng@mx.nthu.edu.tw; Fax: +886-3-5724698; Tel: +886-3-5721454
First published on 3rd December 2015
Abstract
A novel copper promoted synthesis of substituted quinolines from various benzylic azides and internal alkynes has been demonstrated. The reaction features a broad substrate scope, high product yields and excellent regioselectivity. In contrast to the known two-step process of acid promoted [4 + 2] cycloaddition reaction and oxidation, the present methodology allows the synthesis of quinolines in a single step under neutral reaction conditions and can be applied to the synthesis of biologically active 6-chloro-2,3-dimethyl-4-phenylquinoline (antiparasitic agent) and 3,4-diphenylquinolin-2(1H)-one (p38αMAP kinase inhibitor). A plausible reaction mechanism involves rearrangement of benzylic azide to N-arylimine (Schmidt reaction) followed by intermolecular [4 + 2] cycloaddition with internal alkynes.
Introduction
Quinoline and its derivatives are potential heterocyclic scaffolds found in many natural products1–3 and bio-active molecules.4–10 They are also key structures in materials, agrochemicals, dyestuffs, and pharmaceuticals.11–15 In particular, the use of quinoline derivatives as ligands for transition metal complexes for organic synthesis and organic light-emitting materials has attracted great attention for their synthesis (Fig. 1).16–21 | ||
| Fig. 1 Examples of useful quinoline cored compounds. | ||
Many of the classical methods, such as Pfitzinger, Skraup, Friedlander, Doebner von Miller, Conrad–Limbach, and Combes reactions, known for the synthesis of quinolines are limited by harsh reaction conditions, limited substrate scope and low yields.22 Among the various synthetic strategies,23,24 Lewis acid promoted tandem cyclization reaction of N-aryl imines with terminal alkynes under oxidative condition allows access to a variety of 2,4-disubstituted quinolines.25–28 The proposed reaction mechanism involves the nucleophilic addition of a terminal alkyne to imine to form propargylamine intermediate, which undergoes intramolecular cyclization, followed by oxidation to afford quinoline products. Similar cyclization reactions using internal alkynes are difficult and hardly achieved.29 These reactions are not suitable for the synthesis of quinolines without substitution at C-2 position (Scheme 1a).
 | ||
| Scheme 1 Synthetic strategies for substituted quinolines. | ||
Recently, the in situ generation of N-aryliminium ions from benzylic azides by means of strong acids has been utilized for the synthesis of tetrahydroquinoline scaffolds, which can be oxidized to quinolines by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ).30,31 The drawbacks of these reactions are (i) lower product yields, (ii) use of strong acid, (iii) poor regioselectivity and (iv) a two-step process (cyclization and oxidation) (Scheme 1b). Because of the useful applications of quinolines and the lack of simple and high yielding method for their synthesis, the search for new synthetic methodologies are still highly sought after. Our continuing interests in the quinoline synthesis32,33 lead us to envision that the in situ generation of N-aryliminium ion from benzylic azide using a Lewis acid catalyst which is also an oxidant under acid free condition provides a good opportunity to avoid the above mentioned problems at least in part. In this report, we disclose a simple and a novel method for the synthesis of various substituted quinolines from benzylic azides and internal alkynes under neutral reaction conditions using Cu(OTf)2 as both a Lewis acid and oxidant (Scheme 1c).
Results and discussion
Initially, several CuII-salts were screened for the reaction of benzyl azide (1a) and diphenylacetylene (2a) to form 3,4-diphenylquinoline (3aa) (Table 1). Treatment of 1a (0.34 mmol), and 2a (0.28 mmol) in the presence of Cu(OTf)2 (0.56 mmol) in MeNO2 at 100 °C for 24 h gave 3aa in 36% yield (Table 1, entry 2). The product was unambiguously confirmed by its 1H and 13C NMR, HRMS, and X-ray structure analysis.34 Among the Cu(II)-salts and other Lewis acids tested, only Cu(BF4)2·6H2O showed considerable potency to give 3aa in 33% yield (see, Table SI† for the detailed optimization studies). By reversing the ratio of 1a and 2a from 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1.2, 3aa was obtained in 63% yield (entry 7). The yield of 3aa further increased to 77%, as the amount of 2a was raised to 2 equiv. (entry 8). We then optimized the amount of Cu(OTf)2, reaction time and temperature. The optimized reaction conditions with 1a (0.28 mmol), 2a (0.56 mmol), and Cu(OTf)2 (0.70 mmol) in MeNO2 (2 mL) at 80 °C for 24 h provided 3,4-diphenylquinoline (3aa) in 93% isolated yield. The choice of solvent is crucial to obtain a high product yield. Among the several solvents tested, MeNO2 afforded the highest product yield, while PhNO2 and CF3CH2OH also gave 3aa in 60 and 75% yields, respectively (Table 1). The attempt to use Cu(OTf)2 as catalyst by employing 0.5 equivalent of Cu(OTf)2 with a stoichiometric amount of K2S2O8 or (NH4)2S2O8 as oxidants gave the quinoline product in about 50% yield (see Table 1).
:1 to 1:1.2, 3aa was obtained in 63% yield (entry 7). The yield of 3aa further increased to 77%, as the amount of 2a was raised to 2 equiv. (entry 8). We then optimized the amount of Cu(OTf)2, reaction time and temperature. The optimized reaction conditions with 1a (0.28 mmol), 2a (0.56 mmol), and Cu(OTf)2 (0.70 mmol) in MeNO2 (2 mL) at 80 °C for 24 h provided 3,4-diphenylquinoline (3aa) in 93% isolated yield. The choice of solvent is crucial to obtain a high product yield. Among the several solvents tested, MeNO2 afforded the highest product yield, while PhNO2 and CF3CH2OH also gave 3aa in 60 and 75% yields, respectively (Table 1). The attempt to use Cu(OTf)2 as catalyst by employing 0.5 equivalent of Cu(OTf)2 with a stoichiometric amount of K2S2O8 or (NH4)2S2O8 as oxidants gave the quinoline product in about 50% yield (see Table 1).
|
||||
|---|---|---|---|---|
| Entry | Lewis acid (equiv.) | Solvent | Temp. (°C)/time (h) | Yielda (%) |
| a Unless otherwise mentioned, all reactions were performed with 1a (0.34 mmol), 2a (0.28 mmol, limiting reagent), and Lewis acid (as given in the table) in MeNO2 (2 mL) at 100 °C for 24 h. Isolated yields based on the limiting reagents were given. Yield given in the parenthesis was isolated yield. DCE: 1,2-dichloroethane; TFE: 2,2,2-trifluoroethanol.b 1a (0.28 mmol, limiting reagent), and 2a (0.34 mmol) were used.c Reactions were performed using 1a (0.28 mmol, limiting reagent), 2a (0.56 mmol), and Lewis acid (as given in the table) in MeNO2 (2 mL) at the given temperature and time.d Cu(OTf)2 (0.14 mmol) was used along with K2S2O8 or (NH4)2S2O8 (0.32 mmol). | ||||
| 1 | Cu(OAc)2 (2) | MeNO2 | 100/24 | — |
| 2 | Cu(OTf)2 (2) | MeNO2 | 100/24 | 36 |
| 3 | Cu(OCOCF3)2 H2O (2) | MeNO2 | 100/24 | Trace |
| 4 | Cu(BF4)2·6H2O (2) | MeNO2 | 100/24 | 33 |
| 5 | CuCl2 (2) | MeNO2 | 100/24 | — |
| 6 | CuF2 (2) | MeNO2 | 100/24 | — |
| 7 | Cu(OTf)2 (2) | MeNO2 | 100/24 | 63b |
| 8 | Cu(OTf)2 (2) | MeNO2 | 100/24 | 77c |
| 9 | Cu(OTf)2 (3) | MeNO2 | 100/24 | 92c |
| 10 | Cu(OTf)2 (4) | MeNO2 | 100/24 | 87c |
| 11 | Cu(OTf)2 (3) | MeNO2 | 80/24 | 89c |
| 12 | Cu(OTf)2 (3) | MeNO2 | 60/24 | 80c |
| 13 | Cu(OTf)2 (2.5) | MeNO2 | 80/24 | 93c |
| 14 | Cu(OTf)2 (2.5) | MeNO2 | 80/15 | 72c |
| 15 | Cu(OTf)2 (1), 1 atm O2 | MeNO2 | 80/24 | 45c |
| 15 | Cu(OTf)2 (3) | EtOAc | 80/24 | 36c |
| 16 | Cu(OTf)2 (3) | DCE | 80/24 | 22c |
| 17 | Cu(OTf)2 (3) | PhNO2 | 80/24 | 60c |
| 18 | Cu(OTf)2 (3) | TFE | 80/24 | 75c |
| 19 | Cu(OTf)2 (0.5), K2S2O8 | MeNO2 | 100/24 | 49c,d |
| 20 | Cu(OTf)2 (0.5), (NH4)2S2O8 | MeNO2 | 100/24 | 52c,d |

Next, we applied the optimized reaction conditions to a variety of benzylic azides to probe the generality of the reaction (Scheme 2). First, we tested the para substituted benzylic azides 1b–l. Both electron-donating-group (EDG) and electron-withdrawing-group (EWG) substituted substrates were effectively transformed into the respective quinoline products. Thus, the EDG, 4-Me and 4-iPr substituted substrates offered products 3ba and 3ca in 92% and 90% yields, respectively, but the very electron-donation 4-methoxy substituted benzyl azide (1d) yielded only 36% of product 3da. Halo-substituted benzylazides 1e–h are compatible under the reaction conditions to give the expected products 3ea–ha in good yields. EWGs containing benzylic azides 1i–l provided the respective substituted quinolines 3ia–la in 44–90% yields. The steric hindrance of ortho substitution at benzylic azides does not show consistent influence on the yield of this cyclization reaction. We studied the reactions of benzylic azides possessing different ortho substituents such as Me, OMe, Ph, Br, and I with alkyne 2a. The results reveal that most of the reactions afforded the respective quinolines in high yields (Scheme 2, products 3ma–qa). Tetra substituted quinolines 3ra–ta were obtained in good yields by employing disubstituted benzylic azides 1r–t. The reaction of 3-methylbenzylic azide gave an inseparable mixture of regioisomeric quinoline products 3ua + 3ua′ (6:4) in 72% yield. Synthesis of substituted benzo[h]quinoline (3va) and benzo[f]quinoline (3wa) were achieved from 1v and 1w in 74 and 84% yields, respectively. The structure of 3va was further confirmed by X-ray analysis.34 It is worth to mention that the halo substituted quinoline products 3ea–ha, 3pa–qa are useful for further functionalization via cross couplings.
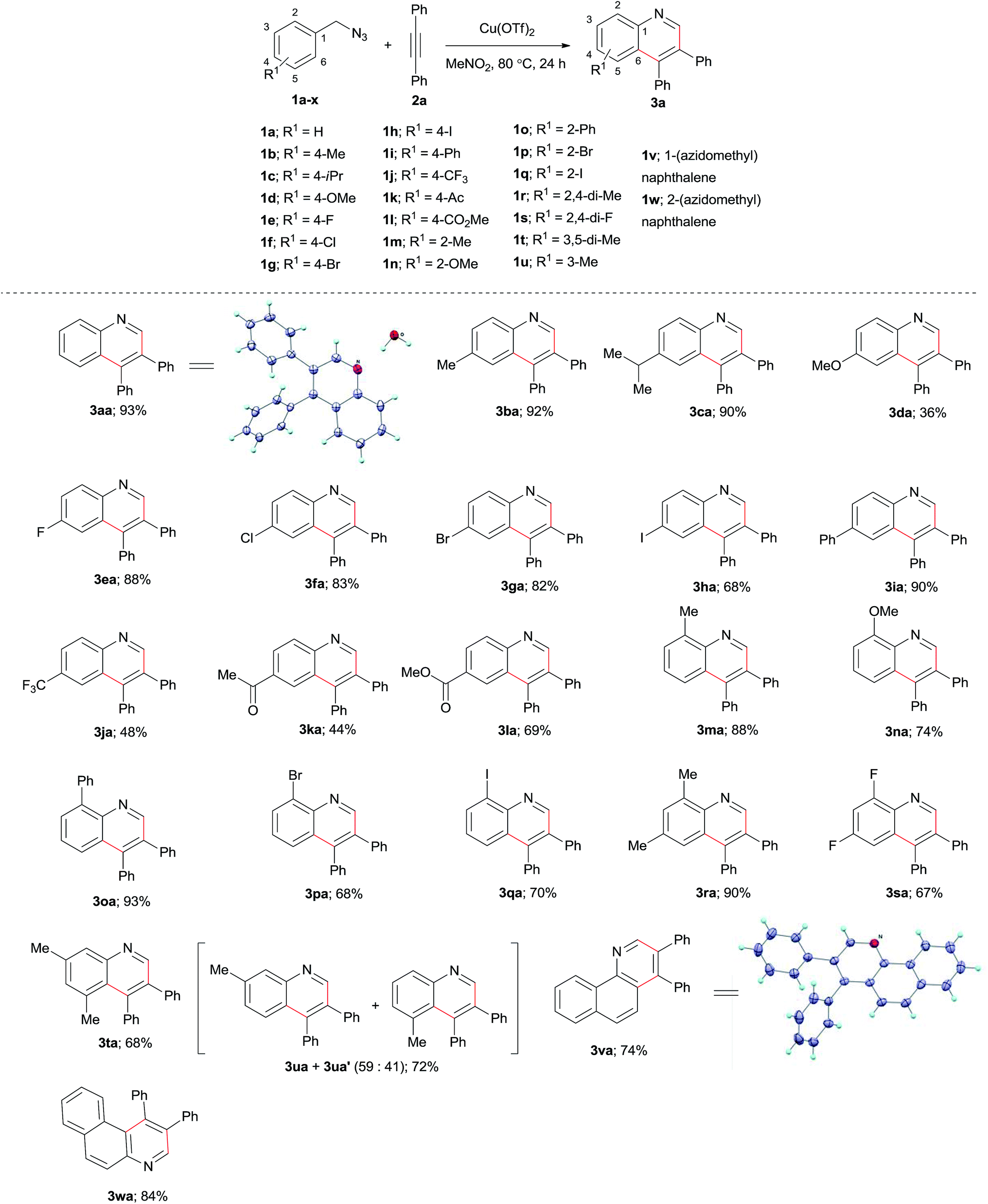 | ||
| Scheme 2 Scope of benzylic azides in the synthesis of substituted quinolines. Conditions: 1 (0.28 mmol), 2a (0.56 mmol), and Cu(OTf)2 (0.70 mmol) in MeNO2 (2 mL) at 80 °C for 24 h. Isolated yields were determined based on 1. | ||
This copper(II) promoted quinoline synthesis was further expanded to a range of symmetrical and unsymmetrical internal alkynes (Scheme 3). Thus, the reaction of p-ditolylacetylene (2b) with 1a gave the expected product 3ab in 87% yield. Similarly, p-F, p-Cl, p-Br substituted diarylacetylenes 2c–e reacted with 1a to provide the corresponding quinolines 3ac–ae in moderate yields. Di(2-thienyl)acetylene also underwent the cyclization reaction to provide 3af in 33% yield. Next, we tested a number of unsymmetrical alkynes 2g–l with 1a and found that the reactions are highly regioselective giving exclusively a single regioisomeric product for each of these reactions. Under the reaction conditions, 1-phenylpropyne (2g) and 1-butynylbenzene (2h) provided the respective quinolines 3ag and 3ah in 87 and 96% yields, respectively. The regioselectivity of the products were confirmed by X-ray structure analysis.34 Electron-deficient alkyne such as ethyl phenylpropiolate (2i) is also active affording the expected quinoline product 3ai in 62% yield. Moreover, 3-tolylpropargyl alcohol (2j), benzyl phenyl acetylene (2k) and 3-tolylpropargyl amine derivative (2l) also operative to produce quinoline derivatives 3aj–3al in good to excellent yields.
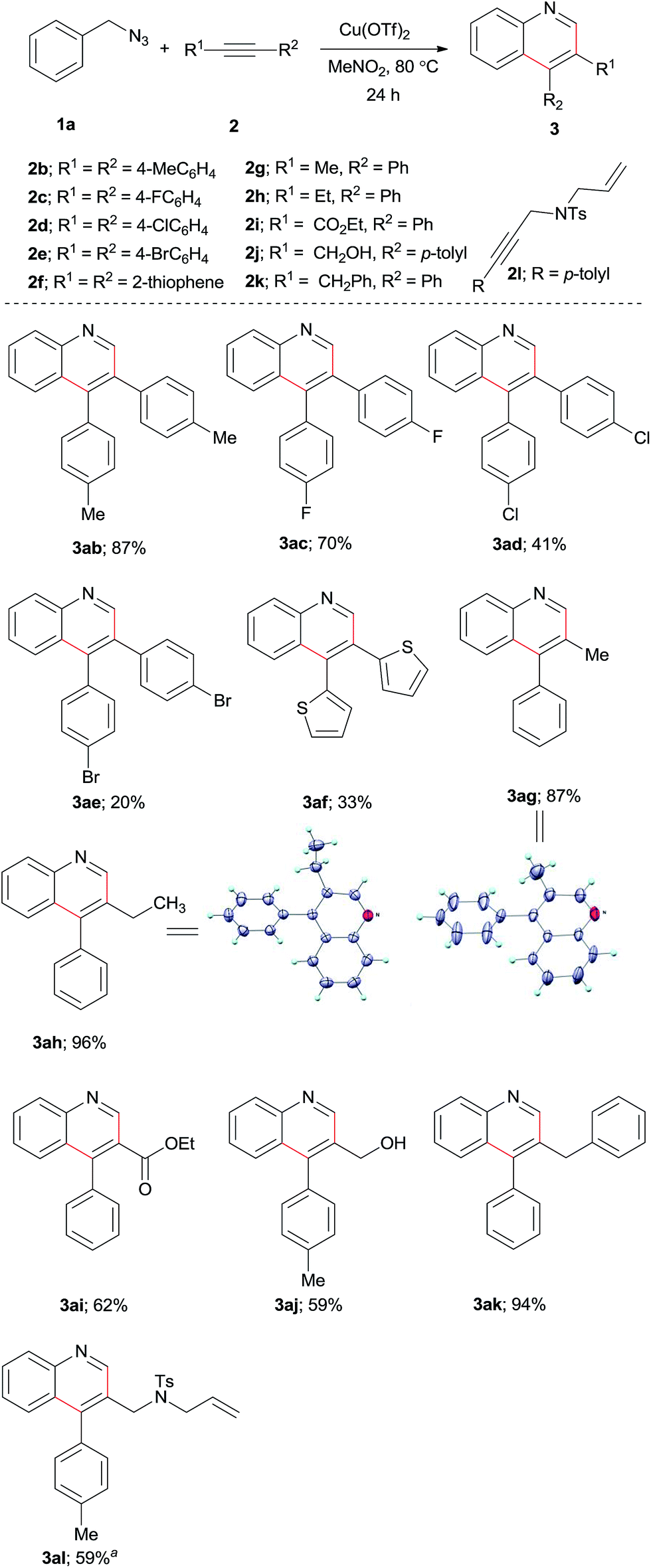 | ||
| Scheme 3 Scope of alkynes in the synthesis of substituted quinolines. Conditions: 1a (0.28 mmol), 2 (0.56 mmol), and Cu(OTf)2 (0.70 mmol) in MeNO2 (2 mL) at 80 °C for 24 h. aCu(BF4)2·6H2O (0.70 mmol) was used instead of Cu(OTf)2. | ||
To demonstrate the synthetic utility of the present copper promoted cyclization reaction, we synthesize biologically active compound 6-chloro-2,3-dimethyl-4-phenylquinoline (4) by the present method. The quinoline compound is known to show effectiveness against Leishmania donovani, Trypanosoma cruzi, T. b. rhodesiense.35 As shown in Scheme 4, compound 4 was conveniently synthesized from 1f and 2g in two steps with 62% overall yield.
 | ||
| Scheme 4 Synthesis of biologically active 6-chloro-2,3-dimethyl-4-phenylquinoline 4. | ||
We also demonstrated the synthesis of 3,4-diphenylquinolin-2(1H)-one (5) from 3,4-diphenylquinoline (3aa) (Scheme 5). Compound 5 is known to be a potential p38αMAP kinase inhibitor.36,37
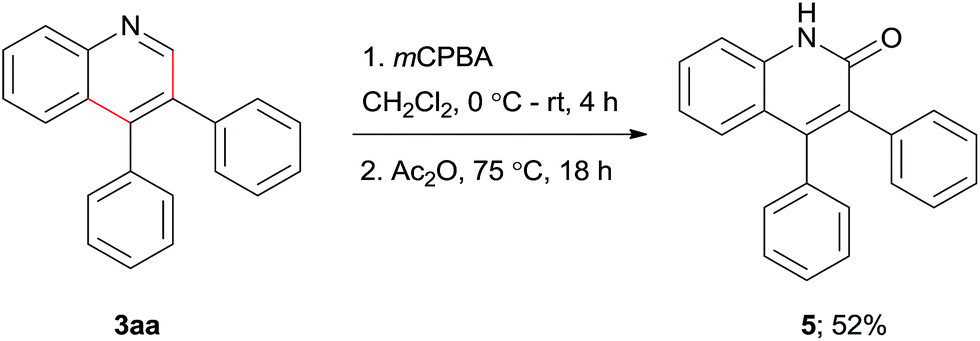 | ||
| Scheme 5 Synthesis of 3,4-diphenylquinolin-2(1H)-one 5. | ||
A plausible reaction mechanism for the copper promoted intermolecular cyclization of benzylic azides and internal alkynes is shown in Scheme 6. The reaction is initiated by CuII assisted rearrangement of benzylic azide into N-aryliminium ion II by the loss of N2 and the migration of the phenyl group.38,39 Next, the intermolecular nucleophilic attack of alkyne to II forms a vinyl cation intermediate III. An intramolecular electrophilic aromatic substitution of intermediate III followed by oxidation afford the final quinoline product. It is worth to mention that the reaction offer high regioselectivity for unsymmetrical alkynes 2h–i (Scheme 2) and good yields for electron withdrawing group substituted benzylic azides (Scheme 1). Presumably, the high regioselectivity is due to the better stabilization of the vinyl cation intermediate III by the phenyl ring than the alkyl or ester group on the alkyne substrate. During this copper promoted cyclization, substituted benzylic azides are turned into an electron-rich substituted aryl amine (see intermediate III). As a result, an electron withdrawing substituent on the phenylamide ring will not completely stop the electrophilic cyclization of III.
 | ||
| Scheme 6 Plausible reaction mechanism. | ||
Conclusions
The copper(II)-promoted synthesis of 3,4-disubstituted quinolines by the intermolecular cyclization reaction of benzylic azides and internal alkynes have been demonstrated. It has been shown that the effectiveness of CuII-salt in the rearrangement of benzylic azide into N-arylimine (Schmidt reaction) which has previously only been achieved by strong acids. The catalytic reaction is highly efficient with a wide range of substituted benzylic azides. Excellent regioselectivity was observed for the reaction with unsymmetrical alkyne giving a single regioisomeric product in high yield. The present reaction system is further applied to the synthesis of two biologically active compounds.Experimental section
General procedure for the synthesis of benzyl azides40
Substituted benzylic bromide (1.0 equiv.) and sodium azide (1.5 equiv.) were dissolved in DMF (2.0 mL mmol−1) and stirred at 30 °C for 12 h. At the end of the reaction, the mixture was diluted with water and extracted with diethyl ether. The combined solution was concentrated in vacuo and the mixture was purified by a silica gel column (hexane/EtOAc, 90:10) to afford benzylic azide.
Typical procedure for the synthesis of substituted quinoline 3aa
A sealed tube containing Cu(OTf)2 (254 mg, 0.70 mmol) and diphenylacetylene 2a (100 mg, 0.56 mmol) was evacuated and purged with nitrogen gas three times. Then, a solution of benzyl azide 1a (38 mg, 0.28 mmol) in MeNO2 (2.0 mL) was added to the system by syringe under a nitrogen atmosphere and the reaction was stirred at 80 °C for 24 h. At the end of the reaction, the mixture was diluted with CH2Cl2 (10 mL), filtered through a Celite pad, which was then washed three times with CH2Cl2 (3 × 20 mL). The combined filtrate was concentrated in vacuo and the mixture was purified by a silica gel column using hexane/EtOAc (95:5) as eluent to afford the desired pure product 3aa in 91% (71 mg) yield.
Yellow solid: mp 135–137 °C; 1H NMR (400 MHz, CDCl3): δ 8.99 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 7.74–7.64 (m, 2H), 7.49–7.43 (m, 1H), 7.37–7.30 (m, 3H), 7.23–7.14 (m, 7H); 13C NMR (100 MHz, CDCl3): δ 151.8 (CH), 147.5 (C), 145.5 (C), 138.1 (C), 136.3 (C), 133.1 (C), 130.5 (2 CH), 130.1 (2 CH), 129.5 (CH), 129.1 (CH), 128.1 (2 CH), 128.0 (2 CH), 127.7 (CH), 127.2 (C), 127.0 (CH), 126.8 (CH), 126.6 (CH); HRMS (FAB) cal for C21H15N [M+] 281.1204, found 281.1204; IR (KBr): 2923, 2854, 1727, 1565, 1488, 1442, 1380, 1272, 1072, 1025, 763 and 701 cm−1.
Procedure for the synthesis of biologically active compound 4 (ref. 41)
Compound 3fg was synthesized in 75% yield from 1-(azidomethyl)-4-chlorobenzene (1f) and phenylpropyne (2g) using a procedure similar to that for the synthesis of quinoline 3aa.Compound 3fg (110 mg, 0.43 mmol) was dissolved in THF (4.0 mL) and MeLi/LiBr (2.2 M in Et2O, 0.87 mmol) was added to the solution at −78 °C. The mixture was allowed to warm to room temperature for 24 h. At the end of the reaction, iodine (328 mg, 1.29 mmol) was added to the mixture at 0 °C and stirred for 1 h at the same temperature. The mixture was then quenched with a saturated sodium thiosulfate solution (10 mL). The resulted biphasic solution was extracted with EtOAc (3 × 30 mL). The combined organic solution was concentrated in vacuo and the mixture was purified by a silica gel column using hexane/EtOAc (95:5) as eluent to afford the desired pure product 4 in 82% (94 mg) yield.
Yellow solid: mp 125–127 °C; 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 8.8 Hz, 1H), 7.54–7.44 (m, 4H), 7.25 (d, J = 2.4 Hz, 1H), 7.21–7.18 (m, 2H), 2.72 (s, 3H), 2.15 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 159.3 (C), 145.5 (C), 144.4 (C), 136.9 (C), 131.2 (C), 130.1 (CH), 129.3 (2 CH), 128.9 (CH), 128.7 (2 CH), 128.5 (C), 128.0 (CH), 127.6 (C), 124.8 (CH), 24.5 (CH3), 17.0 (CH3); HRMS (ESI) [M + H]+ cal for C17H15ClN 268.0893, found 268.0885; IR (KBr): 3062, 2923, 2854, 1727, 1666, 1589, 1481, 1373, 1172, 1072, 948, 825 and 701 cm−1.
Procedure for the synthesis of 3,4-diphenylquinolin-2(1H)-one 5 (ref. 36)
Compound 3aa (100 mg, 0.36 mmol) was dissolved in CH2Cl2 (4.0 mL) and meta-chloroperoxybenzoic acid (134 mg, 0.54 mmol) was added to the solution at 0 °C and stirred for 4 h at the same temperature. After that the reaction was quenched with saturated sodium bicarbonate solution (5 mL). The resulted biphasic solution was extracted with CH2Cl2 (3 × 10 mL) and the combined organic layer was dried over MgSO4 and concentrated by rotary evaporation. The crude product was dissolved in Ac2O (3.0 mL) and heated at 75 °C for 18 h. At the end of the reaction, the mixture was quenched with saturated sodium bicarbonate solution and extracted with CH2Cl2 (3 × 10 mL). The combined solution was dried over MgSO4 and concentrated in vacuo and the resulted residue was purified by a silica gel column using hexane/EtOAc (95:5) as eluent to afford the desired pure product 5 in 52% (55 mg) yield.
Yellow solid: mp 173–175 °C; 1H NMR (400 MHz, d6-DMSO): δ 12.0 (bs, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.32–7.22 (m, 3H), 7.18–7.04 (m, 8H), 6.99 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, d6-DMSO): δ 161.2 (C), 148.1 (C), 138.2 (C), 136.1 (C), 135.7 (C), 131.9 (C), 130.6 (2 CH), 130.1 (CH), 129.5 (2 CH), 127.9 (2 CH), 127.5 (CH), 127.1 (2 CH), 126.8 (CH), 126.5 (CH), 121.7 (CH), 119.9 (C), 115.1 (CH); HRMS (ESI) [M + H]+ cal for C21H16ON 298.1232, found 298.1225; IR (KBr): 3162, 1727, 1643, 1596, 1481, 1442, 1288, 705 and 701 cm−1.
Acknowledgements
We thank the Ministry of Science and Technology of the Republic of China (MOST-103-2633-M-007-001) for support of this research.Notes and references
- J. P. Michael, Nat. Prod. Rep., 2005, 22, 627–646 RSC.
- J. P. Michael, Nat. Prod. Rep., 2007, 24, 223–246 RSC.
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC.
- V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488–1508 CrossRef CAS.
- G. Roma, M. D. Braccio, G. Grossi, F. Mattioli and M. Ghia, Eur. J. Med. Chem., 2000, 35, 1021–1035 CrossRef CAS.
- Y.-L. Chen, K.-C. Fang, J.-Y. Sheu, S.-L. Hsu and C.-C. Tzeng, J. Med. Chem., 2001, 44, 2374–2377 CrossRef CAS.
- A. P. Gorka, A. D. Dios and P. D. Roepe, J. Med. Chem., 2013, 56, 5231–5246 CrossRef CAS PubMed.
- P. Zajdel, A. Partyka, K. Marciniec, A. J. Bojarski, M. Pawlowski and A. Wesolowska, Future Med. Chem., 2014, 6, 57–75 CrossRef CAS PubMed.
- S. Mukherjee and M. Pal, Curr. Med. Chem., 2013, 20, 4386–4410 CrossRef CAS.
- R. Musiol, Curr. Pharm. Des., 2013, 19, 1835–1849 CrossRef CAS PubMed.
- F. M. Hamer, The Cyanine Dyes and Related Compounds, Interscience Publishers, New York, London, Wormerveer printed, 1964 Search PubMed.
- K.-Y. Lu, H.-H. Chou, C.-H. Hsieh, Y.-H. O. Yang, H.-R. Tsai, H.-Y. Tsai, L.-C. Hsu, C.-Y. Chen, I. C. Chen and C.-H. Cheng, Adv. Mater., 2011, 23, 4933–4937 CrossRef CAS PubMed.
- A. R. Surrey and H. F. Hammer, J. Am. Chem. Soc., 1946, 68, 113–116 CrossRef CAS.
- J. Wiesner, R. Ortmann, H. Jomaa and M. Schlitzer, Angew. Chem., Int. Ed., 2003, 42, 5274–5293 CrossRef CAS PubMed.
- M. Takamura, K. Funabashi, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 6801–6808 CrossRef CAS PubMed.
- S. Ernst and W. Kaim, J. Am. Chem. Soc., 1986, 108, 3578–3586 CrossRef CAS.
- P. J. Steel, Coord. Chem. Rev., 1990, 106, 227–265 CrossRef CAS.
- A. Mamo, S. Nicoletti and N. C. Tat, Molecules, 2002, 7, 618–627 CrossRef CAS.
- Y.-Z. Hu, G. Zhang and R. P. Thummel, Org. Lett., 2003, 5, 2251–2253 CrossRef CAS PubMed.
- Y. Fukata, K. Asano and S. Matsubara, Chem. Lett., 2013, 42, 355–357 CrossRef CAS.
- P. W. Yawalkar, S. J. Dhoble, N. Thejo Kalyani, R. G. Atram and N. S. Kokode, Luminescence, 2013, 28, 63–68 CrossRef CAS.
- A. Weissberg and E. C. Taylor, The Chemistry of Heterocyclic Compounds, Quinoline, Parts I–III, ed. G. Jones, Interscience Publications, Wiley, London, 1977, vol. 32 Search PubMed.
- J. B. Bharate, R. A. Vishwakarma and S. B. Bharate, RSC Adv., 2015, 5, 42020–42053 RSC.
- V. Kouznetsov, L. Mendez and C. Gomez, Curr. Org. Chem., 2005, 9, 141–161 CrossRef CAS.
- Y. Zhang, P. Li and L. Wang, J. Heterocycl. Chem., 2011, 48, 153–157 CrossRef CAS.
- F. Xiao, Y. Chen, Y. Liu and J. Wang, Tetrahedron, 2008, 64, 2755–2761 CrossRef CAS.
- K. Cao, F.-M. Zhang, Y.-Q. Tu, X.-T. Zhuo and C.-A. Fan, Chem.–Eur. J., 2009, 15, 6332–6334 CrossRef CAS PubMed.
- A. Kumar and V. Rao, Synlett, 2011, 2011, 2157–2162 CrossRef.
- X. Zhang, B. Liu, X. Shu, Y. Gao, H. Lv and J. Zhu, J. Org. Chem., 2012, 77, 501–510 CrossRef CAS PubMed.
- J. Tummatorn, C. Thongsornkleeb and S. Ruchirawat, Tetrahedron, 2012, 68, 4732–4739 CrossRef CAS.
- J. Tummatorn, P. Poonsilp, P. Nimnual, J. Janprasit, C. Thongsornkleeb and S. Ruchirawat, J. Org. Chem., 2015, 80, 4516–4525 CrossRef CAS PubMed.
- R. P. Korivi and C.-H. Cheng, J. Org. Chem., 2006, 71, 7079–7082 CrossRef CAS PubMed.
- P. Gandeepan, P. Rajamalli and C.-H. Cheng, Asian J. Org. Chem., 2014, 3, 303–308 CrossRef CAS.
- CCDC 1419576–1419579 contains the supplementary crystallographic data for this paper.
- G. C. Muscia, M. Bollini, J. P. Carnevale, A. M. Bruno and S. E. Asís, Tetrahedron Lett., 2006, 47, 8811–8815 CrossRef CAS.
- J. DeRuiter, B. E. Swearingen, V. Wandrekar and C. A. Mayfield, J. Med. Chem., 1989, 32, 1033–1038 CrossRef CAS.
- C. Peifer, R. Urich, V. Schattel, M. Abadleh, M. Röttig, O. Kohlbacher and S. Laufer, Bioorg. Med. Chem. Lett., 2008, 18, 1431–1435 CrossRef CAS PubMed.
- A. Murali, M. Puppala, B. Varghese and S. Baskaran, Eur. J. Org. Chem., 2011, 2011, 5297–5302 CrossRef CAS.
- P. Desai, K. Schildknegt, K. A. Agrios, C. Mossman, G. L. Milligan and J. Aubé, J. Am. Chem. Soc., 2000, 122, 7226–7232 CrossRef CAS.
- O. Berger, A. Kaniti, C. T. van Ba, H. Vial, S. A. Ward, G. A. Biagini, P. G. Bray and P. M. O'Neill, ChemMedChem, 2011, 6, 2094–2108 CrossRef CAS PubMed.
- C. C. C. Johansson, N. Bremeyer, S. V. Ley, D. R. Owen, S. C. Smith and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 6024–6028 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, characterization details and 1H and 13C NMR spectra of new compounds. CCDC 1419576–1419579. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra23065a |
| This journal is © The Royal Society of Chemistry 2015 |