
A protocol of self-assembled monolayers of fluorescent block molecules for trace Zn(ii) sensing: structures and mechanisms
Abstract
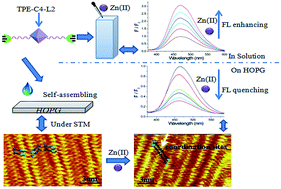
2D epitaxial nanostructures formed by functional molecules at interfaces/surfaces have attracted considerable research interest because of their promising applications in catalysis, photonics, electronic devices, and so on. In this study, we demonstrate for the first time that a monolayer of functional molecules may be used as a fluorescence indicator for heavy metal ions. Self-assembly of bisligand TPE-C4-L2 on highly oriented pyrolytic graphite was performed and the self-assembled monolayer (SAM) was used for trace amounts of Zn(II) sensing with an output signal of fluorescence. In contrast to the Zn(II)-induced fluorescence enhancement of the TPE-C4-L2 block molecule in solution, the self-assembled TPE-C4-L2 film showed quenched fluorescence after Zn(II) was introduced. Using a high-resolution scanning tunneling microscope, the SAM structures of TPE-C4-L2 and its zinc complex were determined. The correlation between their optical properties and structures at the molecular level was also revealed.
 Please wait while we load your content...
Please wait while we load your content...

