
Facile synthesis of porous Fe2TiO5 microparticulates serving as anode material with enhanced electrochemical performances†
Shimei Guoab,
Shenyu Wanga,
Nannan Wua,
Jiurong Liu*a,
Yuxing Nic and
Wei Liu*c
aKey Laboratory for Liquid–Solid Structural Evolution and Processing of Materials, Ministry of Education and School of Materials Science and Engineering, Shandong University, Shandong 250061, China
bCollege of Physics and Electronic Engineering, Qujing Normal University, Yunnan 655011, China
cState Key Laboratory of Crystal Materials, Shandong University, Shandong 250100, China
First published on 26th November 2015
Abstract
Porous iron titanium oxide (Fe2TiO5) microparticulates have been successfully synthesized via a facile hydrothermal route followed by a subsequent calcination process. Polyvinyl-pyrrolidone (PVP), serving as a surfactant, played a pivotal role in controlling the size and inducing the mesoporous structure of Fe2TiO5. The measured specific surface area is 83.1 m2 g−1 and the dominant pore size is ca. 10 nm. When tested as the anode material of lithium-ion batteries (LIBs), the as-prepared porous Fe2TiO5 microparticulates delivered a high reversible capacity of 468.3 mA h g−1 after 100 cycles at a current density of 100 mA g−1, which nearly quadrupled that of porous TiO2 microspheres and doubled that of Fe2O3 nanoparticles. Moreover, the porous Fe2TiO5 microparticulates also showed more superior rate capability and long-term cycling stability with respect to TiO2 and Fe2O3 samples. Even after the rate performance test, a high discharge capacity of 234.1 mA h g−1 was still maintained at a current density of 500 mA g−1 over another 250 cycles. The improved electrochemical performances are mainly attributed to the synergistic effect of TiO2 and Fe2O3 in Fe2TiO5, as well as the mesoporous structure.
1. Introduction
In the past few decades, rechargeable lithium ion batteries (LIBs) have attracted remarkable attention due to their available high voltage, high energy density, no memory effect and long lifespan.1–3 In general, the superior electrochemical performances of LIBs are largely dependent on their electrode materials, namely anode and cathode active material. Especially for anode active material, it has been playing an important role in determining energy density, safety and cycle life of LIBs.4,5 Nowadays, the commercial anode material has been primarily dominated by graphite due to its beneficial layered structure for facile Li+ insertion/extraction, good electrical conductivity, reasonable cost, and abundant resources. However, in the practical application, the potential for lithium insertion/extraction (0–0.25 V vs. Li/Li+) of graphite is close to that of the Li+/Li redox couple, which will result in lithium plating and dendrite formation during the overcharge process, and thus generating serious safety issues (e.g., short out or explosion of LIBs).6–8 In addition, graphite suffers greatly from volume expansion and shrinkage during Li-ion insertion and extraction that leads to the cracking of graphite particles and a loss in electrical contact, and thereby decreasing capacity as well as cycle life.4 Especially, the low theoretical capacity (372 mA h g−1) and poor cycling performance of graphite at high current density have restricted its large-scale application in high-power density batteries.9 Recently, a lot of research attentions have been paid in seeking high capacity, high safety and long cycle-life anode materials to replace graphite.10,11Among various prospective electrode materials, TiO2 has been regarded as a very appealing anode candidate to compete with commercial graphite for LIBs owing to its superior structure stability, long cycle life, low cost and excellent security.12–14 However, it is well known that TiO2 possesses a relatively low theoretical capacity of 335 mA h g−1 even with the maximum accommodation of one Li+ per TiO2 unit (Li1.0TiO2), poor rate capability due to its low electronic conductivity (∼1 × 10−12 to 1 × 10−7 S cm−1), and low lithium ion diffusivity (∼1 × 10−15 to 1 × 10−9 cm2 s−1), which have seriously hindered its potential applications for commercialization.15–17 To address the above issues, a series of Ti-based binary oxides with various of nanostructures, such as SrTiO3 nanoparticles,18 FeTiO3 nanoflowers or nanosheets,19,20 porous TiNb2O7 nanoparticles,21 and Zn2Ti3O8 nanowires,22 have been synthesized as anode materials for LIBs. Benefited from the incorporation of high specific capacity from metal oxides (more than 335 mA h g−1) and electrochemical stability of TiO2, as well as the improved electronic conductivity and lithium ion diffusivity, these binary Ti-based oxide electrodes exhibited the enhanced reversible capacities and superior cycling performance. For example, the mesoporous TiNb2O7 nanocrystals as anode material for LIBs demonstrated a high capacity of 289 mA h g−1 (at 0.1C) and an excellent rate performance of 162 mA h g−1 at 20C and 116 mA h g−1 at 50C (=19.35 A g−1).21 The Si4+ doped NiTiO3 spherical nanoparticles (140–160 nm) serving as anode material maintained a constant capacity ca. 400 mA h g−1 at a current density of 0.4 mA cm−2 up to 25 cycles.23 FeTiO3 nanosheets displayed a stable discharge capacity of ca. 430 mA h g−1 up to 90 cycles at a current density of 100 mA g−1.20
Inspired by the above researches, in this work, a binary Ti-based oxide of Fe2TiO5 porous microparticulates have been synthesized through a facile hydrothermal route followed by a calcination process. Combining the high theoretical specific capacity of Fe2O3 (∼1005 mA h g−1) with the advantages of TiO2 (e.g., superior structure stability and long cycle life),5,12 the as-prepared Fe2TiO5 porous microparticulates exhibited the improved electrochemical performance with respect to pure TiO2, Fe2O3 and the above mentioned binary Ti-based oxides anodes.
2. Experimental
2.1. Materials
Tetrabutyl titanate (TBT, Ti(OC4H9)4, 99.0%), N,N-dimethylformamide (DMF, C5H9NO, 99.0%), isopropanol (IPA, C3H8O, 99.0%), iron(III) acetylacetonate (C15H21FeO6, 98.0%), and polyvinyl-pyrrolidone (PVP, (C6H9NO)n, MW 40,000) were purchased from Sinopharm Chemical Reagent Co., Ltd. Carbon black, Li foil and Celgard 2300 were provided by Hefei Kejing Material Technology Co., Ltd, China. Polyvinylidene fluoride (PVDF), LiPF6 (dissolved in ethylene carbonate, dimethyl carbonate, and ethylene methyl carbonate with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) were purchased from Shenzhen Biyuan Technology Co., Ltd, China. All the chemicals are of analytical grade and were used as received without further purification.
:1:1) were purchased from Shenzhen Biyuan Technology Co., Ltd, China. All the chemicals are of analytical grade and were used as received without further purification.
2.2. Preparation of Fe2TiO5 microparticulates
The preparation of porous Fe2TiO5 microparticulates is illustrated in Fig. 1. In a typical synthesis, 0.5 g polyvinyl-pyrrolidone (PVP) was firstly dissolved in the mixed organic solvent of 10 mL DMF and 30 mL IPA to form a transparent solution. Then, 1 mL TBT and 1 mmol iron(III) acetylacetonate were added to the above solution under magnetic stirring until they were completely dissolved. The obtained bright red solution was transferred into a 65 mL Teflon-lined stainless steel autoclave, and subsequently sealed and heated at 180 °C for 20 h in an oven. After reaction, the reddish brown precipitate was centrifuged and washed using deionized water and ethanol for five times, and then dried in a vacuum oven at 60 °C overnight. The Fe2TiO5 product was obtained by annealing the reddish brown precursor at 500 °C for 2 h in air. As comparison, pure TiO2 and Fe2O3 samples were also prepared in the same procedure only without iron(III) acetylacetonate and PVP or without TBT addition, respectively. | ||
| Fig. 1 Schematic illustration for the fabrication process of porous Fe2TiO5 microparticulates. | ||
2.3. Characterizations
The structure of the resultant products was determined by X-ray powder diffraction (XRD) on a Rigaku D/Max-RC X-ray diffractometer with Ni filtered Cu Kα radiation (λ = 0.1542 nm, V = 40 kV, I = 50 mA) in the range of 10–80° at a scanning rate of 4° min−1. The morphology and microstructure of samples were examined by using a JSM-6700F field emission scanning electron microscope (FE-SEM) at an accelerating voltage of 20 kV and electric current of 1.0 × 10−10 A, and a JEOL JEM-2100 high resolution transmission electron microscope (HR-TEM) operated at 200 kV. The element contents were examined by energy-dispersive X-ray spectroscopy (EDS) detector attached to the FE-SEM. The N2 adsorption/desorption isotherms of porous products were measured at 77 K on a Quadrasorb-SI instrument. The specific surface area was calculated with the Brunauer–Emmett–Teller (BET) model and the pore size distribution was determined using the Barrett–Joyner–Halenda (BJH) method. The composition was determined by X-ray photoelectron spectroscopy (XPS) on a Kratos Analytical spectrometer, using Al Kα (hν = 1486.6 eV) radiation as the excitation source under the anode voltage of 12 kV and emission current of 10 mA.2.4. Electrochemical measurements
To prepare the working electrode, the active material, carbon black, and polyvinylidene fluoride (PVDF) with a weight ratio of 7:2:1 were mixed in N-methyl-2-pyrrolidinone (NMP) to form a homogenous slurry, which was coated on a copper foil substrate, followed by drying in a vacuum oven at 120 °C for 12 h. The typical loading amount of active materials is 1.5–2.0 mg cm−2. The CR2025-type cells were assembled using Li foil as counter and reference electrode, Celgard 2300 as separator, and 1 M LiPF6 (dissolved in ethylene carbonate, dimethyl carbonate, and ethylene methyl carbonate with a volume ratio of 1:1:1) as the electrolyte. The assembly was performed in a glove-box filled with argon atmosphere. The performance of the cells was evaluated galvanostatically in the voltage range from 0.02 to 3 V at various current densities on a LAND CT2001A battery test system. Cyclic voltammogram (CV) was obtained by a PARSTAT 2273 electrochemistry workstation at a scan rate of 0.3 mV s−1 and the potential vs. Li/Li+ ranging from 0.01 to 3 V. Electrochemical impedance spectra were tested on the same instrument with AC signal amplitude of 10 mV in the frequency range from 100 kHz to 0.01 Hz. The data were adopted to draw Nyquist plots using real part Z′ as X-axis, and imaginary part Z′′ as Y-axis.
3. Results and discussion
XRD measurement was carried out to investigate the crystal phase of the as-prepared product. As shown in Fig. 2a, no obvious diffraction peak is observed in the Fe2TiO5 precursor indicating that the as-prepared precursor is amorphous. After being annealed at 500 °C for 2 h in air, all the identified peaks can be assigned to the orthorhombic pseudobrookite Fe2TiO5 (JCPDS no. 41-1432, a0 = 9.797 Å, b0 = 9.981 Å, c0 = 3.73 Å),24 and no any peak of other phase is detected (Fig. 2b), suggesting that the precursor has been completely converted into pure Fe2TiO5 after annealing process.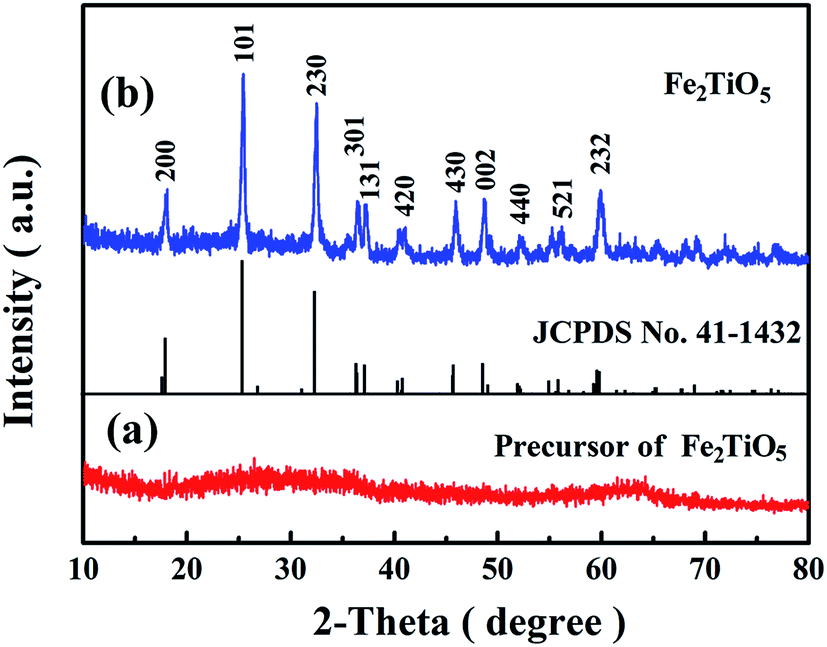 | ||
| Fig. 2 XRD patterns of (a) Fe2TiO5 precursor and (b) Fe2TiO5 microparticulates. | ||
To further characterize the composition of Fe2TiO5 product, X-ray photoelectron spectroscopy (XPS) measurement was performed to determine the elements, chemical bonding, and their corresponding valence state. From the survey scan spectrum (Fig. 3a), the peaks of Fe 2p, Ti 2p, O 1s and C 1s were detected indicating the existence of Fe, Ti, O and C elements in the as-prepared Fe2TiO5 product. The C 1s signal can be attributed to the carbon contamination due to the ambient exposure of sample, which is usually found in oxides and consistent with our previous research.25 The deconvoluted O 1s peaks at 531.5 and 532 eV (Fig. 3b) are assigned to C–O and Fe–O–Ti, respectively, according to the electronegativity successive decrease of C, Fe, and Ti elements. For the Fe 2p spectrum, two distinct peaks were observed around the binding energies of 725.2 and 711.9 eV (Fig. 3c), which are assigned to Fe 2p1/2 and Fe 2p3/2, respectively. The fingerprint shakeup satellite peak of Fe2O3 appears at ca. 719.9 eV, suggesting that the valence of Fe in product is Fe3+.26–28 In Fig. 3d, the Ti 2p double peaks centered at ca. 458.3 and 464.2 eV stem from Ti 2p3/2 and Ti 2p1/2, respectively. The splitting binding energy between Ti 2p1/2 and Ti 2p3/2 core levels is ca. 5.9 eV, indicating a normal state of Ti4+ in product.29–31
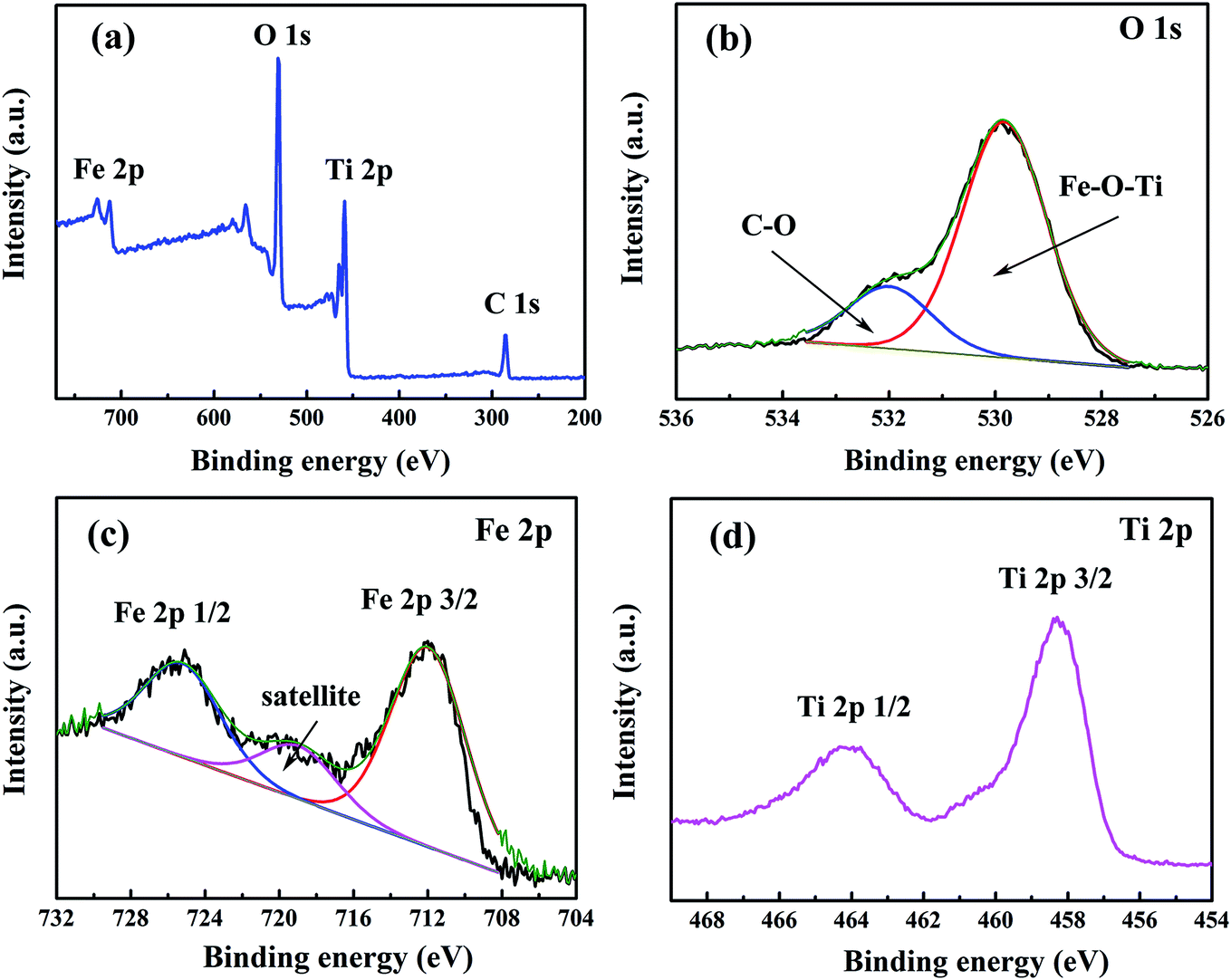 | ||
| Fig. 3 XPS spectra of (a) survey scan, (b) O 1s, (c) Fe 2p and (d) Ti 2p of Fe2TiO5 microparticulates. | ||
The low-magnification SEM image reveals that the synthesized Fe2TiO5 microparticulates with the size of ca. 200 nm are agglomerated together to form larger cluster and have rough surface (Fig. 4a). Combining with HR-TEM observation (Fig. 4b), it can be confirmed that the as-prepared Fe2TiO5 has a porous architecture comprised of numerous nano-grains (ca. 10 nm) and pores. From the higher resolution image (Fig. 4c), two sets of lattice fringe spacings of 0.44 and 0.33 nm (inset of Fig. 4c) correspond to the (210) and (111) plane of pseudobrookite Fe2TiO5 (JCPDS no. 41-1432), respectively, consistent with the previous reports.24,32 In Fig. 4d, the energy-dispersive spectrum (EDS) of Fe2TiO5 also confirms the presence of Fe, O and Ti elements agreement with the above XPS analysis (Fig. 3a). As comparison, the SEM images of as-synthesized TiO2 and Fe2O3 samples are shown in Fig. S1a and b (ESI†). TiO2 exhibits the morphology of porous microspheres with the diameter of ca. 500–800 nm (Fig. S1a†), while the synthesized Fe2O3 is nanoparticles with a narrow size distribution of about 100 nm (Fig. S1b†).
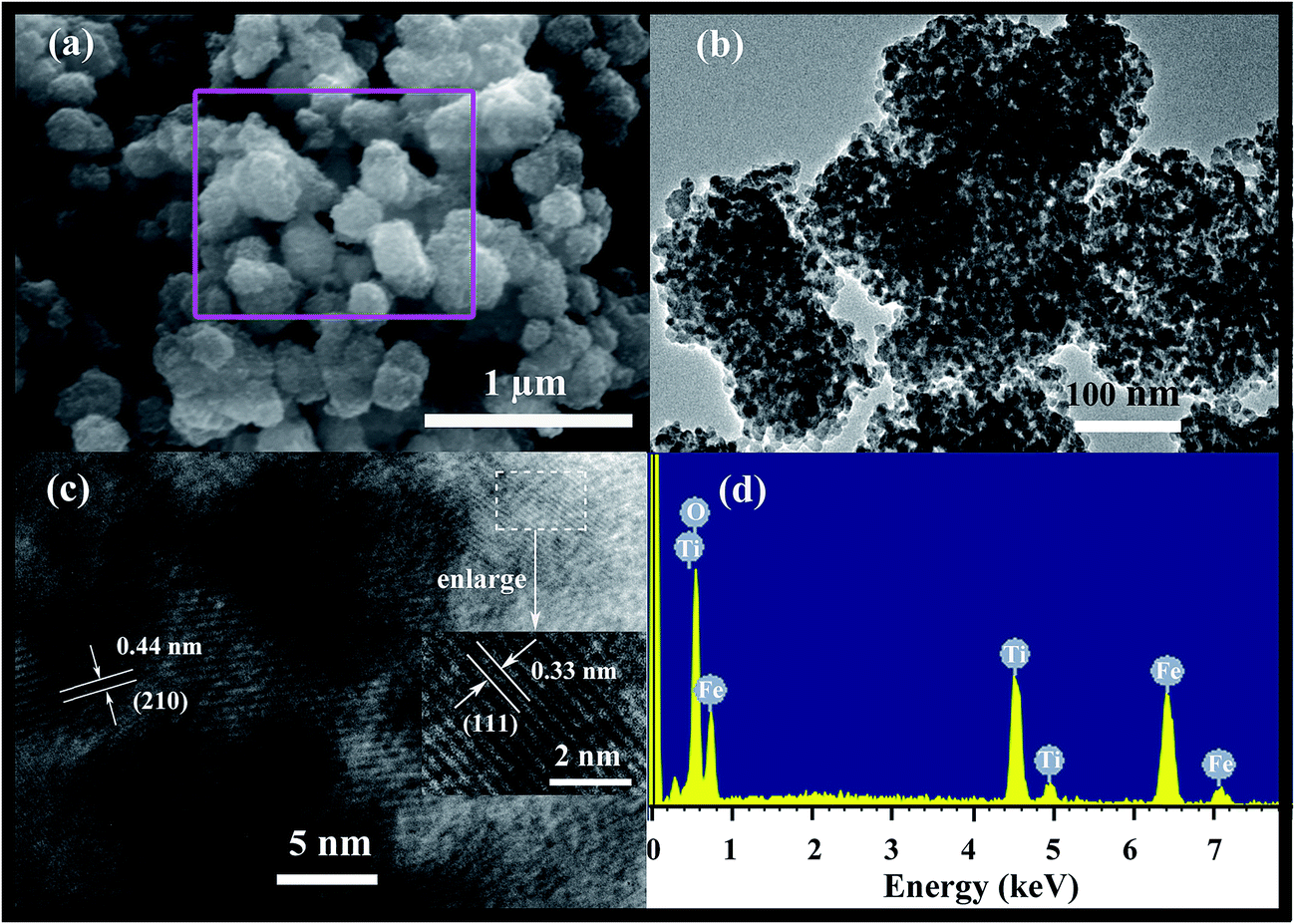 | ||
| Fig. 4 SEM images (a), TEM images (b), HR-TEM images (c), and (d) the corresponding EDS spectrum from the marked areas in (a) of as-prepared Fe2TiO5 microparticulates. | ||
To investigate the effect of PVP on the morphology and structure of resultant product, the control experiments were operated with different addition amount of PVP while the other experimental parameters were kept the same as those mentioned in the aforementioned typical synthesis. Without PVP addition, the synthesized Fe2TiO5 has a spherical morphology with large size distribution (1–5 μm) (Fig. 5a). From the high magnification FE-SEM image (inset of Fig. 5a), it is observed that Fe2TiO5 microspheres have smooth surface and solid structure, and no pores are found at the surface or interior of these microspheres. After 0.2 g PVP is added in the reaction, the morphology remains unchanged, while the obtained microspheres have a more narrow size distribution (less than 1 μm) as shown in Fig. 5b. The high magnification FE-SEM image (inset of Fig. 5b) indicates the as-prepared Fe2TiO5 microsphere has a compact structure, and no obvious pores are observed except that the microsphere surface tends to rough. When the content of PVP increases to 0.5 g, the formed Fe2TiO5 microparticulates exhibit a porous structure and smaller size (Fig. 3). The spherical morphology disappeared due to the agglomeration among particles. With increasing the content of PVP to 1.0 g, the solution becomes very viscous, and the reddish brown precipitate of precursor cannot be obtained any more. Thus, PVP is believed to play an important role in controlling the morphology and size distribution and inducing the pore structure of Fe2TiO5 product. As a kind of nonionic surfactant, PVP can physically absorb on the surface of product subunits to prohibit the grain growth leading to a narrow size distribution.33,34 In addition, due to the steric effect of PVP molecules, the grains are loosely aggregated to form larger particles. After annealed at 500 °C in air, PVP molecules are oxidized into volatile CO2 and H2O, which are released to generate many voids in the interior as well as at surface of Fe2TiO5 microparticulates (Fig. 1).35
 | ||
| Fig. 5 FE-SEM images of Fe2TiO5 fabricated with different amount of PVP: (a) 0 g and (b) 0.2 g. | ||
Porous structure can facilitate an efficient contact of the internal active materials with electrolyte, leading to a fast diffusion of Li+ ions. Meanwhile, the high specific surface area and porosity are able to favorably alleviate the volume variation during the Li+ insertion/extraction, resulting in a relatively high reversible capacity and cycling stability.36,37 Therefore, nitrogen absorption–desorption measurements were performed to investigate the porosity and surface area of Fe2TiO5 microparticulates and pure TiO2 porous microspheres. As shown in Fig. 6a and b, the nitrogen adsorption/desorption isotherms of both samples exhibit a type IV nitrogen adsorption branch with a type-H3 hysteresis loop over the pressure range above 0.45P/P0, indicating the presence of mesopores in Fe2TiO5 microparticulates and TiO2 microspheres.8,9,37,38 From the pore size distribution curve of Fe2TiO5 microparticulates (inset of Fig. 6a), the pores with a size of ca. 10 nm are dominant consistent with the HR-TEM observation (Fig. 4b), and TiO2 microspheres also exhibit similar pore size distribution (inset of Fig. 6b). The BET surface area of Fe2TiO5 porous microparticulates is 83.1 m2 g−1 and the pore volume is calculated to be 0.23 cm3 g−1, which are both higher than the corresponding values of 58.4 m2 g−1 and 0.14 cm3 g−1 for TiO2 microspheres. The high specific surface area and pore volume are beneficial to the enhancement of cycling and rate performances of Fe2TiO5 microparticulates anode.
 | ||
| Fig. 6 N2 adsorption–desorption isotherms of porous TiO2 microspheres (a) and Fe2TiO5 microparticulates (b). The insets are pore size distribution curves. | ||
To reveal the electrochemical reaction of the as-synthesized Fe2TiO5 porous microparticulates as LIBs anode, cyclic voltammetry (CV) was carried out at room temperature in the range of 0.01–3.0 V at a scan rate of 0.3 mV s−1. Li metal was used as the counter and reference electrodes. For comparison, pure TiO2 porous microspheres and Fe2O3 nanoparticles were also employed as anode materials. The CV curves of TiO2 porous microspheres (Fig. 7a) and Fe2O3 nanoparticles (Fig. 7b) are in good agreement with those of previously reported anatase TiO2 and α-Fe2O3.16,38–40 However, it is worth noting that both of these two materials show a large decrease of cathodic current in the second CV curve with respect to the first one, suggesting the irreversible lithium insertion/extraction reaction and a large capacity loss during the initial two cycles.13 On the contrary, the CV curves of Fe2TiO5 porous microparticulates (Fig. 7c) exhibit minor shrink during the initial two cycles comparing with the TiO2 and Fe2O3 samples indicating its better reversibility. As shown in Fig. 7c, during the first discharge process, there are four clear cathodic peaks observed at ca. 1.8, 1.7, 1.2 and 0.6 V, respectively. According to the reaction mechanism of TiO2, Fe2O3 and previous researches of other multi-component oxides, the two coterminal irreversible peaks centered at ca. 1.8 V and 1.7 V, which disappear in the second cycle, can be attributed to the insertion of Li+ into Fe2TiO5 resulting in the crystal structure destruction, and the lithium storage in TiO2 to form LixTiO2 (TiO2 + xLi+ + xe− → LixTiO2).10,16,41 The following peak at ca. 1.2 V corresponds to the lithium intercalation into Fe2O3 (Fe2O3 + 2Li+ + 2e− → Li(Fe2O3)),10,42,43 while the peak centered at about 0.6 V could be ascribed to the further reduction of Fe3+ into Fe0 (Li(Fe2O3) + 4Li+ + 4e− → 2Fe0 + 3Li2O), and the formation of amorphous Li2O and solid–electrolyte interface (SEI) layer.10,39,40 In the subsequent charge process, only one broad anodic peak from 1.5 V to 2.5 V (centered at about 2.1 V) is observed, which can be assigned to the oxidation of Fe0 to Fe2+ and further oxidation to Fe3+ (Fe0 → Fe2+ at ca. 1.6 V and Fe2+ → Fe3+ at ca. 1.9 V), as well as the extraction of Li ions from LixTiO2 (at ca. 2.1 V).42,43 After the first cycle, the cathodic peaks at ca.1.8 V, 1.7 V and 1.2 V are replaced by a broad cathodic peak ranged from 1.1 V to 1.8 V in the subsequent cycles. The sharp cathodic peak at ca. 0.6 V in the first cathodic profile disappeared and only a weak shoulder peak was observed at ca. 0.5 V in the subsequent cycles, indicated that the active material was polarized and the SEI film was formed in the first cycle, corresponding to the large initial irreversible capacity.39,44 From the second cycle onward, the CV curves are nearly overlapped, demonstrating that the electrochemical reaction tends to be stable.43,45 In addition, the electrochemical reactions are also confirmed in the discharge–charge voltage profiles of Fe2TiO5 porous microparticulates. As shown in Fig. 7d, three short potential plateaus appear at ca. 1.8 (I), 1.2 (II) and 0.6 V (III) in the first discharge curve and a slope from 1.5 V to 2.5 V is observed in the subsequent charge, consistent with the above CV analysis. When the first cycle is completed, the cell exhibits the initial discharge and charge capacities of 940.2 and 506.9 mA h g−1, respectively, corresponding to the initial coulombic efficiency (the ratio of charge capacity to discharge capacity) of 54%. The large capacity loss of the as-prepared sample in the first cycle can be attributed to some lithium insertion into irreversible sites, the inevitable formation of solid electrolyte interface (SEI) film and Li2O, and the active material loss by the disconnection of electrical contact during Li insertion that have been found in most anode materials.46,47
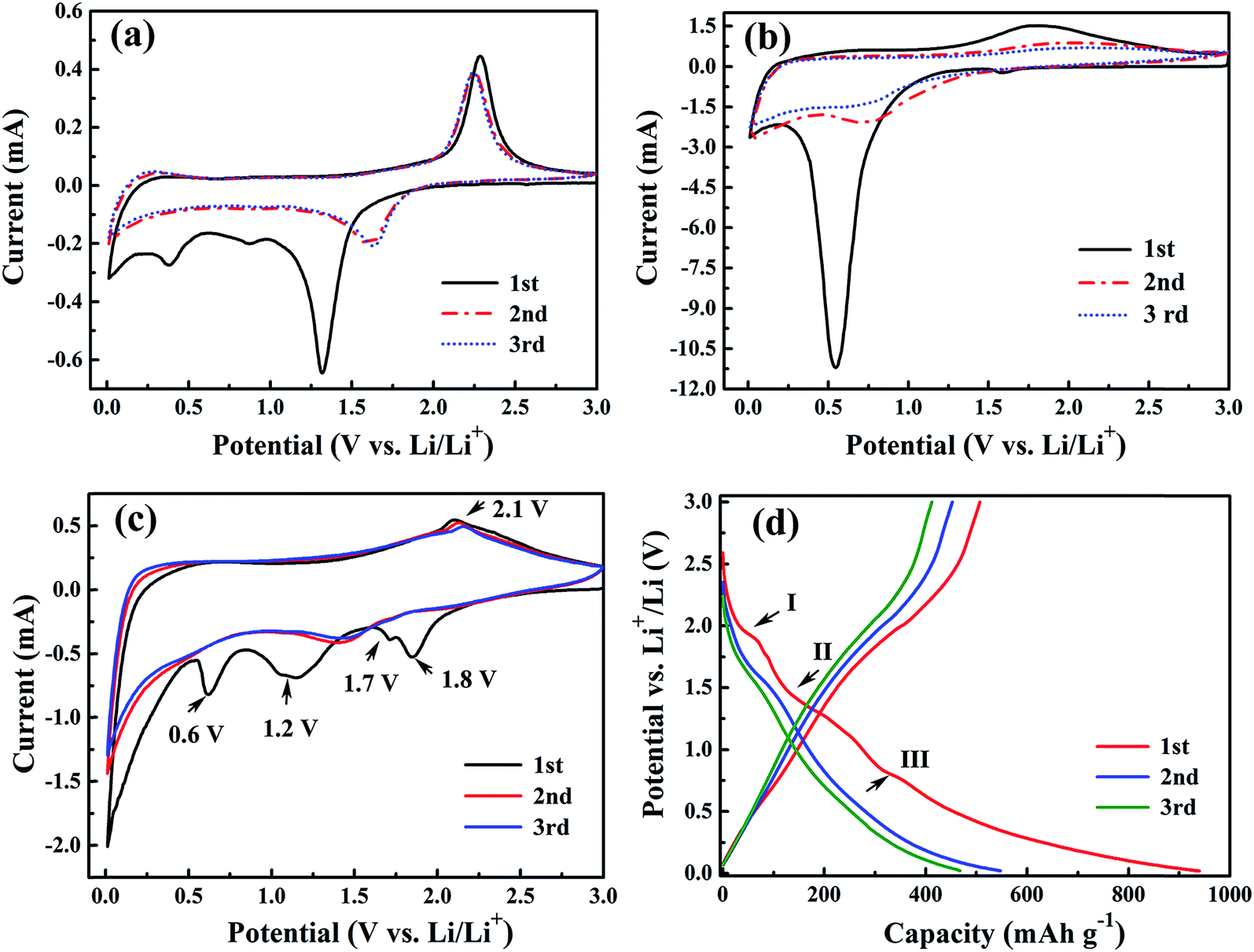 | ||
| Fig. 7 Cyclic voltammetry (CV) curves of TiO2 microspheres (a), Fe2O3 nanoparticles (b) and Fe2TiO5 microparticulates (c) at a scan rate of 0.3 mV s−1 in the range of 0.01–3.0 V. Galvanostatic discharge/charge curves of the initial three cycles for Fe2TiO5 microparticulates (d). | ||
The cycling and rate performances of the as-synthesized Fe2TiO5 porous microparticulates, TiO2 porous microspheres and Fe2O3 nanoparticles (Fig. S1a and S1b, ESI†) are depicted in Fig. 8. As shown in Fig. 8a, the reversible capacity of Fe2TiO5 anode is found to be 468.3 mA h g−1 after 100 cycles at a current density of 100 mA g−1 and the coulombic efficiency remains more than 95% after the initial three cycles. In comparison, the first discharge capacity (279.6 mA h g−1) of TiO2 at the current density of 100 mA g−1 is much lower than that (940.2 mA h g−1) of Fe2TiO5, and the capacity is maintained at a low value of ca. 126.5 mA h g−1 in the successive cycling until 100th cycle. For Fe2O3 nanoparticles, although Fe2O3 anode exhibits the highest initial discharge capacity of 1295.3 mA h g−1 among three samples, it suffers from severe capacity fading in the subsequent cycling and only maintains a reversible capacity of 224.1 mA h g−1 after 100 cycles, which is less than half of porous Fe2TiO5 microparticulates. From the rate capability shown in Fig. 8b, compared with TiO2 and Fe2O3 anodes, the Fe2TiO5 one also exhibits the higher reversible capacities of 363.7, 286, 239.3, 172.6, and 117.5 mA h g−1 at the current density of 100, 200, 400, 800 and 1600 mA g−1, respectively, indicating that Fe2TiO5 also has more superior rate capability than other two samples. The detailed rate capability data of TiO2 and Fe2O3 samples are listed in Table S1 (ESI†). When the current density was returned to 100 mA g−1, the discharge capacities of TiO2 and Fe2TiO5 samples approximately returned back to their initial values and maintained stable cycling performance, suggesting that the high current charge/discharge process did little to break down the integrity of the electrodes.48,49 It is noted that, although the Fe2O3 electrode recovers its initial capacity at first when the current density is returned to 100 mA g−1, the electrode undergoes a capacity fading from 50 to 80 cycles, and only achieves a stable reversible capacity of ca. 126 mA h g−1 finally. To further evaluate the applicability of porous Fe2TiO5 microparticulates as anode materials in LIBs, the long cycling performance has also been investigated at a higher current density of 500 mA g−1 after the rate performance test (100 cycles). As shown in Fig. 8c, a reversible capacity of 234 mA h g−1 was maintained without obvious capacity fading even after another 250 cycles. It is worth noting that, as shown in Fig. 8a and c, the reversible capacities of Fe2TiO5 and Fe2O3 electrodes both firstly decrease in the initial several cycles and then increase for the following cycles. This phenomenon is commonly found in both Fe-based transition metal oxides and other metal oxides.45,50 It can be attributed to the gradual improvement of lithium ion accessibility and additional capacity storage contributed by an organic polymeric/gel-like layer formed at low potential.10,45,50 In addition, the presence of Fe nanoparticles at Fe2TiO5 or Fe2O3 interface caused by some irreversible electrochemical reaction is also a possible reason. It can improve the conductivity of electrode and the reversibility reaction of active material resulting in the enhanced capacity.10,51 The more superior cycling and rate performances of Fe2TiO5 than TiO2 microspheres and Fe2O3 nanoparticles demonstrate that the binary Ti-based oxide (Fe2TiO5) anode significantly exhibits the improved electrochemical performance as expected due to the synergistic effect of superior electrochemical stability of TiO2 and high capacity of Fe2O3. In addition, the reversible capacity of the as-prepared porous Fe2TiO5 microparticulates is also higher than those of the reported Fe2TiO5 nanoparticles prepared by ball milling (55.8 mA h g−1 after 50 cycles at a current density of 36 mA g−1) and hydrothermal process (151.3 mA h g−1 after 50 cycles at a current density of 36 mA g−1)24 mainly attributing to the high specific surface area and porosity of Fe2TiO5 microparticulates, which facilitate the efficient contact of internal active materials with electrolyte and the diffusion of Li+ ions, and alleviate the volume variation during the Li+ insertion/extraction, resulting in a relatively high reversible capacity and cycling stability.36,37 Moreover, comparing with other binary Ti-based oxide anodes, such as FeTiO3 nanoflowers (ca. 200 mA h g−1 after 50 cycles at a current density of 50 mA g−1),19 FeTiO3 nanosheets (ca. 430 mA h g−1 after 90 cycles at a current density of 100 mA g−1),20 and TiNb2O7 nanocrystals (ca. 289 mA h g−1 after 10 cycles at a current density of 38.7 mA g−1),21 the as-synthesized porous Fe2TiO5 microparticulates also exhibit higher reversible capacity and better long-term cycling performance even at high current density (500 mA g−1), further confirming the superiority of the as-synthesized porous Fe2TiO5 microparticulates for LIBs anode.
 | ||
| Fig. 8 (a) Cycling performances of TiO2 microspheres, Fe2O3 nanoparticles and Fe2TiO5 microparticulates, and coulombic efficiency of Fe2TiO5 microparticulates at the current density of 100 mA h g−1. (b) Rate capabilities of TiO2, Fe2O3 and Fe2TiO5 at different cycling rates. (c) Long-term cycling performance of Fe2TiO5 microparticulates at the current density of 500 mA g−1 after rate performance test. | ||
To further clarify the electrochemical performance of Fe2TiO5 anode, electrochemical impedance spectroscopy (EIS) measurements were performed from 100 kHz to 0.01 Hz. Fig. 9 presents the Nyquist plots of Fe2TiO5 porous microparticulates, TiO2 porous microspheres and Fe2O3 nanoparticles after 100 charge/discharge cycles at a current density of 100 mA g−1. The EIS data are analyzed by fitting to an equivalent electrical circuit shown in the inset of Fig. 9, in accordance with the Li-ion insertion/extraction mechanism in electrode.2,13,52 As can be seen in Fig. 9, the Nyquist plots of Fe2TiO5 and TiO2 are both comprised of a depressed semicircle in the high- and medium-frequency region and an inclined line in the low frequency region, while Fe2O3 sample displays two arcs in the high- and medium-frequency region and a short linear tail in the low frequency. The diameter of semicircle in high- and medium-frequency region of each cell is related to the electrolyte resistance (Re), the SEI resistance (Rsf) and the charge transfer resistance (Rct), while the inclined line in low frequency region represents the Warburg impedance (Zw) that is derived from the lithium ion diffusion in electrode materials.10,43 Obviously, the diameter of semicircle for Fe2TiO5 sample (ca.142 Ω) is much smaller than that of TiO2 (ca. 322 Ω), indicating a lower internal resistance of Fe2TiO5. Comparing with Fe2O3 nanoparticles, the Fe2TiO5 anode also displays a smaller internal resistance (Re + Rsf + Rct) as listed in Table S2 (ESI†), suggesting more efficient transfer of electrons and Li ions between the interface of Fe2TiO5 microparticulates and electrolyte, which is beneficial to the capacity enhancement of Fe2TiO5 anode. In addition, the largest slope of inclined line in low frequency region for Fe2TiO5 anode demonstrates faster diffusion of Li ions in Fe2TiO5 than in TiO2 and Fe2O3 samples due to the porous structure with higher specific surface area and larger pore volume (Fig. 4 and 6), which improves the diffusion of electrolyte and the interfacial contact between active material and electrolyte and shortens the lithium diffusion path.33,36 Therefore, the lower internal resistance and faster diffusion rate of lithium ions in Fe2TiO5 porous microparticulates than in TiO2 and Fe2O3 samples might be the main reasons for the enhanced cycling performance and rate capability of Fe2TiO5 anode.
 | ||
| Fig. 9 Experimental and fitted Nyquist plots of TiO2 microspheres, Fe2O3 nanoparticles and Fe2TiO5 microparticulates after 100 cycles at the current density of 100 mA h g−1. The inset is the corresponding equivalent circuit. | ||
4. Conclusions
In summary, porous Fe2TiO5 microparticulates have been synthesized as anode material for Li-ion battery by a facile approach, in which PVP is a key factor in controlling the particle size and inducing the pore structure of Fe2TiO5. Comparing with the as-prepared TiO2 porous microspheres and Fe2O3 nanoparticles, porous Fe2TiO5 microparticulates exhibited much higher reversible capacity and superior long-term cycling performance even at high current density due to the combination of electrochemical stability of TiO2 and high capacity of Fe2O3. The experimental results confirm that the porous Fe2TiO5 microparticulates have lower internal resistance and faster diffusion rate of lithium ions than TiO2 and Fe2O3 samples, which are also beneficial to enhance the cycling performance and rate capability of Fe2TiO5 anode.Acknowledgements
This work was supported by the Fundamental Research Funds of Shandong University (2015JC016, 2015JC036) and the National Natural Science Foundation of China (No. 51572157). The authors also acknowledge the financial supports from the Science and Technology Development Plan (2014GGX102004) and Natural Science Fund for Distinguished Young Scholars of Shandong (JQ201312).References
- Q. Wang, Z. Wen and J. Li, Adv. Funct. Mater., 2006, 16, 2141–2146 CrossRef CAS.
- Y. Wang, M. Wu and W. F. Zhang, Electrochim. Acta, 2008, 53, 7863–7868 CrossRef CAS.
- Y. Guo, J. Hu and L. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- G. Zhu, Y. Wang and Y. Xia, Energy Environ. Sci., 2012, 5, 6652–6667 CAS.
- Y. Luo, J. Luo, J. Jiang, W. Zhou, H. Yang, X. Qi, H. Zhang, H. J. Fan, D. Y. W. Yu, C. M. Li and T. Yu, Energy Environ. Sci., 2012, 5, 6559–6566 CAS.
- F. Wu, X. Li, Z. Wang and H. Guo, Nanoscale, 2013, 5, 6936–6943 RSC.
- W. Li, F. Wang, Y. Liu, J. Wang, J. Yang, L. Zhang, A. A. Elzatahry, D. Al-Dahyan, Y. Xia and D. Zhao, Nano Lett., 2015, 15, 2186–2193 CrossRef CAS PubMed.
- V. Etacheri, Y. Kuo, A. van der Ven and B. M. Bartlett, J. Mater. Chem. A, 2013, 1, 12028–12032 CAS.
- X. Li, Y. Zhang, T. Li, Q. Zhong, H. Li and J. Huang, J. Power Sources, 2014, 268, 372–378 CrossRef CAS.
- J. Luo, X. Xia, Y. Luo, C. Guan, J. Liu, X. Qi, C. F. Ng, T. Yu, H. Zhang and H. J. Fan, Adv. Energy Mater., 2013, 3, 737–743 CrossRef CAS.
- Y. Luo, J. Luo, W. Zhou, X. Qi, H. Zhang, D. Y. W. Yu, C. M. Li, H. J. Fan and T. Yu, J. Mater. Chem. A, 2013, 1, 273–281 CAS.
- L. Xin, Y. Liu, B. Li, X. Zhou, H. Shen, W. Zhao and C. Liang, Sci. Rep., 2014, 4, 4479–4485 Search PubMed.
- Z. Wen, S. Ci, S. Mao, S. Cui, Z. He and J. Chen, Nanoscale Res. Lett., 2013, 8, 499–504 CrossRef PubMed.
- H. Wang, D. Ma, X. Huang, Y. Huang and X. Zhang, Sci. Rep., 2012, 2, 701–708 Search PubMed.
- X. Li, Y. Chen, H. Yao, X. Zhou, J. Yang, H. Huang, Y. Mai and L. Zhou, RSC Adv., 2014, 4, 39906–39911 RSC.
- W. Luo, X. Hu, Y. Sun and Y. Huang, J. Mater. Chem., 2012, 22, 4910–4915 RSC.
- B. Li, Q. Zhang, C. Zhang, S. Kang, X. Li and Y. Wang, Int. J. Electrochem. Sci., 2013, 8, 8414–8421 CAS.
- D. C. Johnson and A. L. Prieto, J. Power Sources, 2011, 196, 7736–7741 CrossRef CAS.
- T. Tao, A. M. Glushenkov, M. M. Rahman and Y. Chen, Electrochim. Acta, 2013, 108, 127–134 CrossRef CAS.
- X. Guan, J. Zheng, M. Zhao, L. Li and G. Li, RSC Adv., 2013, 3, 13635–13641 RSC.
- C. Jo, Y. Kim, J. Hwang, J. Shim, J. Chun and J. Lee, Chem. Mater., 2014, 26, 3508–3514 CrossRef CAS.
- Z. Hong, M. Wei, Q. Deng, X. Ding, L. Jiang and K. Wei, Chem. Commun., 2010, 46, 740–742 RSC.
- V. D. Nithya, R. K. Selvan, K. Karthikeyan and Y. S. Lee, J. Nanosci. Nanotechnol., 2015, 15, 694–702 CrossRef CAS.
- K. Min, K. Park, A. Lim, J. Kim and D. Kim, Ceram. Int., 2012, 38, 6009–6013 CrossRef CAS.
- J. Liu, Y. Mao, E. Lan, D. R. Banatao, G. J. Forse, J. Lu, H. Blom, T. O. Yeates, B. Dunn and J. P. Chang, J. Am. Chem. Soc., 2008, 130, 16908–16913 CrossRef CAS PubMed.
- T. Fujii, F. de Groot, G. A. Sawatzky, F. C. Voogt, T. Hibma and K. Okada, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 3195–3202 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef.
- J. Shao, J. Zhang, J. Jiang, G. Zhou and M. Qu, Electrochim. Acta, 2011, 56, 7005–7011 CrossRef CAS.
- Y. Wang, T. Chen and Q. Mu, J. Mater. Chem., 2011, 21, 6006–6013 RSC.
- Z. Ali, S. N. Cha, J. I. Sohn, I. Shakir, C. Yan, J. M. Kim and D. J. Kang, J. Mater. Chem., 2012, 22, 17625–17629 RSC.
- H. Xia, W. Xiong, C. K. Lim, Q. Yao, Y. Wang and J. Xie, Nano Res., 2014, 7, 1797–1808 CrossRef CAS.
- M. Guo, J. Zhao, X. Xu, G. Liu and X. Wang, Ceram. Int., 2014, 40, 5825–5830 CrossRef CAS.
- L. Li, Y. Cheah, Y. Ko, P. Teh, G. Wee, C. Wong, S. Peng and M. Srinivasan, J. Mater. Chem. A, 2013, 1, 10935–10941 CAS.
- Y. Li, H. Zhang, X. Li, H. Zhang and W. Wei, J. Power Sources, 2013, 233, 202–208 CrossRef CAS.
- D. Wang, P. Yang and X. Cheng, Nanosci. Nanotechnol. Lett., 2015, 7, 358–364 CrossRef.
- M. A. Rahman, Y. C. Wong, G. Song and C. Wen, J. Porous Mater., 2015, 22, 1313–1343 CrossRef CAS.
- S. Guo, G. Lu, S. Qiu, J. Liu, X. Wang, C. He, H. Wei, X. Yan and Z. Guo, Nano Energy, 2014, 9, 41–49 CrossRef CAS.
- J. S. Chen, H. Liu, S. Z. Qiao and X. W. D. Lou, J. Mater. Chem., 2011, 21, 5687–5692 RSC.
- Y. Fu, Q. Wei, X. Wang, H. Shu, X. Yang and S. Sun, J. Mater. Chem. A, 2015, 3, 13807–13818 CAS.
- J. S. Cho, Y. J. Hong and Y. C. Kang, ACS Nano, 2015, 9, 4026–4035 CrossRef CAS PubMed.
- F. Lin, H. Song, S. Tian, X. Chen, J. Zhou and F. Wang, Electrochim. Acta, 2012, 83, 305–310 CrossRef CAS.
- J. Morales, L. Sanchez, F. Martin, F. Berry and X. L. Ren, J. Electrochem. Soc., 2005, 152, A1748–A1754 CrossRef CAS.
- M. V. Reddy, T. Yu, C. Sow, Z. X. Shen, C. T. Lim, G. V. S. Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2792–2799 CrossRef CAS.
- Z. Xiao, C. Haoxin, X. Yaping and G. Jinxue, J. Mater. Chem. A, 2014, 2, 3912–3918 Search PubMed.
- L. Gao, H. Hu, G. Li, Q. Zhu and Y. Yu, Nanoscale, 2014, 6, 6463–6467 RSC.
- S. Kim, K. Kim, S. Lee, S. Kim, J. S. Kim and J. K. Lee, Curr. Appl. Phys., 2013, 13, 1923–1927 CrossRef.
- D. Yuan, W. Yang, J. Ni and L. Gao, Electrochim. Acta, 2015, 163, 57–63 CrossRef CAS.
- X. Gu, L. Chen, Z. Ju, H. Xu, J. Yang and Y. Qian, Adv. Funct. Mater., 2013, 23, 4049–4056 CrossRef CAS.
- Y. Liu, X. Zhao, F. Li and D. Xia, Electrochim. Acta, 2011, 56, 6448–6452 CrossRef CAS.
- F. Wu, R. Huang, D. Mu, B. Wu and S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19254–19264 CAS.
- L. Pan, X. Zhu, X. Xie and Y. Liu, Adv. Funct. Mater., 2015, 25, 3341–3350 CrossRef CAS.
- W. Yue, S. Tao, J. Fu, Z. Gao and Y. Ren, Carbon, 2013, 65, 97–104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22930h |
| This journal is © The Royal Society of Chemistry 2015 |