
Simultaneous determination of dexamethasone and lenalidomide in rat plasma by solid phase extraction and ultra-performance liquid chromatography-tandem mass spectrometry: application to pharmacokinetic studies†
Hadir M. Maher*ab,
Nourah Z. Alzomana,
Mona M. Alshehria,
Haya I. Aljohara,
Shereen Shehataa,
Manal Alossaimid and
Norah O. Abanmyc
aCollege of Pharmacy, Department of Pharmaceutical Chemistry, King Saud University, Riyadh 11495, P.O. Box 22452, Saudi Arabia. E-mail: hadirrona@yahoo.com; Fax: +966-118054589; Tel: +966-118052929
bFaculty of Pharmacy, Department of Pharmaceutical Analytical Chemistry, University of Alexandria, El-Messalah, Alexandria 21521, Egypt
cCollege of Pharmacy, Department of Clinical Pharmacy, King Saud University, Riyadh 11495, P.O. Box 22452, Saudi Arabia
dPharmaceutical Analysis Department, University of Strathclyde, UK
First published on 9th November 2015
Abstract
An ultra-performance liquid chromatography method with tandem mass spectrometric detection (UPLC-MS/MS) has been developed and validated for the simultaneous determination of lenalidomide (LND) and dexamethasone (DEX) in rat plasma using domperidone (DOM) as an internal standard (IS). Sample preparation was performed using solid phase extraction methodology (SPE) followed by chromatographic analysis on a C 18 column (100 × 1.0 mm, i.d., 1.7 μm particle size) and a mobile phase composed of (0.1% formic acid in water) and (0.1% formic acid in acetonitrile) in the ratio of (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80, v/v) at the flow rate of 0.2 mL min−1. Electrospray ionization (EI) in the positive ionization mode using multiple reaction monitoring (MRM) was applied to detect the transitions of DEX at m/z 393 > 147, LND at m/z 260 > 149 (LND), and DOM at m/z 426 > 175. The method was validated over the concentration range of 0.01–5 ng mL−1 with a very low limit of quantitation of 0.01 ng mL−1 for both LND and DEX. Recoveries of both analytes from plasma samples ranged from 86–106% throughout their linear ranges. Intra-day and inter-day precision was evaluated and in all cases, the RSD values were within the acceptance values (<15%). The applicability of the method was extended to the determination of the pharmacokinetics of both LND and DEX, following their oral administration to rats, either alone or in combination, suggesting the applicability of the method in further clinical studies.
:80, v/v) at the flow rate of 0.2 mL min−1. Electrospray ionization (EI) in the positive ionization mode using multiple reaction monitoring (MRM) was applied to detect the transitions of DEX at m/z 393 > 147, LND at m/z 260 > 149 (LND), and DOM at m/z 426 > 175. The method was validated over the concentration range of 0.01–5 ng mL−1 with a very low limit of quantitation of 0.01 ng mL−1 for both LND and DEX. Recoveries of both analytes from plasma samples ranged from 86–106% throughout their linear ranges. Intra-day and inter-day precision was evaluated and in all cases, the RSD values were within the acceptance values (<15%). The applicability of the method was extended to the determination of the pharmacokinetics of both LND and DEX, following their oral administration to rats, either alone or in combination, suggesting the applicability of the method in further clinical studies.
1. Introduction
Multiple myeloma (MM) is a hematological malignancy characterized by excessive proliferation of plasma cells in the bone marrow, with subsequent over-production of a monoclonal protein in most patients.1–3 MM accounts for approximately 10% of hematological malignancies and 1% of all malignancies.2 Although MM is responsive to initial chemotherapy, it remains incurable and almost all patients show eventual relapse.1,3 The most widely occurring challenges during MM treatment are disease recurrence, immunosuppression, and treatment-related toxicities. Novel, more effective treatment approaches have been developed in order to improve outcomes and extend survival for patients with relapsed or refractory multiple myeloma (RRMM). Biologically-based treatment strategies such as thalidomide, and lenalidomide (LND), are now available which specifically target myeloma cell interactions within the bone marrow microenvironment. These interactions are the fundamental basis for the growth and survival of malignant cells. Thus, these agents are powerful tools in prolonging the duration of action and overcoming drug resistance in patients with MM.1,3–5LND is a potent immunomodulatory agent, belonging to a novel class of drugs called immunomodulatory compounds that are used to treat hematologic malignancies such as MM6 and mantle cell lymphoma (MCL).7 LND (Fig. 1) was designed to have a more manageable safety profile compared with its parent compound, thalidomide, and to provide more potent anti-inflammatory and immunomodulatory effects.1,8 The efficacy of LND in MM has been confirmed. LND has a dual mechanism of action,6 involving both a direct tumoricidal activity and immunomodulation, which may result in rapid and sustained control of MM, respectively.9,10 The tumoricidal effect of LND occurs through several mechanisms, including induction of tumor suppressor genes, disruption of stromal support, and activation of caspases.11 The immunomodulatory effects of LND, including T-cell and natural killer (NK)-cell activation, and increased expression of death effector molecules, lead to enhanced immune cell function.12–15 Clinically, these dual effects result in high- and rapid-response rates, and sustained disease control.
 | ||
| Fig. 1 Chemical structures of the studied drugs; DEX, (a), LND, (b), and DOM (IS), (c). | ||
A wide range of novel combination regimens that include LND are still in the development phase. They aim at further improving treatment options for patients with MM.16 Dexamethasone (DEX) (Fig. 1) synergistically enhances the anticancer effects of LND.17 LND, in combination with DEX, have been approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of patients with RRMM who have received at least one prior line of therapy.2,3,5 LND along with DEX is also used in newly diagnosed MM.18 A combination of LND with DEX is of high effectiveness in the treatment of MM resulting in an improved overall survival, response rate, and time to progression in RRMM. In addition, LND plus DEX is a potential therapy in the maintenance of myeloma.2
The primary adverse effects associated with LND are myelosuppression (including neutropenia, thrombocytopenia and, to a lesser extent, anaemia) and venous thromboembolism (VTE), particularly when combined with DEX in the absence of thromboprophylaxis.19 Patients receiving a combination of LND and a high-dose DEX are susceptible to febrile neutropenia and VTE.20 The combination of LND and a low-dose DEX in patients with newly diagnosed MM is associated with reduced risk of VTE relative to high-dose DEX. Also, clinical data proved that LND plus low-dose DEX is associated with better short-term overall survival and with lower toxicity compared with LND plus high-dose DEX in patients with newly diagnosed MM.2,6,19,21
Renal impairment (RI) is a common complication affecting patients with MM. Since LND is excreted renally, RI has a significant effect on the drug pharmacokinetics by reducing the renal clearance rate, with increased plasma concentrations of LND and prolonged plasma half-life.22,23 Moreover, a significant increase of up to 185–420% in the area under the drug plasma concentration–time curve has been reported in patients with RI.23 Thus, dose adjustments are required when LND is given to patients with RI. Consequently, lower starting doses are required in patients with RI to avoid over-exposure and an increased risk of adverse events (especially myelosuppression), while maintaining good therapeutic index.
Treatment with LND/DEX is effective and well tolerated in RRMM patients with RI. Clinical data support the efficacy and safety of appropriately dose-adjusted regimens in patients with RRMM, provided that the dose of LND is properly reduced according to the renal function.22,23 Also, corticosteroids overdose may result in severe adverse effects including, osteoporosis and fractures, hyperglycemia/diabetes, Cushingoid appearance and weight gain, and myopathy. In addition to the possibility of cataracts and glaucoma, immunosuppression, and psychiatric disturbances.24 Thus developing analytical methods capable of monitoring plasma levels of both LND and DEX is of a potential concern in patients with MM especially those with associated RI.
To facilitate pharmacokinetic studies with this combination, the current method has been developed with a particular focus on achieving superior sensitivity and specificity.
Hyphenated LC-tandem MS techniques provide the great advantages of high sensitivity and selectivity achieved by MS/MS spectra that are characteristics to particular target compounds. Also, compared with conventional HPLC, UPLC permits the use of short, narrow bore columns, packed with ultra-small particle size, resulting in improved resolution with short analysis time. These features are highly important for high throughput analysis in addition to diminishing matrix effects during the analysis of biological matrices.25 Thus UPLC-MS/MS has been employed in this study.
Only few reports have been found in the literature dealing with the determination of LND in biological samples, namely HPLC-UV,26 HPLC-fluorimetry,27 HPLC-MS,28,29 LC-MS/MS,30–33 and UPLC-MS/MS.34 Analysis of DEX in plasma samples has been previously performed by numerous LC methods namely, HPLC-UV,35 LC-MS,36,37 LC-MS/MS,38–40 UPLC-MS,41 and UPLC-MS/MS.42
Some of the reported methods involved tedious liquid–liquid extraction (LLE) procedures,29,31,35,37,38,40,41 not highly advanced detection,26,27,35 and relatively long run time26,27,29,31–42 that negatively affect the accuracy of the results and limit their throughput in routine analysis of biological specimens. However, our proposed method utilized solid phase extraction (SPE) as an advantageous sample purification technique compared with LLE, along with the super selectivity potential of MS/MS detection, and within very short runtime (1.5 min). Moreover, comparing the method sensitivity with previously published bioanalytical methods, the proposed method is the most sensitive method, up to date, described for the quantitation of LND and DEX. This improved sensitivity is extremely important to determine terminal phase concentrations with accurate estimation of different pharmacokinetic parameters. Thus this method is considered superior and can be readily applied as a highly sensitive, specific analytical method that can be readily applied in accurate high throughput clinical studies.
To our knowledge, no method has been found in the literature so far dealing with the simultaneous analysis of LND and DEX in actual plasma samples. So this work aims at developing and validating a sensitive and specific UPLC-MS/MS method for the simultaneous determination of both LND and DEX in rat plasma. The application of this method was extended to the determination of the pharmacokinetics of each drug alone and when given in combination.
2. Experimental
2.1. Materials and reagents
Reference standard samples of DEX were obtained from Saudi Food and Drug Authority (Riyadh, Saudi Arabia). LND was purchased from LC Laboratories (Woburn, MA, USA). Domperidone (DOM) reference standard was obtained from Amoun Pharmaceutical Company (El-Obour City, Cairo, Egypt).Revlimid® capsules (Celgene Corporation, NJ, USA) labeled to contain 10 mg LND per capsule were used. Dexamethasone tablets Oradexon (dexamethasone tablets, USP) (Pharma Science INC, Montreal, Canada) labeled to contain 0.5 mg DEX were also used in the study.
HPLC grade solvents were used. Methanol, acetonitrile (Panreac, E.U.), and formic acid (Sigma Aldrich, Chemie GmbH, Steinheim, Germany) were used in the study.
Ultrapure water was obtained through Milli-Q Advantage water purification system (Millipore, Molsheim, France) with 0.22 μm filter.
2.2. Instrumentation
UPLC-MS/MS analysis was performed using a Waters Model Xevo TQ-S separation system (Singapore). The instrument was supplied with sample manager (Aquity™ Ultra-performance LC) and binary solvent manager (Aquity™ Ultra-performance LC). Detection was carried out using electrospray ionization mass spectrometry (Zspray™ ESI-APCI-ESCI, Ultra-performance LC) with multiple reaction monitoring (MRM)-mode using a triple-quadrupole mass spectrometric detector (STEP WAVE™, Ultra-performance LC). The instrument was controlled by the Masslynx™ Version 4.1 software (Micromass).Solid-phase extraction (SPE) was performed in a J.T. Bakers vacuum system. Two types of Spe-ed cartridges (Applied Separations, Allentown, PA) were tried, namely, octadecyl C 18 (200 mg, 3 mL) and octyl C 8 (200 mg, 3 mL).
Nitrogen evaporator N-EVAP 112 with heating system OA-SYS (Organomation Associates, Inc, MA, USA) was used in sample preparation.
For sample filtration, disposable syringe filters (CHROMAFIL® Xtra PA-20/25 polyamide filters, pore size: 0.2 μm, filter-Ɵ: 25 mm (MACHEREY NAGEL, GmbH & Co. KG, Duren, Germany)) were used.
2.3. Chromatographic conditions
The separation was performed using Acquity UPLC BEH™ C 18 column (100 × 1.0 mm, i.d., 1.7 μm particle size) (Waters, Ireland). The mobile phase was composed of two solvent systems namely, the aqueous phase, A (0.1% formic acid in water) and the organic modifier, B (0.1% formic acid in acetonitrile). Isocratic elution was applied using a mobile phase of A:B (20:80, v/v). Column temperature was maintained at 45 °C while the auto-sampler temperature was kept at 10 °C throughout the runtime (1.5 min). The flow rate was 0.2 mL min−1 and the injection volume was 5 μL using partial loop mode.
2.4. Mass spectrometric conditions
Electrospray ionization (EI) was operated in the positive ionization mode. Quantitation was performed using multiple reaction monitoring (MRM) of the transitions m/z 393 > 147 (DEX), m/z 260 > 149 (LND), and m/z 426 > 175 (DOM) being used as the internal standard (IS). To obtain optimum MS parameters, the collision energy, cone voltage, and capillary voltage were set at (32 eV, 62 V, 4 kV for DEX), (14 eV, 14 V, 3.5 kV for LND), and (27 eV, 50 V, 3 kV for DOM), respectively. Nitrogen was used as the desolvating gas at a flow rate of 800 L h−1. Cone gas flow was adjusted at 150 L h−1. Desolvation temperature of 400 °C was applied whereas the source temperature was adjusted to 150 °C. The MS analyzer parameters were as follows: LM1 and HM1 resolution of 2.8 and 14.5, respectively, for ion energy 1, and LM2 and HM2 resolution of 2.8 and 14.5, respectively, for ion energy 2. The dwell time of 0.0255 s was used.2.5. Preparation of stock and standard solutions
Stock solutions of 1 mg mL−1 of each of DEX, LND, and DOM were prepared in methanol. Further dilutions were made using the same solvent to obtain standard solutions of 10, 1.0, and 0.1 ng mL−1 of DEX and LND. However, DOM stock solution was diluted with methanol to give a final concentration of 0.1 ng mL−1.2.6. Spiking of plasma samples and construction of matrix-based calibration graphs
Matrix-based calibration graphs of each of DEX and LND were obtained by spiking drug free rat plasma samples with predetermined amounts of each drug, along with DOM (IS). The calibration graphs were constructed by spiking portions of 0.5 mL plasma separately with different volumes of standard solutions of both DEX and LND to get final concentrations of 0.01–5 ng mL−1 of each drug. Standard solutions of 10 ng mL−1 of DEX and LND were used for spiking plasma samples of high concentration levels within the calibration set (5–0.5 ng mL−1). However, standard solutions of 1.0 ng mL−1 of both drugs were used to prepare spiked samples with middle concentration ranges (0.5–0.05 ng mL−1). Meanwhile, samples spiked with low concentration levels (0.05–0.01 ng mL−1) were prepared from standard solutions containing 0.1 ng mL−1 of both drugs. A volume of 250 μL of 0.1 ng mL−1 DOM was separately added to each sample. Spiked samples were then made up to final volumes of 1.5 mL with methanol. Blank samples were simply prepared by adding 1.0 mL methanol to 0.5 mL plasma samples.Methanol was used for protein precipitation. Each mixture was then centrifuged at 6000 rpm for 5 min. The supernatant was transferred into another tube and the precipitate was extracted with further 500 μL methanol. The supernatant and the washing were purified by passing through a C 18 Spe-ed cartridge used for solid phase extraction, previously conditioned with 5.0 mL methanol followed by 5.0 mL water. The retained drugs were eluted with 0.5 mL methanol and the eluate was evaporated to dryness under nitrogen using nitrogen evaporator. The residue was further reconstituted in 0.5 mL acetonitrile. Portions of 5 μL volume were then injected in triplicates into the UPLC-MS/MS system under the above mentioned chromatographic and mass spectrometric conditions to obtain the calibration graphs of each compound. Alternatively, the corresponding regression equations were derived.
2.7. Animal treatment and preparation of treated rat plasma samples
All animal experimentation was conducted in accordance with the accepted standards of human animal care as per the WHO regulations in Saudi Arabia. All experiments were performed in compliance with the relevant laws and institutional guidelines. Four groups of five male Wistar rats each (average weight 250 g) were housed under standard laboratory conditions of average temperature (24–27 °C), relative humidity (40–70%), and light cycle (12–14 h per day), in a well-ventilated place for 2 weeks prior to treatment. The first group (gp. I) did not receive any medication and was used as the control. The second to fourth groups were the treated groups being subjected to oral administration of 5 mg kg−1 dose of DEX (gp. II), 2 mg kg−1 dose of LND (gp. III), or a mixture of DEX (5 mg kg−1 dose) and LND (2 mg kg−1 dose) for group IV. Tablets of LND and DEX were prepared in suspension form by triturating an accurately weighed amount of each powdered tablet with methyl cellulose solution (0.5% w/v, in water) to give final concentrations of 2.5 mg mL−1 DEX and 2 mg mL−1 LND. Oral doses of 0.5 mL (DEX) or 0.25 mL (LND) of the corresponding drug suspension were administered using a gavage needle to the rat groups (II, III, IV) after a total fasting time of 12 h. The rats were fed 4 h after dosing.Blood samples (0.8 mL) from control and treated groups were collected from orbital plexus in EDTA-K2 tubes containing dipotassium salt of EDTA as an anticoagulant. Blood samples were collected at different time intervals (0.0, 0.25, 0.5, 0.8, 1.0, 2.0, 4.0, and 24.0 h) following drug administration. Plasma was separated from blood samples by centrifugation at 4 °C for 30 min at 4000 rpm, and then kept at −70 °C till further analysis. Portions of 0.5 mL volumes of each plasma sample were separately transferred into clean tubes and then spiked with a constant volume of 0.25 mL of DOM, IS (0.1 ng mL−1). Plasma samples were then treated exactly as described under the construction of matrix-based calibration graphs.
3. Results and discussion
3.1. LND/DEX combinations
It was initially proven that LND plus high-dose DEX showed high response rate with lower toxicity compared with thalidomide-high dose DEX combinations in patients with newly diagnosed MM.43 Later on, the efficacy of LND oral combinations with low-dose DEX was evaluated in comparison with those with high-dose DEX. During the clinical implementation, patients taking LND plus high-dose DEX were assigned to LND (25 mg on days 1–21) plus DEX (40 mg on days 1–4, 9–12, and 17–20 of a 28 day cycle). However, patients taking LND plus low-dose DEX were given LND with the same dose schedule as those taking high-dose DEX, along with DEX (40 mg on days 1, 8, 15, and 22 of a 28 day cycle). The results showed that a better overall survival and less adverse effects were noticed with low-dose DEX compared with high-dose DEX.21 Most adverse effects, namely venous thromboembolism, infection, pneumonia, and hyperglycemia, were significantly reduced with low-dose DEX.20 This finding suggested that LND combinations with low-dose DEX is particularly beneficial in elderly people and those more sensitive to side effects in order to reduce toxicities while maintaining efficacy.20As previously mentioned, patients with RI and receiving a combination therapy of LND and DEX should be extremely cautious with LND dosage.19,20,23 RI results in a significant alteration of the pharmacokinetics of LND with a remarkable increase in the area under the plasma concentration–time curve.20 It was also reported that patients with RI and suffering from RRMM are susceptible to increased incidence of myelosuppression-related toxicities with unadjusted doses of LND. Thus based on the measured creatinine clearance (CrCl) rate, dose reductions were calculated. The recommendations for LND dosing, instead of the 25 mg regular dosing, in patients with RI are as follows: 10 mg every 24 h for moderate RI (CrCl ≥ 30 to < 60 mL min−1); 15 mg every 48 h for severe RI (CrCl < 30 mL min−1, and not requiring dialysis); and down to 5 mg every 24 h in the end stage renal disease (CrCl < 30 mL min−1, and requiring dialysis).19,20,23 Reduced doses of LND in LND/DEX combinations did not affect the efficacy of this drug regimen in case of RI compared with patients without RI and receiving normal LND doses. It was also shown that renal function could be even improved with LND/low-dose DEX combinations following LND dosage adjustment.23 A recent study showing that a regimen of adjusted dose LND (15 mg on days 1–12) and low-dose DEX (20 mg day−1 on days 1–4, 9–12, and 17–20) is well-tolerated and highly effective in patients with RRMM and RI (CrCl ≤ 60 mL min−1) or aged people more than 60 years.23
Based on these collected data and with reference to different LND reduced dosages, the experimental design of our work was assigned to the administration of 2 mg kg−1 LND to the rats compared with 5 mg kg−1 DEX as a possible intermediate ratio of LND/DEX drug regimens.
3.2. Selection of internal standard
In spite the fact that the most suitable IS in LC-MS/MS analysis in biological samples is a deuterated form of the analyte.44 However, due to the high cost, the deuterated form was not employed in this work. In this study, different compounds (ibuprofen, pseudoephedrine, chlorpheniramine, and DOM) were tried as IS. DOM was finally selected due to its similarity in the chromatographic behavior and extraction efficiency to the cited analytes.3.3. Optimization of chromatographic conditions
Various combinations of methanol, acetonitrile, and formic acid were tried as different mobile phases. Initially, mobile phases with different ratios of an aqueous phase, A (water with 0.1% formic acid) and organic modifiers, B (methanol or acetonitrile, each mixed with 0.1% formic acid) were investigated regarding peak shape, peak symmetry and sharpness, sensitivity, and analysis time. The percentage of organic modifier in the mobile phase had a significant effect on the shape, sharpness, and response of DEX peaks. Best results were obtained using methanolic modifiers in a ratio of 80% along with the aqueous phase. Moreover, the percentage of formic acid in the mobile phase was examined in the range (0.01–0.15%). It was noticed that formic acid was essential to get sharp DEX peaks and that increasing formic acid content in the mobile phase resulted in decreased retention times for DOM with best results achieved with 0.1% formic acid. Final analysis was performed with an isocratic elution using a mobile phase of A:B (20:80) for 1.5 min. Using this system, sharp and symmetric peaks within short runtime were obtained (DEX, LND eluted at 0.38 min, and DOM (IS) at 0.62 min).
3.4. Optimization of mass spectrometric conditions
UPLC-MS/MS parameters were carefully optimized for the determination of DEX and LND. Standard solutions of DEX, LND, and DOM (IS) (2.0 ng mL−1) were directly infused along with the mobile phase into the mass spectrometer with ESI as ionization mode. Tuning of the mass spectrometer was done in both positive and negative ion modes. The positive ionization mode was selected for final analysis since it gave good response for the analytes and IS. In the precursor ion full scan spectra, [M + H]+ fragments were the most abundant at m/z 393.23 (DEX), m/z 260.13 (LND), and at m/z 426.26 (DOM). The product ions selected for quantitation were at m/z 147.05 (DEX), m/z 149.06 (LND), and at m/z 175.10 (DOM) (Fig. 2). Different parameters were optimized in order to get the highest response of the protonated molecules of the studied compounds. They include capillary voltage, cone voltage, desolvation temperature, ESI source temperature, and desolvation gas flow rate. Generally, the precursor ion intensities increase gradually with increasing the capillary voltage or cone voltage till certain values after which they decrease dramatically. It was noticed that optimum capillary voltages were 4, 3.5, 3 kV for DEX, LND, and DOM, respectively, while the selected values for cone voltages were 62, 14, 50 V for DEX, LND, and DOM, respectively. To maximize the response of the daughter fragment ions, the collision energy was optimized for each compound. An increase in the collision energy resulted in an increase in the intensity of the particular fragment ion till a certain value after which a decrease in the intensity would be observed. Collision energies of 32, 14, 27 eV produced maximum intensities of the selected daughter ions for DEX, LND, and DOM, respectively.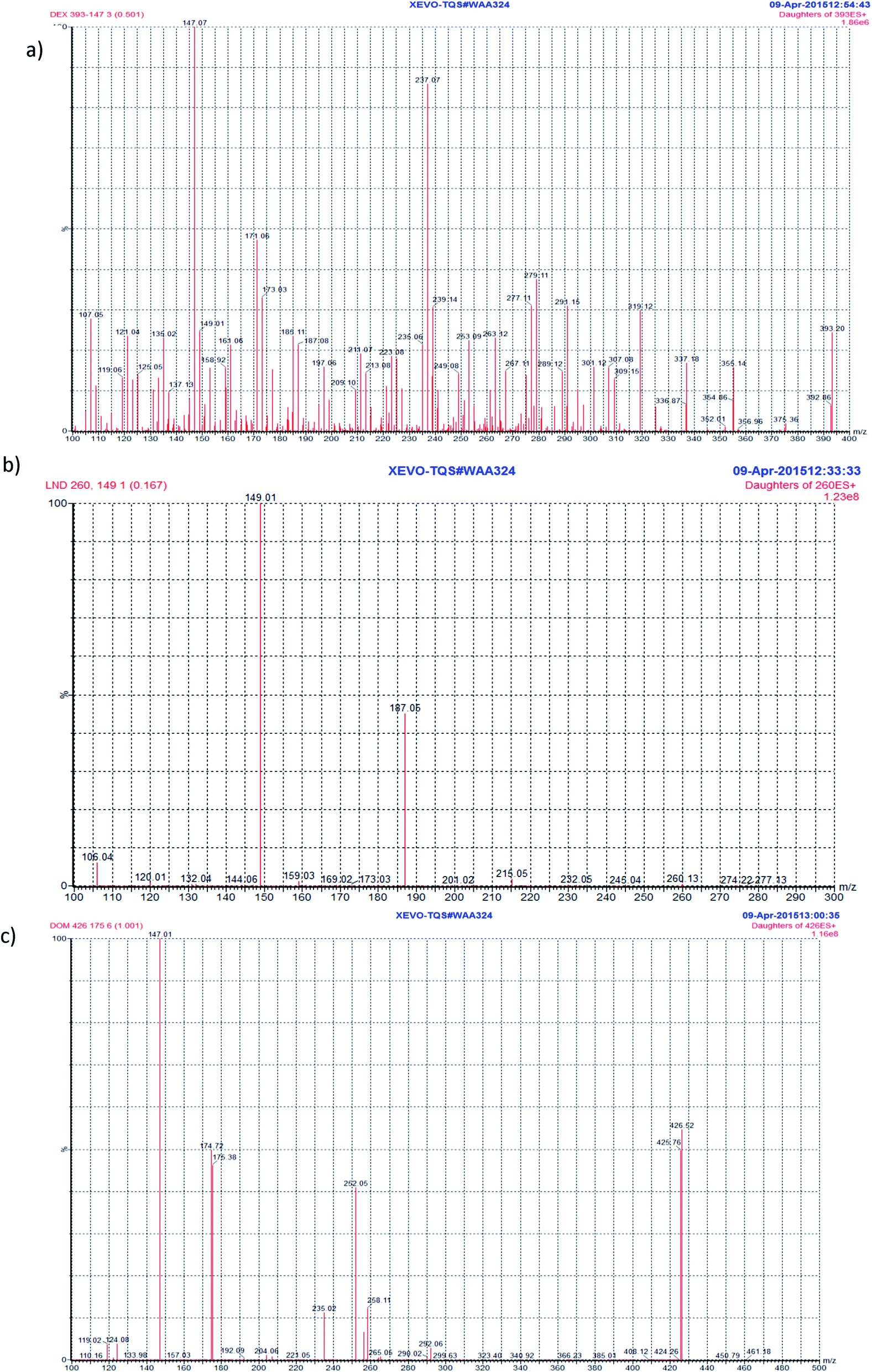 | ||
| Fig. 2 Product ion spectra of DEX, (a), LND, (b), and DOM (IS), (c). | ||
3.5. Extraction and purification optimization
Methanol (100%) was used for the extraction of the drugs from plasma samples since it provided excellent recovery for all drugs (86–106%). It is also very important in protein precipitation of plasma samples. Thus, methanol served to produce clear supernatant solutions.Different procedures were tested during the clean-up optimization. Supernatants were subjected to different treatments using each of the two solid-phase extraction (SPE) Spe-ed cartridges namely, octadecyl C 18 (200 mg, 3 mL) and octyl C 8 (200 mg, 3 mL). Washing the cartridges with water directly before the elution of the analytes with methanol was tested. It was noticed that this step had a negative effect on the intensity of the recovered peaks of both DEX and LND. Thus, this step was omitted in the final procedure and the analytes were directly eluted from the cartridges with methanol following the loading of the extracts. SP extraction recovery of each type of cartridge (C8, C18) was assessed by triplicate analysis of plasma samples spiked with DEX and LND at 0.1 ng mL−1 concentration level. The average recovery reported with C18 cartridge was 88.41 ± 5.88 for DEX and 86.23 ± 8.25 for LND. However, these values were less with C 8 cartridge, being 68 ± 7.21 with DEX and 74.54 ± 6.11 with LND. Based on the analytes' recovery, octadecyl C 18 cartridges were used in actual analysis. It is noteworthy to mention that SPE with C 18 cartridges has been previously reported for the analysis of LND26,28,30 and DEX.42
3.6. Method validation
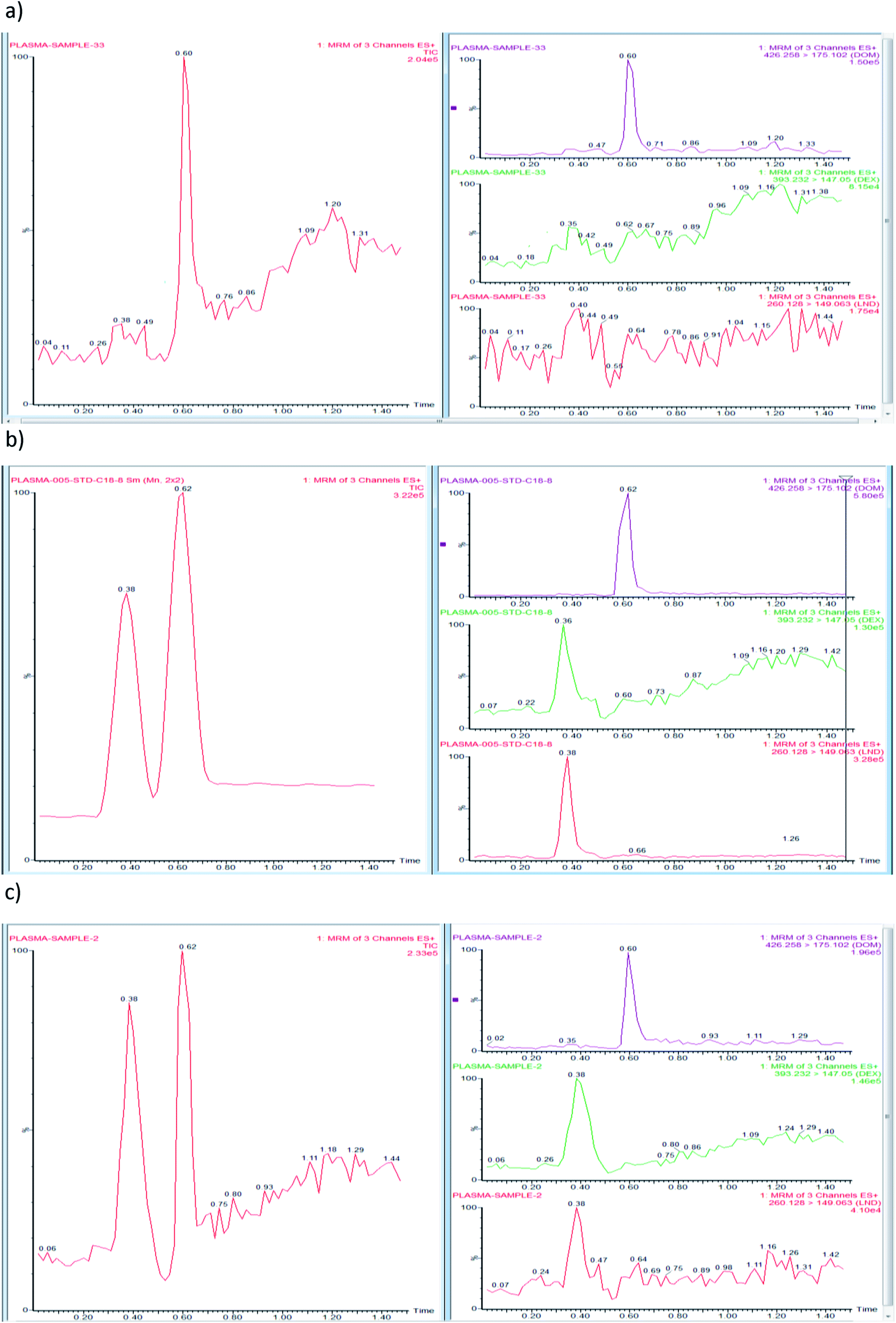 | ||
| Fig. 3 Representative multiple reaction monitoring chromatograms of a blank plasma sample with DOM (IS), (a), plasma sample spiked with a standard mixture of DEX, LND, and DOM (0.05 ng mL−1), (b) and plasma sample taken 1 h following oral administration of DEX (5 mg kg−1 dose) and LND (2 mg kg−1 dose) and spiked with DOM (IS) (0.05 ng mL−1), (c). | ||
| Parameter | DEX | LND |
|---|---|---|
| a LOQ: limit of quantitation.b LOD: limit of detection.c Sa: standard deviation of intercept.d Sb: standard deviation of slope.e Sy/x: standard deviation of residuals.f F: variance ratio, equals the mean of squares due to regression divided by the mean of squares about regression (due to residuals). | ||
| Linearity range (ng mL−1) | 0.01–5 | 0.01–5 |
| LOQa (ng mL−1) | 0.01 | 0.01 |
| LODb (ng mL−1) | 0.004 | 0.004 |
| Intercept | 0.1257 | 0.4506 |
| Slope | 2.2179 | 7.4828 |
| Correlation coefficient | 0.9990 | 0.9980 |
| Sac | 0.0838 | 0.3510 |
| Sbd | 0.0425 | 0.1779 |
| Sy/xe | 0.2128 | 0.8913 |
| Ff | 2726.98 | 1769.69 |
| Concentration added (ng mL−1) | Intra-day | Inter-day | |||
|---|---|---|---|---|---|
| Mean recovery (%) ± RSDa | Er (%) | Mean recovery (%) ± RSDb | Er (%) | ||
| a Mean recovery (%) ± RSD of three determinations.b Data are based on assay of three replicates on three different days. | |||||
| DEX | 3 | 87.23 ± 8.25 | −12.77 | 87.99 ± 11.01 | −12.01 |
| 0.1 | 93.56 ± 5.16 | −6.44 | 92.67 ± 6.18 | −7.33 | |
| 0.025 | 89.78 ± 9.89 | −10.22 | 101.73 ± 7.53 | 1.73 | |
| LND | 3 | 92.01 ± 6.98 | −7.99 | 104.99 ± 9.52 | 4.99 |
| 0.1 | 87.63 ± 9.07 | −12.37 | 86.89 ± 11.94 | −13.11 | |
| 0.025 | 106.44 ± 10.34 | 6.44 | 104.67 ± 12.28 | 4.67 | |
The negligible matrix effect could be confirmed by low degree of matrix ionization indicating the absence of interfering components in the matrix that could affect the ionization of the analytes or the IS. This allowed the possibility of the determination of the analytes at low concentrations.
3.7. Pharmacokinetic studies
The developed UPLC-MS/MS method has been used to investigate the pharmacokinetics of DEX and LND, either alone or in combination, following their oral administration to rats. This was performed by analyzing actual rat plasma samples taken at different time intervals following drug administration. The resulting mean plasma concentration versus time profiles for DEX and LND are shown in Fig. 4. The main pharmacokinetic parameters like maximum plasma concentration Cmax, time to maximum concentration tmax, and half-life t1/2 were calculated from the curves. It was found that following oral administration of 5 mg per kg body weight of DEX to rats resulted in mean values of Cmax, tmax, and t1/2 of 1.35 ng mL−1, 1.00 h, and 5.25 h, respectively, when given alone (gp. II), and 1.46 ng mL−1, 1.00 h, and 7.50 h, respectively, when given in combination (gp. IV). Also, oral administration of LND (2 mg per kg body weight) to rats resulted in mean values of Cmax (3.79 ng mL−1), tmax (0.50 h), and t1/2 (0.71 h). However, these values were found to be 3.11 ng mL−1, 0.50 h, and 0.71 h, respectively, when LND was given concomitantly with DEX (gp. IV). Thus this method could be used successfully for the simultaneous determination of LND and DEX when taken in combinations in the management of MM. Practical work revealed that plasma levels of DEX when given alone were slightly lower than those recorded after its co-administration with LND (in the same dosages) and the opposite was noticed for LND. These findings suggested that co-administration of DEX and LND may affect plasma levels of both drugs, compared with single administration, with subsequent influence on their toxicities and therapeutic effects.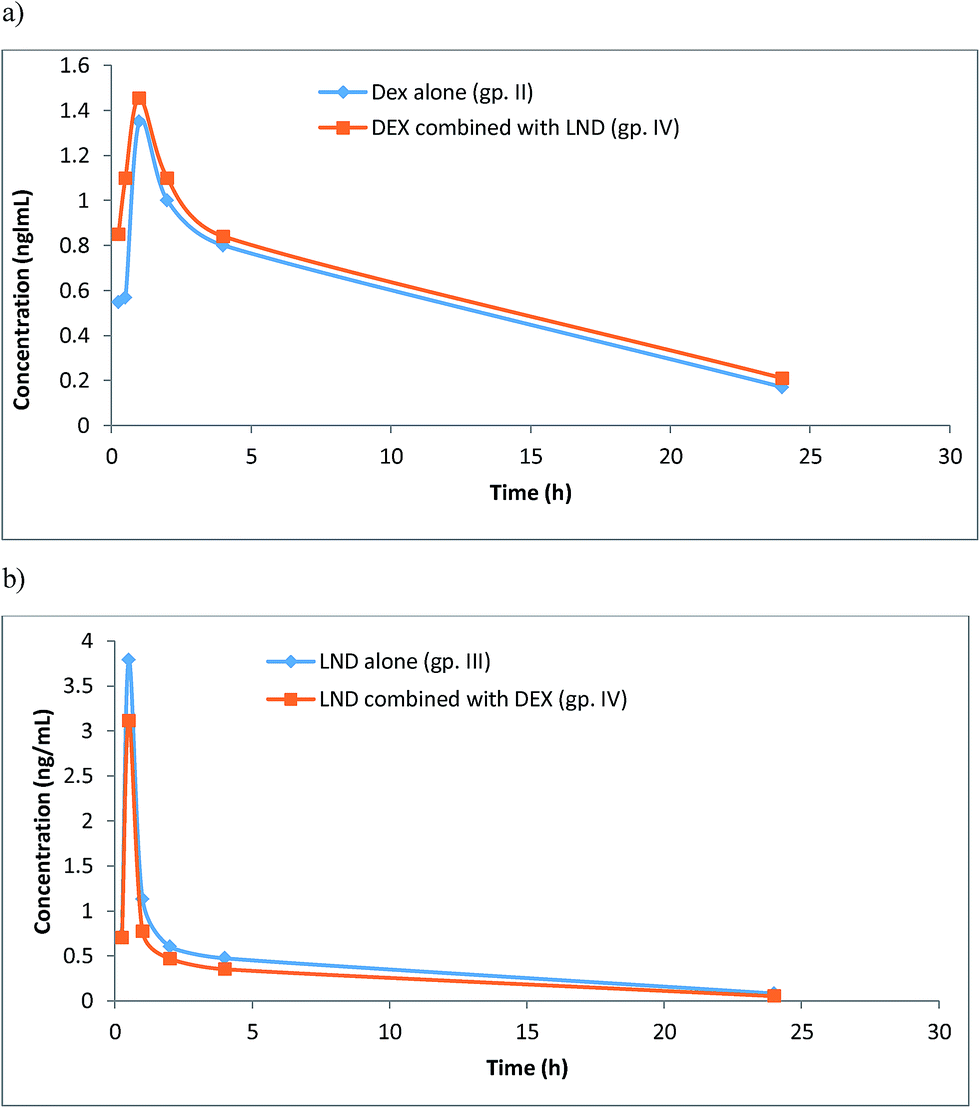 | ||
| Fig. 4 Plasma concentration time plots after oral administration of DEX (5 mg kg−1) (gp. II), (a), LND (2 mg kg−1) (gp. III), (b), compared with their combination (gp. IV). | ||
Since the present study was performed with rats, and not humans, and since some differences could exert when comparing their pharmacokinetics,47 the results gave a suggestion of possible drug interaction in clinical studies. Thus careful monitoring of plasma levels of both drugs should be conducted with concomitant administration of both DEX and LND in patients with MM, especially those suffering from RI. This is extremely important to ensure particular drug plasma levels required to produce the required therapeutic effect and to control drug toxicities.
4. Conclusion
An UPLC-MS/MS method was developed and validated for the simultaneous determination of LND and DEX in rat plasma. This method is the most sensitive reported to date with limit of quantitation of 0.01 ng mL−1 for both compounds. This high sensitivity is advantageous in accurate measurement of low levels of the drugs in plasma samples and is thus beneficial in current and future clinical studies. The method also applied SPE technique as an advanced clean-up procedure. Moreover, the analysis run-time was short, not exceeding 1.5 min, which guarantees the high throughput of the assay. The proposed method enabled determination of the pharmacokinetics of both DEX and LND following their oral administration to rats, whether alone or in combination. This issue is extremely important in therapeutic drug monitoring (TDM) of patients with MM and suffering from RI. Preliminary results in rats suggested the possibility of drug-induced interaction which encourages further clinical studies on RI-MM patients receiving such combination.Acknowledgements
The authors would like to extend their appreciation to the Deanship of Scientific Research at King Saud University for its funding of this research through the research group project No. RGP-VPP-331.References
- A. Palumbo, J. San Miguel, P. Sonneveld, P. Moreau, J. Drach, G. Morgan and H. Einsele, Cancer Treat. Rev., 2008, 34, 283–291 CrossRef CAS PubMed.
- D. Wei, Y. Mingxi, X. Jianwang, Z. Hao, L. Ming and L. Pei, J. Med. Coll. PLA, 2011, 26, 29–33 CrossRef.
- A. Palumbo, M. Dimopoulos, J. San Miguel, J. Harousseau, M. Attal, M. Hussein, S. Knop, H. Ludwig, M. Lilienfeld-Toal and P. Sonneveld, Blood Rev., 2009, 23, 87–93 CrossRef CAS PubMed.
- S. Kumar and S. V. Rajkumar, Eur. J. Cancer, 2006, 42, 1612–1622 CrossRef CAS PubMed.
- E. Kastritis and M. A. Dimopoulos, Expert Opin. Pharmacother., 2007, 8, 497–509 CrossRef CAS PubMed.
- M. A. Dimopoulos, E. Terpos and R. Niesvizky, Critical Reviews in Oncology/Hematology, 2013, 88, S23–S35 CrossRef PubMed.
- R. Dawar and F. Hernandez-Ilizaliturri, Best Pract. Res., Clin. Haematol., 2012, 25, 185–190 CrossRef CAS PubMed.
- G. W. Muller, R. Chen, S. Y. Huang, L. G. Corral, L. M. Wong, R. T. Patterson, Y. Chen, G. Kaplan and D. I. Stirling, Bioorg. Med. Chem. Lett., 1999, 9, 1625–1630 CrossRef CAS PubMed.
- D. Verhelle, L. G. Corral, K. Wong, J. H. Mueller, L. Moutouh-de Parseval, K. Jensen-Pergakes, P. H. Schafer, R. Chen, E. Glezer, G. D. Ferguson, A. Lopez-Girona, G. W. Muller, H. A. Brady and K. W. Chan, Cancer Res., 2007, 67, 746–755 CrossRef CAS PubMed.
- L. Escoubet-Lozach, I. L. Lin, K. Jensen-Pergakes, H. A. Brady, A. K. Gandhi, P. H. Schafer, G. W. Muller, P. J. Worland, K. W. Chan and D. Verhelle, Cancer Res., 2009, 69, 7347–7356 CrossRef CAS PubMed.
- T. Hideshima and K. C. Anderson, Nat. Rev. Cancer, 2002, 2, 927–937 CrossRef CAS PubMed.
- J. B. Marriott, I. A. Clarke, K. Dredge, G. Muller, D. Stirling and A. G. Dalgleish, Clin. Exp. Immunol., 2002, 130, 75–84 CrossRef CAS PubMed.
- D. H. Chang, N. Liu, V. Klimek, H. Hassoun, A. Mazumder, S. D. Nimer, S. Jagannath and M. V. Dhodapkar, Blood, 2006, 108, 618–621 CrossRef CAS PubMed.
- F. Davies and R. Baz, Blood Rev., 2010, 24, S13–S19 CrossRef PubMed.
- M. Tufail, D. S. Siegel, L. McBride, E. Bilotti, E. Bello, P. Anand, K. Olivo, U. Bendarz, A. McNeill and D. H. Vesole, Clin. Lymphoma, Myeloma Leuk., 2012, 12, 186–190 CrossRef CAS PubMed.
- M. Cives, V. Simone, O. Brunetti, V. Longo and F. Silvestris, Critical Reviews in Oncology/Hematology, 2013, 85, 9–20 CrossRef PubMed.
- Z. Qiana, L. Zhang, Z. Cai, L. Sun, H. Wang, Q. Yi and M. Wang, Leuk. Res., 2011, 35, 380–386 CrossRef PubMed.
- E. Shannon, F. Sandoval, N. Greig and P. Stagg, Int. Immunopharmacol., 2012, 12, 441–446 CrossRef CAS PubMed.
- M. V. Mateos, Cancer Treat. Rev., 2010, 36, S24–S32 CrossRef PubMed.
- A. Palumbo, M. Dimopoulos, J. San Miguel, J. L. Harousseau, M. Attal, M. Hussein, S. Knop, H. Ludwig, M. von Lilienfeld-Toal and P. Sonneveld, Blood Rev., 2009, 23, 87–89 CrossRef CAS PubMed.
- S. V. Rajkumar, S. Jacobus, N. S. Callander, R. Fonseca, D. H. Vesole, M. E. Williams, R. Abonour, D. S. Siegel, M. Katz and P. R. Greipp, Lancet Oncol., 2010, 11, 29–37 CrossRef CAS PubMed.
- K. Oehrlein, C. Langer, I. Sturm, W. Pönisch, C. Hahn-Ast, S. Kuhn and K. C. Weisel, Clin. Lymphoma, Myeloma Leuk., 2012, 3, 191–196 CrossRef PubMed.
- M. A. Dimopoulos, E. Terpos, H. Goldschmidt, A. Alegre, T. Mark and R. Niesvizky, Cancer Treat. Rev., 2012, 38, 1012–1019 CrossRef CAS PubMed.
- D. Liu, A. Ahmet, L. Ward, P. Krishnamoorthy, E. D. Mandelcorn, R. Leigh, J. P. Brown, A. Cohen and H. Kim, Allergy, Asthma, Clin. Immunol., 2013, 9, 30–55 CrossRef PubMed.
- X. Wang, F. Qin, L. Jing, Q. Zhu, F. Li and Z. Xiong, Biomed. Chromatogr., 2013, 27, 371–376 CAS.
- I. Rapado, J. J. Lahuerta, L. Montejano, M. Á. Montalbán, L. Pares and J. Martínez-López, Mod. Chemother., 2012, 1, 11–13 CrossRef.
- N. Y. Khalil, I. A. Darwish, T. A. Wani and A. A. Al-Majed, Chem. Cent. J., 2013, 7, 52 CrossRef PubMed.
- K. E. Warren, S. Goldman, I. F. Pollack, J. Fangusaro, P. Schaiquevich, C. F. Stewart, D. Wallace, S. M. Blaney, R. Packer, T. MacDonald, R. Jakacki, J. M. Boyett and L. E. Kun, J. Clin. Oncol., 2010, 29, 324–329 CrossRef PubMed.
- T. M. Tohnya, K. Hwang, E. R. Lepper, H. A. Fine, W. L. Dahut, J. Venitz, A. Sparreboom and W. D. Figg, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 811, 135–141 CrossRef CAS.
- M. S. Hasnain, S. Rao, M. K. Singh, N. Vig, A. Gupta, A. Ansari, P. Sen, P. Joshib and S. A. Ansari, Analyst, 2013, 138, 1581–1588 RSC.
- S. Veeraraghavan, S. Thappali, S. Viswanadha, S. Nalla, S. Chennupati, M. Golla, S. Vakkalanka and M. Rangasamy, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 949–950, 63–69 CrossRef CAS PubMed.
- N. Chen, L. Wen, H. Lau, S. Surapaneni and G. Kumar, Cancer Chemother. Pharmacol., 2012, 69, 789–797 CrossRef CAS PubMed.
- Q. Liu, K. L. Farley, A. J. Johnson, N. Muthusamy, C. C. Hofmeister, K. A. Blum, L. J. Schaaf, M. R. Grever, J. C. Byrd, J. T. Dalton and M. A. Phelps, Ther. Drug Monit., 2008, 5, 620–627 CrossRef PubMed.
- M. Iqbal, T. A. Wani, N. Y. Khalil and I. A. Darwish, Chem. Cent. J., 2013, 7, 7 CrossRef PubMed.
- C. Queckenberg, B. Wachall, V. Erlinghagen, P. Di Gion, D. Tomalik-Scharte, M. Tawab, K. Gerbeth and U. Fuhr, Clin. Ther., 2011, 33, 1831–1841 CrossRef CAS PubMed.
- I. Panderi, A. Gerakis, V. Zonaras, L. Athanasiou and M. Kazanis, Anal. Chim. Acta, 2004, 504, 299–306 CrossRef CAS.
- G. Damonte, A. Salis, L. Rossi, M. Magnani and U. Benatti, J. Pharm. Biomed. Anal., 2007, 43, 376–380 CrossRef CAS PubMed.
- Y. Yang, H. Li, K. Gao, M. Liu, Y. Sun, T. Yan, J. P. Fawcett, Y. Cui and J. Gua, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 862, 119–124 CrossRef CAS PubMed.
- M. Zhang, G. A. Moore, B. P. Jensen, E. J. Begg and P. A. Bird, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 17–24 CrossRef CAS PubMed.
- A. Watteyn, H. Wyns, E. Plessers, E. Russo, S. de Baere, P. de Backer and S. Croubels, Vet. J., 2013, 195, 216–220 CrossRef CAS PubMed.
- M. E. Touber, M. C. van Engelen, C. Georgakopoulus, J. A. van Rhijn and M. W. F. Nielen, Anal. Chim. Acta, 2007, 586, 137–146 CrossRef CAS PubMed.
- B. C. McWhinney, S. E. Briscoe, J. P. J. Ungerer and C. J. Pretorius, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2863–2869 CrossRef CAS PubMed.
- J. A. Zonder, J. Crowley and M. A. Hussein, et al., ASH Annual Meeting Abstracts, 2007, 110, A77 Search PubMed.
- X. Wang, F. Qin, L. Jing, Q. Zhu, F. Li and Z. Xiong, Biomed. Chromatogr., 2013, 27, 371–376 CAS.
- P. Armitage and G. Berry, Statistical Methods in Medical Research, Blackwell, Oxford, UK, 3rd edn, 1994, p. 283 Search PubMed.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Prentice Hall, Harlow, UK, 4th edn, 2000, pp. 111–118 Search PubMed.
- N. Li, Y. Deng, D. Wang, Y. Qiao and F. Li, Talanta, 2013, 104, 109–115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22339c |
| This journal is © The Royal Society of Chemistry 2015 |