DOI:
10.1039/C5RA22310E
(Paper)
RSC Adv., 2015,
5, 101193-101199
Smallest deltahedra silicon dicarbide: C2Si32−†
Received
24th October 2015
, Accepted 15th November 2015
First published on 17th November 2015
Abstract
The composition and valence electrons of molecules usually have a great impact on the eventual topology. With a high tendency to form sp/sp2-hybridized multiple bonding of C2, the main-group dicarbides (C2Xn) usually adopt a non-closo (or open) topology. By contrast, naked Zintl ions (e.g., Si52−) usually feature deltahedral structures. In this paper, we report an unexpected example of a pentatomic carbon-silicon cluster C2Si32− which has the global minimum C2Si32−-01 featuring a closo-structure with all deltahedras. The global minimum nature of C2Si32−-01 was confirmed by various sophisticated methods including G3B3, G4, CBS-QB3 and W1BD as well as CCSD(T) extrapolated up to the complete basis set limit based on the CCSD(T)/aug-cc-pVTZ, CCSD(T)/aug-cc-pVQZ and CCSD(T)/aug-cc-pV5Z calculations. The AdNDP analysis revealed that C2Si32−-01 possesses a lone pair of electrons on each of the three silicon atoms, four 2c–2e bonds and four 3c–2e bonds, among which the two 2c–2e bonds between the two carbons indicate the existence of a multiply bonded C2 (1.320 Å) that carries the most negative charges. With a total of 22 valence electrons, C2Si32−-01 formally resembles the known Wade–Mingos clusters with (n + 1) polyhedral skeletal electron pairs (PSEPs). Replacement of Si2 by the highly electron-withdrawing C2 does not break the deltahedral topology of the Zintl ion Si52−. To the best of our knowledge, C2Si32− represents the smallest deltahedral main-group dicarbide and also the first deltahedral main-group dicarbide with (n + 1) PSEPs. To direct its organometallic applications, we designed the hetero-deckered sandwich compounds CpMg(C2Si32−)MgCp, in which the C2Si32−-01 unit can be nicely maintained.
1. Introduction
In general, the topological structure of a chemical compound depends on its inherent elemental composition and electronic structure. Various conceptual rules have been developed to effectively connect the topology and electron number.1 Among them is the famous Wade–Mingos rule, which is usually applied to judge the structures of boranes and carboranes by the skeletal electron pairs, i.e., the n-vertex clusters with (n + 1) polyhedral skeletal electron pairs (PSEPs) prefer to be arranged into closo-deltahedra.1a,b On the other hand, the Zintl ions composed of heavier main-group or transition metal elements feature deltahderal structures, and their topology can in most cases be accounted for by the Wade–Mingos rule.2
The ligand-free dicarbides (C2Xn) usually contain a multiply bonded C2 unit. The simplest tri-atomic dicarbides, i.e., C2X, have been the earlier focus of numerous studies.3–6 They have played important roles in various fields. For example, the well-known C2Ca can not only be used as the basic material to synthesize the organic compounds, but also be applied to the nonferrous metals and steel industry.4 The actinide dicarbides as the appropriate fuels of nuclear reactors, have also received much attention.6 Besides, some of them have been detected in space.3
Concerning the topological structures, the simplest C2X usually take the chainlike or T-shaped ground states.3,5,6 With more than one X-atom, the structure and bonding within the dicarbide are expected to be complicated since the C–C, C–X and X–X bonds may undergo interactive competition. It has now been found that some transition metals (e.g., X = Co,7a Fe,7b Ni (ref. 7b)) can effectively allow the multiply bonded C2 to be positioned as one edge of a closo-structure, forming the deltahedral topology. However, in spite of a large body of published research articles on the main-group dicarbides,8,9 only the hexatomic cluster C2Si4 has been firmly shown to have a deltahedral structure.8 The rather limited examples of the main-group dicarbides must be ascribed to the strong preference of the C2-moiety to drive the whole or part of the molecule to be open in chainlike or planar forms.
Can other ligand-free main-group dicarbides possess a deltahedral structure besides C2Si4? In this paper, based on a global isomeric search strategy and various sophisticated quantum chemical methods including G3B3, G4, CBS-QB3, W1BD and CCSD(T)/CBS, we report the smallest main-group dicarbide molecule C2Si32− with all deltahedras. With 22 valence electrons, the pentatomic C2Si32− formally has (n + 1) PSEPs, differing from C2Si4 with (n) PSEPs. Using C2Si32− as a decker, we designed novel hetero-deckered sandwich compounds.
2. Theoretical methods
In order to explore the global structure of C2Si32−, we introduced a locally developed grid-based comprehensive isomeric search strategy for both the singlet and triplet systems.10 For each system, a three-dimensional cubic boxes consisting of the uniformly distributed 125 candidate sites were constructed. The two C and three Si-atoms were placed into these candidate sites to get diversified structural forms. Eventually, a total of 138 effective grid-based structures were generated as the input for B3LYP11/6-31G(d)12 optimizations followed by the harmonic vibrational calculations. All the B3LYP/6-31G(d) local minimum structures without imaginary frequencies were further subject to the B3LYP/aug-cc-pVTZ13 re-optimization followed by the frequency calculations. The single-point energies were performed at the CCSD(T)14/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ level with inclusion of the zero-point vibrational energy (ZPVE) correction from the B3LYP/aug-cc-pVTZ calculations. For the key isomers of C2Si32−, we applied the composite G3B3,15 G4,16 CBS-QB3 (ref. 17) and W1BD18 calculations that have been developed for reliable “model chemistry”. In addition, the CCSD(T) calculations with extrapolation up to the complete basis set limit (denote as CCSD(T)/CBS19) were carried out based on the aug-cc-pVTZ, aug-cc-pVQZ13 and aug-cc-pV5Z13c,d-CCSD(T) single-point energies. The natural charge populations and Wiberg bond index were obtained at the B3LYP/aug-cc-pVTZ level using the natural bond orbital (NBO) analysis. Chemical bonding partition of global minimum structure was performed utilizing the Adaptive Natural Density Partitioning (AdNDP) method.20 For the hetero-deckered sandwich compound, we used the PBE0 (ref. 21)/cc-pVTZ//B3LYP/6-31G(d) to calculate the single-point energies. The calculation in tetrahydrofuran (THF) solution were carried out using the SMD solvent model of Gaussian09.22 All calculations were performed with the Gaussian03 (ref. 23) and Gaussian09 (ref. 24) program packages.
3. Results and discussions
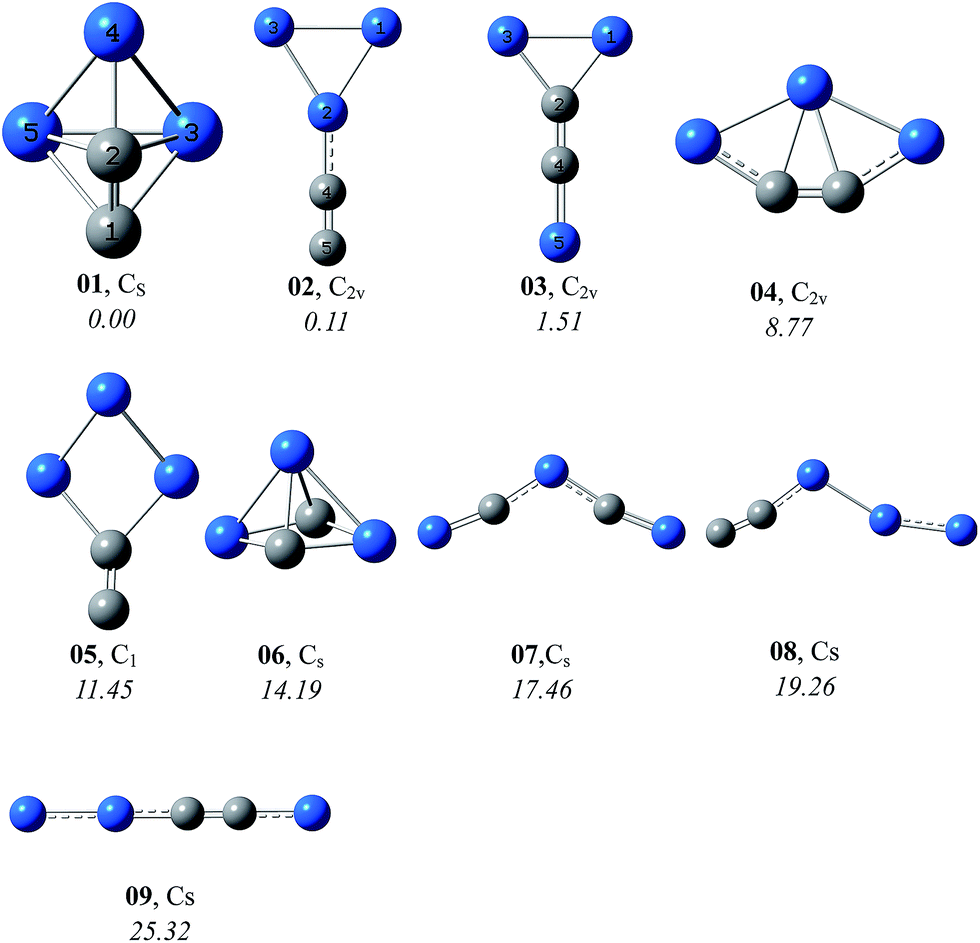
By means of the grid isomeric search, a total of 20 for singlet and 36 for triplet C2Si32− were obtained at the B3LYP/aug-cc-pVTZ level. For simplicity and for easier discussion, we only presented the selected singlet (in Fig. 1) and triplet (in Fig. 2) isomers within 30 kcal mol−1. Note that the total energy of the closo isomer 01 was set to be 0.00 kcal mol−1 as the energy reference. In general, the singlet isomers are more stable than the triplet ones. The lowest-energy triplet isomer 301 is higher than the lowest-energy singlet isomer by 15.04 kcal mol−1, and is the seventh in the energy order of all the singlet and triplet isomers. So in the following context we only discuss the properties of singlet isomers. The information of all the isomers can be found in ESI† (SI).
 |
| | Fig. 1 Representative singlet isomers (ΔE < 30 kcal mol−1) of C2Si32− with their symmetry and relative energies (kcal mol−1) at CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ+ZPVE level. The gray and blue balls are used for carbon and silicon atoms, respectively. | |
 |
| | Fig. 2 Representative triplet isomers (ΔE < 30 kcal mol−1) of C2Si32− with their symmetry and relative energies (kcal mol−1) at CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ+ZPVE level. The total energy of the singlet isomer 01 was set to be 0.00 kcal mol−1 as the energy reference. The left superscript number “3” denotes the triplet state. The gray and blue balls are used for carbon and silicon atoms, respectively. | |
3.1 Structural properties
At the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ+ZPVE level, the lowest-energy isomer 01 has a closo-structure, which is a result of the vertical attack of the C2-moiety to the Si3 ring, forming six deltahedras. Interestingly, the two carbon atoms occupy different sites, one is equatorial whereas the other is apex. In topology, 01 is quite analogous to the isoelectronic triagonal bipyramidal cluster Si52−, which was synthesized in form of the (Rb-crypt)2Si5·4NH3 crystal.25 The equatorial Si–Si (2.503 Å) and axial Si–Si (2.393 Å) bond lengths of C2Si32−-01 are in accordance with the experimental values 2.535 and 2.350 Å of Si52−,25 respectively. Additionally, the C–C distance of 01, i.e., 1.320 Å, lies around the experimental value of the conventional C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (1.34 Å)26 and is supported by the large Wiberg bond index (WBI) of C–C (1.938). In Table 1, the natural charge population analysis indicates that the negative charge of C2Si32− is mainly distributed on C2 (−1.589e) with minor on Si3 (−0.411e).
C double bond (1.34 Å)26 and is supported by the large Wiberg bond index (WBI) of C–C (1.938). In Table 1, the natural charge population analysis indicates that the negative charge of C2Si32− is mainly distributed on C2 (−1.589e) with minor on Si3 (−0.411e).
Table 1 The natural charge (e) of the three lowest-energy isomers of C2Si32− at the B3LYP/aug-cc-pVTZ level
| |
1 |
2 |
3 |
4 |
5 |
| 01 |
−0.639 |
−0.950 |
−0.127 |
−0.156 |
−0.127 |
| 02 |
−0.527 |
0.345 |
−0.527 |
−0.974 |
−0.316 |
| 03 |
−0.073 |
−0.991 |
−0.073 |
−0.803 |
−0.060 |
The species 02 and 03 in C2v symmetry both exhibit a planar structure. The C2 moiety connects terminally with the Si3 ring in 02, while it is inserted into the Si3 ring in 03. Alternatively, 03 can be viewed as a result of the SiC-terminal connection with the CSi2 ring. The C–C bond length of 02 is very short (1.252 Å) with the WBI value 2.725, indicative of the triple bonding. In 03, the internal C–C bond distance is 1.340 Å (WBI is 1.608), consistent with a double bond. The natural charge population analysis of 02 and 03 are similar to that of 01, i.e., the negative charge is mostly localized on C2. The almost doubly bonded C2 unit in 03 carries more charge (−1.794e) than the triply bonded one in 02 (−1.290e).
3.2 Energetics
As shown in Fig. 1, the relative energies of the former three isomers 01, 02 and 03 are close within 2 kcal mol−1, i.e., 0.00, 0.11 and 1.51 kcal mol−1, respectively at the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ+ZPVE level. 01 and 02 are almost isoenergetic. It's thus quite desirable to study their energetic competition at more sophisticated levels (see Table 2). First, we carried out the composite G3B3, G4, CBS-QB3 and W1BD calculations, which have been well defined for the purpose of accurately predicting the thermochemical properties of chemical systems. All the four composite methods showed the lowest-energy nature of 01. Interestingly, the energetic competition between the two planar isomers 02 and 03 are not consistent at different levels. G3B3 and G4 place 03 to be more stable than 02, whereas CBS-QB3 and W1BD show 02 to be more stable than 03. Moreover, the relative energy of 02 with reference to 01 at the G3B3, G4 and CBS-QB3 levels are higher than that at W1BD by more than 2 kcal mol−1. The W1BD method was designed to be computationally much more expensive and more accurate than the CBS-QB3 and G3 methods. It should be noted that the above four composite methods comprising a set of optimal parameters have been largely tested on diversified chemical systems. Alternatively, the CCSD(T) method extrapolated to the complete basis set (namely CCSD(T)/CBS) is also an easy-to-use and a powerful tool for predicting energetics of chemical systems especially in the case where the exotic structures that have not been included in the test of composite methods or the desired basis sets are not involved in the composite methods. Using the aug-cc-pVTZ, aug-cc-pVQZ and aug-cc-pV5Z-CCSD(T) calculations based on the B3LYP/aug-cc-pVTZ geometries, we derived the CCSD(T)/CBS relative energies of 02 and 03 as 0.49 and 2.95 kcal mol−1, respectively above 01. It's of interest to note that the CCSD(T)/CBS results are close to the W1BD values. We thus concluded that the global minimum of C2Si32− is a closo-structure 01 with all deltahedras.
Table 2 Relative energies (kcal mol−1) of the three lowest-energy isomers of C2Si32− at different levels
| |
01 |
02 |
03 |
| G3B3 |
0.00 |
2.77 |
1.02 |
| G4 |
0.00 |
3.73 |
1.78 |
| CBS-QB3 |
0.00 |
3.64 |
4.14 |
| W1BD |
0.00 |
0.69 |
2.17 |
| CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ+ZPVE |
0.00 |
0.11 |
1.51 |
| CCSD(T)/aug-cc-pVQZ//B3LYP/aug-cc-pVTZ+ZPVE |
0.00 |
0.26 |
1.96 |
| CCSD(T)/aug-cc-pV5Z//B3LYP/aug-cc-pVTZ+ZPVE |
0.00 |
0.38 |
2.44 |
| CCSD(T)_CBS |
0.00 |
0.49 |
2.95 |
In consideration of the reliability of the B3LYP functional used in the electronically complex systems, we utilized the other two functionals (M06-2X27 and PBE0) to assess the computed structural parameters of the key isomers. The results indicated that the M06-2X and PBE0 functionals produce essentially the same structure for the former three low-lying isomers with only minor difference in bond lengths (see Table 3).
Table 3 The bond lengths (Å) of the 01, 02 and 03 at the B3LYP/aug-cc-pVTZ, M06-2X/aug-cc-pVTZ and PBE0/aug-cc-pVTZ
| 01 |
C1–C2 |
C1–Si3 |
C1–Si5 |
C2–Si3 |
C2–Si4 |
C2–Si5 |
Si3–Si4 |
Si3–Si5 |
Si4–Si5 |
| B3LYP |
1.320 |
2.176 |
2.176 |
2.143 |
1.842 |
2.143 |
2.503 |
2.393 |
2.503 |
| M06-2X |
1.318 |
2.117 |
2.117 |
2.165 |
1.843 |
2.165 |
2.467 |
2.368 |
2.467 |
| PBE0 |
1.331 |
2.127 |
2.127 |
2.113 |
1.834 |
2.113 |
2.471 |
2.370 |
2.471 |
| 02 |
Si1–Si2 |
Si1–C3 |
Si2–C3 |
C3–C4 |
C4–Si5 |
| B3LYP |
2.286 |
1.876 |
1.876 |
1.340 |
1.703 |
| M06-2X |
2.275 |
1.859 |
1.859 |
1.351 |
1.689 |
| PBE0 |
2.272 |
1.866 |
1.866 |
1.344 |
1.696 |
| 03 |
Si1–Si2 |
Si1–Si3 |
Si2–Si3 |
Si3–C4 |
C4–C5 |
| B3LYP |
2.405 |
2.251 |
2.251 |
1.792 |
1.252 |
| M06-2X |
2.376 |
2.234 |
2.234 |
1.796 |
1.248 |
| PBE0 |
2.385 |
2.242 |
2.242 |
1.789 |
1.252 |
3.3 Bonding characteristics
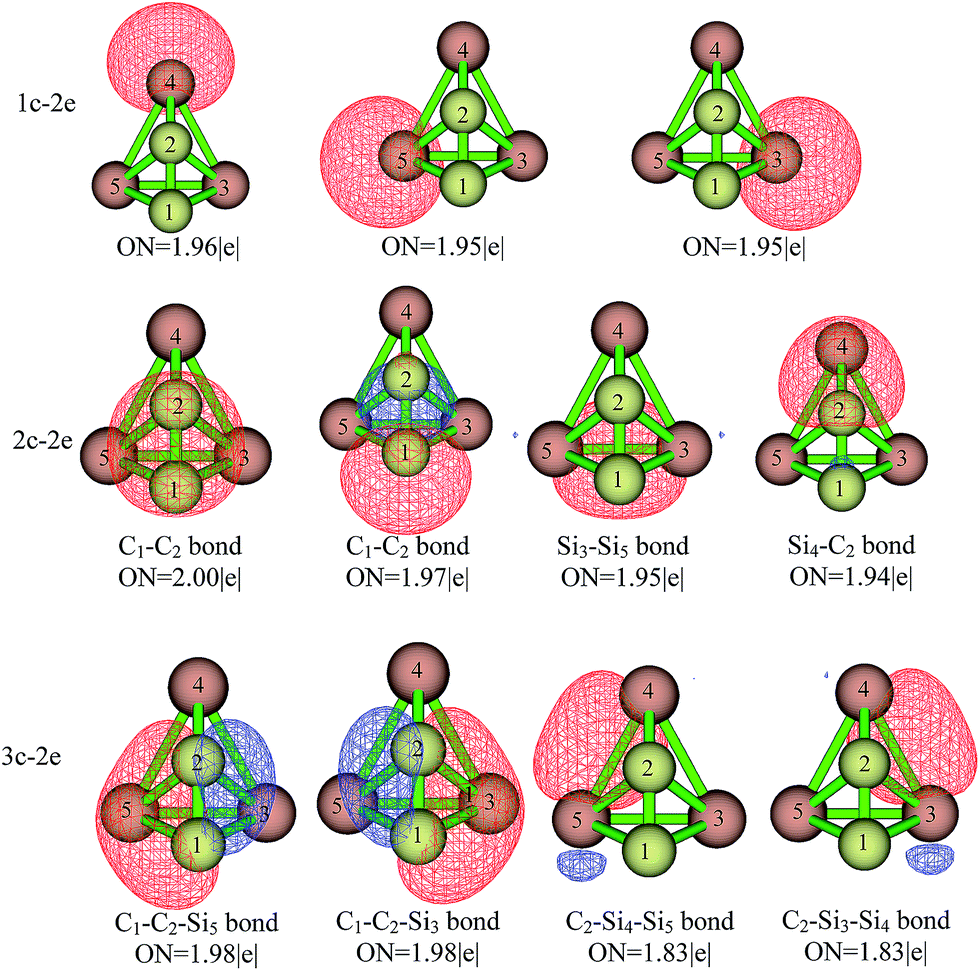
To explore in detail the bonding situation within isomer C2Si32−-01, we applied the AdNDP analysis.20 The deduced localized molecular orbitals of 01 were given in Fig. 3. In 01, there are three silicon lone pairs, four 2c–2e, bonds and four 3c–2e bonds. The occupation numbers (ON) of the silicon lone pairs in equator and apex are 1.96|e| and 1.95|e|, respectively. For the 2c–2e bonds, the ON (2.00|e| and 1.97|e|) of two C1–C2 bonds indicate that 01 retains the multiple bonding within C2. The other two 2c–2e bonds are Si3–Si5 (ON = 1.95|e|) and C2–Si4 (ON = 1.94|e|). In addition, 01 possesses four 3c–2e bonds, i.e., C1–C2–Si3, C1–C2–Si5, C2–Si3–Si4 and C2–Si4–Si5. The former C1–C2–Si3, C1–C2–Si5 have higher occupancy (ON = 1.98|e|) compared to the latter two identical 3c–2e bonds (ON = 1.83|e|). The existence of four 3c–2e bonds within C2Si32−-01 is reminiscent of the well-known boranes and carboranes that are electron deficient and typically characterized by the 3c–2e bonding.1a–c
 |
| | Fig. 3 The localized molecular orbitals of C2Si32-01 obtained by AdNDP analysis. The ON denotes the occupation numbers on the localized orbital. | |
3.4 Sandwich compounds based on C2Si32−
The dianionic C2Si32− belongs to the family of charged ligand-free clusters, which can play important roles in the organometallic or cluster-assembled chemistry.28 They can effectively take part in the “sandwiching” interaction with metal ions together with the counter deckers, forming novel sandwich compounds. And vice versa, during the sandwiching, the charged ligand-free clusters can be stabilized electronically (by removal of the Coulomb repulsion) and sterically (by crowded substituents) by the metal ions and counter deckers.
In the sandwiching stabilization of C2Si32−, either C2Si32− itself or other stable anionic clusters can be the counter decker, resulting in the “homo-deckered” or “hetero-deckered” sandwich compounds, respectively. To avoid the cluster fusion between nonstoichiometric deckers, the hetero-deckered sandwich strategy is a more favorable choice by introducing a stable decker such as cyclopentadienyl anion (Cp−).29 Nowadays, Cp− is one of the versatile ligand in organometallic chemistry, and has been considered as the most commonly used reagent for the synthesis of many metallocenes that are important in a wide range of applications.30
As an example, we studied CpM(C2Si32−)MCp with M being the alkali-earth Mg. To reduce the computational cost, the PBE0 method was chosen for sandwich-like compounds since the relative energies of C2Si32− at PBE0/cc-pVTZ//B3LYP/aug-cc-pVTZ+ZPVE are generally in accordance with the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ+ZPVE values as listed in ESI† (SI). Fig. 4 shows the representative isomers obtained at the PBE0/cc-pVTZ//B3LYP/6-31G(d)+ZPVE level. Others isomers can be found in ESI† (SI). The hetero-deckered sandwich compound of Mg-1 (closo-C2Si32−) is more stable than the second low-lying isomer Mg-2 (open-C2Si32−) by 16.70 kcal mol−1. From the structural parameters listed in Table 4, we can see that the six bond distances of the closo-C2Si32− vary little before and after the hetero-deckered sandwiching. Thus, the dianionic closo-01 can be nicely retained within the sandwich compounds.
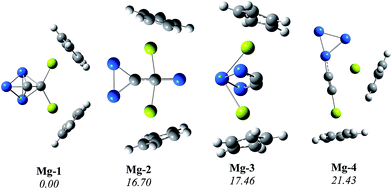 |
| | Fig. 4 Representative isomers of CpMg(C2Si32−)MgCp with their relative energies (kcal mol−1) at the PBE0/cc-pVTZ//B3LYP/6-31G(d)+ZPVE level. The gray, blue, white and yellow balls are used for carbon, silicon, hydrogen and magnesium, respectively. | |
Table 4 The bond lengths (Å) of the closo-C2Si32− in the 01 and Mg-1
| |
C1–C2 |
C1–Si3 |
C1–Si5 |
C2–Si3 |
C2–Si4 |
C2–Si5 |
Si3–Si4 |
Si3–Si5 |
Si4–Si5 |
| 01 |
1.320 |
2.176 |
2.176 |
2.143 |
1.842 |
2.143 |
2.503 |
2.393 |
2.503 |
| Mg-1 |
1.355 |
2.200 |
2.200 |
2.150 |
1.771 |
2.150 |
2.577 |
2.316 |
2.577 |
To investigate whether the designed sandwich compound Mg-1 is stable or not against fragmentation, we studied various dissociation pathways as follows:
| | |
CpMg(C2Si3)2−MgCp → CpMgCCMgCp + Si3
| (1) |
| | |
CpMg(C2Si3)2−MgCp → CpMgMgCp + C2Si3
| (2) |
| | |
CpMg(C2Si3)2−MgCp → CpMgCp + C2Si3Mg
| (3) |
The easiest pathway is (3) with the dissociation energy of 57.25 kcal mol−1. The pathways (1) and (2) need to absorb much more energy. The corresponding endothermicities are 80.41 and 88.23 kcal mol−1. Clearly, due to the very high dissociation energies, the hetero-deckered sandwich compounds CpMg(C2Si32−)MgCp are quite stable against fragmentation.
Due to the exposure of the highly electron-withdrawing C2 moiety within CpMg(C2Si32−)MgCp, the solvent effect might have an influence on the isomeric stability. The tetrahydrofuran (THF) solution was chosen for our study. The relative energies of Mg-1, Mg-2, Mg-3 and Mg-4 in gas phase are 0.00, 16.70, 17.46 and 21.43 kcal mol−1, respectively. Their corresponding energies in the THF solution are 0.00, 20.36, 24.54 and 16.20 kcal mol−1. Although the relative energy order of the latter three isomers in solution is different from that in gas phase, the global minimum structure of CpMg(C2Si32−)MgCp in both phases remains to be Mg-1 with closo C2Si32−.
3.5 Implications
Besides the easy accessibility for spectroscopic study, small-sized clusters are relevant to the growth or assembly into larger systems. The p-block first-row elements (e.g., B, C, N) are prone to form strongly π-bonded structure. Thus, when incorporated into the heavier main-group clusters, they have strong trend to destabilize the formation of a closo or deltahedral structure, especially in small clusters.31 Understandably, the structure of C2Xn is usually fully or partially open instead of being closo, resulting in the formation of the multiply bonded C2. Thus, our computational finding at various sophisticated levels that the global structure of the small C2Si32− features a closo polyhedra is quite unexpected.
In sharp contrast, the deltahedra is known to be the characteristics of the Zintl ions. Deltahedral Zintl ions composed of the group 14 elements have been known for more than 100 years.32 Many such compounds have been prepared including the smallest E52− (E = Si, Ge, Sn, Pb).33 The bonding of these ligand-free clusters cannot be accounted for by classical octet rule, i.e., 2c–2e bond. Instead, they feature the delocalized 3c–2e bonds and their topologies can usually be explained in terms of the Wade–Mingos rule as for boranes and carboranes. Clearly, replacement of Si2 moiety within Si52− by the highly electron-withdrawing C2 does not break the closo topology. To our best knowledge, C2Si32− is the smallest main-group dicarbide. The carried two additional electrons render C2Si32− to be applicable in organometallic chemistry, e.g., to form sandwich compounds. Also, with the multiply bonded C2, the C2Si32− compounds are expected to have particular applications such as in catalysis and cycloaddition reactions.
4. Conclusions
In this paper, we reported the smallest silicon dicarbide with all deltahedras, which is also the first main-group dicarbide with (n + 1) polyhedral skeletal electron pairs (PSEPs). In addition, to counteract the coulomb repulsion within the dianion C2Si32−, we introduced the Cp− to design the hetero-deckered sandwich compounds, in which the closo unit 01 can be nicely kept. Due to the strong preference of the C2-moiety in making the whole or partial molecule be open, the main-group dicarbides with no ligands usually adopt a non-closo topology. The presently disclosed closo-C2Si32− really represents an exception, and its sandwich-like compounds warmly welcome future laboratory studies.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 21273093, 21473069, 21073074). The authors are very thankful for the reviewers' helpful comments and suggestions.
References
-
(a) K. Wade, Chem. Commun., 1971, 792 RSC;
(b) D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311 CrossRef CAS;
(c) A. C. Tang and Q. S. Li, Int. J. Quantum Chem., 1986, 29, 579 CrossRef CAS;
(d) P. V. R. Schleyer and A. I. Boldyrev, J. Chem. Soc., Chem. Commun., 1991, 22, 1536 RSC.
-
(a) S. C. Sevov and J. M. Goicoechea, Organometallics, 2006, 25, 5678 CrossRef CAS;
(b) R. S. P. Turbervill and J. M. Goicoechea, Chem. Rev., 2014, 114, 10807 CrossRef CAS PubMed.
-
(a) K. Tucker, M. Kutner and P. Thaddeus, Astrophys. J., 1974, 193, L115 CrossRef CAS;
(b) A. Largo and C. Barrientos, Chem. Phys., 1989, 138, 291 CrossRef CAS;
(c) J. El-Yazal, J. M. L. Martin and J.-P. François, J. Phys. Chem. A, 1997, 101, 8319 CrossRef CAS;
(d) D. Halfen, D. Clouthier and L. M. Ziurys, Astrophys. J., Lett., 2008, 677, L101 CrossRef CAS;
(e) A. Largo, C. Barrientos, X. Lopez and J. M. Ugalde, J. Phys. Chem., 1994, 98, 3985 CrossRef CAS;
(f) F. X. Sunahori, J. Wei and D. J. Clouthier, J. Am. Chem. Soc., 2007, 129, 9600 CrossRef CAS PubMed;
(g) S. Saito, K. Kawaguchi, S. Yamamoto, M. Ohishi, H. Suzuki and N. Kaifu, Astrophys. J., 1987, 317, L115 CrossRef CAS;
(h) A. J. Schoeffler, H. Kohguchi, K. Hoshina, Y. Ohshima and Y. Endo, J. Chem. Phys., 2001, 114, 6142 CrossRef CAS;
(i) J. Cernicharo, C. Kahane, J. Gomez-Gonzalez and M. Guélin, Astron. Astrophys., 1986, 167, L9 CAS;
(j) J. Flores and A. Largo, Chem. Phys., 1990, 140, 19 CrossRef CAS;
(k) D. Michalopoulos, M. Geusic, P. Langridge-Smith and R. Smalley, J. Chem. Phys., 1984, 80, 3556 CrossRef CAS;
(l) L. Snyder, C. Henkel, J. Hollis and F. Lovas, Astrophys. J., 1985, 290, L29 CrossRef CAS;
(m) P. Thaddeus, S. Cummins and R. Linke, Astrophys. J., 1984, 283, L45 CrossRef CAS.
-
(a) S. Bresler, G. Zakharov and S. Kirillov, Polym. Sci. U.S.S.R., 1962, 3, 832 CrossRef;
(b) H. S. Knight and F. T. Weiss, Anal. Chem., 1962, 34, 749 CrossRef CAS;
(c) J. J. Mu and R. A. Hard, Ind. Eng. Chem. Res., 1987, 26, 2063 CrossRef CAS;
(d) C. Jaturapitakkul and B. Roongreung, J. Mater. Civ. Eng., 2003, 15, 470 CrossRef CAS.
-
(a) A. Largo, P. Redondo and C. Barrientos, J. Am. Chem. Soc., 2004, 126, 14611 CrossRef CAS PubMed;
(b) V. M. Rayón, P. Redondo, C. Barrientos and A. Largo, Chem.–Eur. J., 2006, 12, 6963 CrossRef PubMed;
(c) B. Xiao, J. Feng, J. Chen and L. Yu, Chem. Phys. Lett., 2007, 448, 35 CrossRef CAS;
(d) V. M. Rayón, P. Redondo, C. Barrientos and A. Largo, J. Chem. Phys., 2010, 133, 124306 CrossRef PubMed.
-
(a) S. Imoto and K. Fukuya, J. Less-Common Met., 1986, 121, 55 CrossRef CAS;
(b) M. F. Zalazar, V. C. M. Rayón and A. Largo, J. Phys. Chem. A, 2012, 116, 2972 CrossRef CAS PubMed;
(c) X. Wang, L. Andrews, D. Ma, L. Gagliardi, A. P. Gonçalves, C. C. L. Pereira, J. Marçalo, C. Godart and B. Villeroy, J. Chem. Phys., 2011, 134, 244313 CrossRef PubMed;
(d) X. Wang, L. Andrews, P. Å. Malmqvist, B. O. Roos, A. P. Gonçalves, C. C. L. Pereira, J. Marçalo, C. Godart and B. Villeroy, J. Am. Chem. Soc., 2010, 132, 8484 CrossRef CAS PubMed;
(e) P. Pogány, A. Kovács, Z. Varga, F. M. Bickelhaupt and R. J. M. Konings, J. Phys. Chem. A, 2012, 116, 747 CrossRef PubMed;
(f) I. Shein and A. Ivanovskii, J. Nucl. Mater., 2009, 393, 192 CrossRef CAS;
(g) A. Kovács and R. Konings, J. Nucl. Mater., 2008, 372, 391 CrossRef;
(h) D. D. Jackson, G. W. Barton, O. H. Krikorian and R. S. Newbury, J. Phys. Chem., 1964, 68, 1516 CrossRef CAS;
(i) S. K. Gupta and K. A. Gingerich, J. Chem. Phys., 1980, 72, 2795 CrossRef CAS;
(j) P. Pogány, A. Kovács, D. Szieberth and R. J. Konings, Struct. Chem., 2012, 23, 1281 CrossRef;
(k) N. Sasaki, K. Kubo and M. Asano, J. Nucl. Sci. Technol., 1971, 8, 614 CrossRef;
(l) W. Huang and L. Yang, Can. J. Phys., 2014, 93, 409 CrossRef;
(m) D. Wandner, P. Link, O. Heyer, J. Mydosh, M. A. Ahmida, M. M. Abd-Elmeguid, M. Speldrich, H. Lueken and U. Ruschewitz, Inorg. Chem., 2010, 49, 312 CrossRef CAS PubMed;
(n) S. K. Gupta and K. A. Gingerich, J. Chem. Phys., 1979, 71, 3072 CrossRef CAS.
-
(a) J.-Y. Yuan, H.-G. Xu and W.-J. Zheng, Phys. Chem. Chem. Phys., 2014, 16, 5434 RSC;
(b) B. Zhang, B. Cao, C. Chen, J. Zhang and H. Duan, J. Cluster Sci., 2013, 24, 197 CrossRef CAS.
-
(a) G. Froudakis, A. Zdetsis, M. Mühlhäuser, B. Engels and S. D. Peyerimhoff, J. Chem. Phys., 1994, 101, 6790 CrossRef;
(b) A. Nakajima, T. Taguwa, K. Nakao, M. Gomei, R. Kishi, S. Iwata and K. Kaya, J. Chem. Phys., 1995, 103, 2050 CrossRef CAS;
(c) S. Hunsicker and R. O. Jones, J. Chem. Phys., 1996, 105, 5048 CrossRef CAS;
(d) Z. Y. Jiang, X. H. Xu, H. S. Wu, F. Q. Zhang and Z. H. Jin, Chem. Phys., 2003, 290, 223 CrossRef CAS;
(e) H. Wang, W.-C. Lu, J. Sun, Z.-S. Li and C.-C. Sun, Chem. Phys. Lett., 2006, 423, 87 CrossRef CAS;
(f) J. L. Deng, K. H. Su, X. Wang, Q. F. Zeng, L. F. Cheng, Y. D. Xu and L. T. Zhang, Eur. Phys. J. D, 2008, 49, 21 CrossRef CAS;
(g) M. Savoca, A. Lagutschenkov, J. Langer, D. J. Harding, A. Fielicke and O. Dopfer, J. Phys. Chem. A, 2013, 117, 1158 CrossRef CAS PubMed.
-
(a) M. M. Ghouri, L. Yareeda and D. S. Mainardi, J. Phys. Chem. A, 2007, 111, 13133 CrossRef CAS PubMed;
(b) S. Becker and H. J. Dietze, Int. J. Mass Spectrom. Ion Processes, 1988, 82, 287 CrossRef CAS;
(c) R.-X. Wang, D.-J. Zhang, R.-X. Zhu and C.-B. Liu, Acta Chim. Sin., 2007, 65, 2092 CAS;
(d) Y. Pei and X. C. Zeng, J. Am. Chem. Soc., 2008, 130, 2580 CrossRef CAS PubMed;
(e) C. Wang, W. Cui, J. Shao, X. Zhu and X. Lu, Comput. Theor. Chem., 2013, 1006, 19 CrossRef CAS;
(f) G. Pascoli and H. Lavendy, Int. J. Mass Spectrom., 2001, 206, 153 CrossRef CAS;
(g) M. Chen, L. Zheng, Q. Zhang, M. Liu and C. Au, J. Phys. Chem. A, 2003, 107, 10111 CrossRef CAS;
(h) M. Chen, H. Liang, M. Liu, Q. Zhang and C. Au, Int. J. Mass Spectrom., 2004, 232, 165 CrossRef CAS;
(i) X. Li, L.-S. Wang, N. A. Cannon and A. I. Boldyrev, J. Chem. Phys., 2002, 116, 1330 CrossRef CAS;
(j) F. Y. Naumkin, J. Phys. Chem. A, 2008, 112, 4660 CrossRef CAS PubMed;
(k) Y.-B. Wu, H.-G. Lu, S.-D. Li and Z.-X. Wang, J. Phys. Chem. A, 2009, 113, 3395 CrossRef CAS PubMed;
(l) F. Dong, S. Heinbuch, Y. Xie, J. J. Rocca and E. R. Bernstein, Phys. Chem. Chem. Phys., 2010, 12, 2569 RSC;
(m) B. I. Loukhovitski, A. S. Sharipov and A. M. Starik, J. Phys. Chem. A, 2015, 119, 1369 CrossRef CAS PubMed.
-
(a) C. B. Shao and Y. H. Ding, Grid-Based Comprenhensive Isomeric Search Algorithm, Jilin University, Changchun, China, 2010 Search PubMed;
(b) Z. H. Cui, M. Contreras, Y. H. Ding and G. Merino, J. Am. Chem. Soc., 2011, 133, 13228 CrossRef CAS PubMed;
(c) S. M. Gao and Y. H. Ding, RSC Adv., 2012, 2, 11764 RSC;
(d) X. Y. Tang, Z. H. Cui, C. B. Shao and Y. H. Ding, Int. J. Quantum Chem., 2012, 112, 1299 CrossRef CAS;
(e) S. M. Gao and Y. H. Ding, RSC Adv., 2012, 2, 11764 RSC;
(f) C. Guo, Z. H. Cui and Y. H. Ding, Struct. Chem., 2013, 24, 263 CrossRef CAS;
(g) C. Guo, Z. H. Cui and Y. H. Ding, Int. J. Quantum Chem., 2013, 113, 2213 CrossRef CAS;
(h) C. Guo, C. Wang and Y. H. Ding, Struct. Chem., 2014, 25, 1023 CrossRef CAS;
(i) X. Y. Zhang and Y. H. Ding, Comput. Theor. Chem., 2014, 1048, 18 CrossRef CAS;
(j) X. Y. Zhang and Y. H. Ding, RSC Adv., 2015, 5, 27134 RSC.
-
(a) R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989 Search PubMed;
(b) A. D. Becke, J. Chem. Phys., 1992, 96, 2155 CrossRef CAS;
(c) J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS.
-
(a) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D.J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS;
(b) T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007 CrossRef;
(c) V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223 CrossRef CAS;
(d) V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976 CrossRef CAS.
-
(a) R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS;
(b) D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358 CrossRef CAS;
(c) D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 2975 CrossRef CAS;
(d) N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 064107 CrossRef PubMed.
-
(a) J. Cizek, Adv. Chem. Phys., 1969, 14, 35 CrossRef CAS;
(b) G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS;
(c) G. E. Scuseria, C. L. Janssen and H. F. Schaefer, J. Chem. Phys., 1988, 89, 7382 CrossRef CAS;
(d) G. E. Scuseria and H. F. Schaefer, J. Chem. Phys., 1989, 90, 3700 CrossRef CAS.
- A. G. Baboul, L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 1999, 110, 7650 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
-
(a) J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822 CrossRef CAS;
(b) J. A. Montgomery Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532 CrossRef.
-
(a) J. M. L. Martin and G. de Oliveira, J. Chem. Phys., 1999, 111, 1843 CrossRef CAS;
(b) S. Parthiban and J. M. L. Martin, J. Chem. Phys., 2001, 114, 6014 CrossRef CAS;
(c) E. C. Barnes, G. A. Petersson, J. A. Montgomery Jr, M. J. Frisch and J. M. Martin, J. Chem. Theory Comput., 2009, 5, 2687 CrossRef CAS.
- K. A. Peterson, D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1994, 100, 7410 CrossRef CAS.
-
(a) D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207 RSC;
(b) D. Y. Zubarev and A. I. Boldyrev, J. Org. Chem., 2008, 73, 9251 CrossRef CAS PubMed;
(c) D. Y. Zubarev and A. I. Boldyrev, J. Phys. Chem. A, 2009, 113, 866 CrossRef CAS PubMed.
-
(a) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS;
(b) C. Adamo, M. Cossi and V. Barone, J. Mol. Struct.: THEOCHEM, 1999, 493, 145 CrossRef CAS;
(c) M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
-
(a) J. M. Goicoechea and S. C. Sevov, J. Am. Chem. Soc., 2004, 126, 6860 CrossRef CAS PubMed;
(b) D. Y. Zubarev, A. I. Boldyrev, X. Li, L.-F. Cui and L.-S. Wang, J. Phys. Chem. A, 2005, 109, 11385 CrossRef CAS PubMed.
- J. Dowling and B. P. Stoicheff, Can. J. Phys., 1959, 37, 703 CrossRef CAS.
-
(a) E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 1996 CrossRef CAS;
(b) M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, J. Phys. Chem. A, 2013, 117, 12590 CrossRef CAS PubMed.
-
(a) J. Frunzke, M. Lein and G. Frenking, Organometallics, 2002, 21, 3351 CrossRef CAS;
(b) E. Urne˙žius, W. W. Brennessel, C. J. Cramer, J. E. Ellis and P. v. R. Schleyer, Science, 2002, 295, 832 CrossRef PubMed;
(c) R. Wolf, A. W. Ehlers, M. M. Khusniyarov, F. Hartl, B. de Bruin, G. J. Long, F. Grandjean, F. M. Schappacher, R. Pöttgen and J. C. Slootweg, Chem.–Eur. J., 2010, 16, 14322 CrossRef CAS PubMed;
(d) S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630 CrossRef CAS PubMed.
-
(a) L. M. Yang, Y. H. Ding and C. C. Sun, ChemPhysChem, 2006, 7, 2478 CrossRef CAS PubMed;
(b) L. M. Yang, Y. H. Ding and C. C. Sun, Chem.–Eur. J., 2007, 13, 2546 CrossRef CAS PubMed;
(c) L. M. Yang, J. Wang, Y. H. Ding and C. C. Sun, Organometallics, 2007, 26, 4449 CrossRef CAS;
(d) L. M. Yang, Y. H. Ding, W. Q. Tian and C. C. Sun, Phys. Chem. Chem. Phys., 2007, 9, 5304 RSC;
(e) L. M. Yang, Y. H. Ding and C. C. Sun, J. Am. Chem. Soc., 2007, 129, 658 CrossRef CAS PubMed;
(f) L. M. Yang, Y. H. Ding and C. C. Sun, J. Am. Chem. Soc., 2007, 129, 1900 CrossRef CAS PubMed;
(g) L. M. Yang, Y. H. Ding and C. C. Sun, Theor. Chem. Acc., 2008, 119, 335 CrossRef CAS;
(h) L. M. Yang, J. Wang, Y. H. Ding and C. C. Sun, J. Phys. Chem. A, 2007, 111, 9122 CrossRef CAS PubMed;
(i) L. M. Yang, Y. H. Ding and C. C. Sun, J. Phys. Chem. A, 2007, 111, 10675 CrossRef CAS PubMed.
-
(a) X. Wu and X. C. Zeng, J. Am. Chem. Soc., 2009, 131, 14246 CrossRef CAS PubMed;
(b) D. Senthilnathan, S. Vaideeswaran, P. Venuvanalingam and G. Frenking, J. Mol. Model., 2011, 17, 465 CrossRef CAS PubMed.
-
(a) G. H. Chen, Y. H. Ding, X. R. Huang and C. C. Sun, J. Phys. Chem. A, 2005, 109, 5619 CrossRef CAS PubMed;
(b) G. H. Chen, Y. H. Ding, X. H. Huang, H. X. Zhang, Z. S. Li and C. C. Sun, J. Phys. Chem. A, 2002, 106, 10408 CrossRef CAS;
(c) G. Maier, H. P. Reisenauer and J. Glatthaar, Organometallics, 2000, 19, 4775 CrossRef CAS;
(d) M. Zhou, L. Jiang and Q. Xu, J. Phys. Chem. A, 2004, 108, 9521 CrossRef CAS;
(e) M. J. Maclean, P. C. H. Eichinger, T. Wang, M. Fitzgerald and J. H. Bowie, J. Phys. Chem. A, 2008, 112, 12714 CrossRef CAS PubMed.
-
(a) A. C. R. Joanis, C. R. Hebd. Seances Acad. Sci., 1891, 113, 795 Search PubMed;
(b) E. Zintl and A. Z. Harder, Z. Phys. Chem., Abt. A, 1931, 154, 47 CAS;
(c) E. Zintl and W. Z. Dullenkorf, Z. Phys. Chem., Abt. B, 1932, 16, 183 Search PubMed.
-
(a) J. D. Corbett, Struct. Bonding, 1997, 87, 157 CrossRef CAS;
(b) T. F. Fässler, Coord. Chem. Rev., 2001, 215, 377 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22310e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.