
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using the hydrolysis of anhydrides to control gel properties and homogeneity in pH-triggered gelation†
Emily R. Drapera,
Laura L. E. Mearsa,
Ana M. Castillaa,
Stephen M. Kingb,
Tom O. McDonalda,
Riaz Akhtar*c and
Dave J. Adams*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: d.j.adams@liverpool.ac.uk
bISIS Facility, Rutherford Appleton Laboratory, Science and Technology Facilities Council, Didcot, Oxfordshire OX11 0QX, UK
cCentre for Materials and Structures, School of Engineering, University of Liverpool, Liverpool L69 3GH, UK. E-mail: r.akhtar@liverpool.ac.uk
First published on 29th October 2015
Abstract
The pH of an aqueous solution of a low molecular weight gelator can be adjusted through the hydrolysis of a number of anhydrides to the corresponding acids. The rate of hydrolysis and hence of pH change can be used to control the rate of gel formation. This rate does not affect the primary assembly of the low molecular weight gelator, but does affect the mechanical properties of the resulting gels, as well as the homogeneity and reproducibility of the gels. The mechanical properties are compared by both rheology and dynamic nanoindentation.
Introduction
Low molecular weight gelators (LMWGs) self-assemble in solution to give self-supporting gels.1–3 Gelation is a result of the assembly of the LMWG into one dimensional fibres, which form cross-links by entanglement to give a three dimensional network.4 The properties of the gels are controlled most simplistically by the mechanical properties of the fibres, the number of cross-links, and the distribution of the fibres and cross-links within the sample. However, it is often difficult to understand the relative importance of each of these parameters.To form gels, a trigger has to be applied to the LMWG solution such that the solubility of the LMWG is decreased. This can be done in a number of ways, for example using a heat–cool cycle, adjusting the solvent quality by adding an anti-solvent, or changing a property such as the pH or ionic strength of the solution. The mechanical properties of the final gels can be strongly affected by the process by which gelation is triggered. We recently reviewed this for a small number of LMWGs,5 and it is clear that depending on whether a pH-trigger or a solvent-trigger, for example, is used can lead to gels with very different properties even though the same gelator is used. There are many reports discussing the differences in the properties of gels formed from different gelators.6 However, the importance of the triggering method and process of assembly are aspects that are rarely discussed in the literature.7
Functionalised dipeptides (for example 1, Scheme 1) are an interesting class of LMWG, which can be used to form hydrogels using a range of triggers.8–10 One trigger that is widely used is pH.8,11–19 At high pH, the carboxylic acid at the C-terminus is deprotonated, meaning the dipeptide is often dispersed as colloidal aggregates in solution. On lowering the pH, the carboxylic acid is protonated and hence the solubility of the dipeptide is significantly decreased, resulting in many cases (although not all) in gel formation at suitable concentrations of the dipeptide.
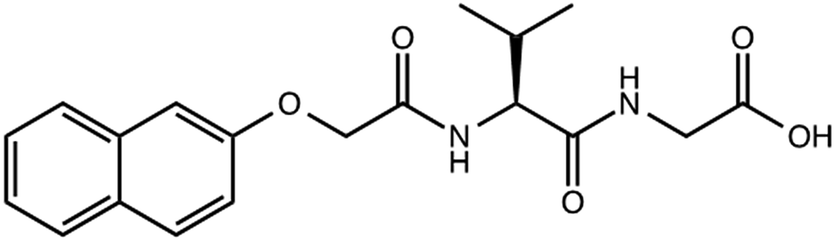 | ||
| Scheme 1 Structure of gelator 1. | ||
The properties of the gels depend on how the pH change is triggered. For example, for gels formed from Fmoc-dipeptides we have previously reported that significantly more homogeneous gels can be formed when the pH is adjusted using the hydrolysis of glucono-δ-lactone (GdL) to gluconic acid (Scheme 2)20 as opposed to using a mineral acid such as hydrochloric acid (HCl).13 GdL hydrolyses slowly, with the mixing time being significantly shorter than the rate of hydrolysis, which means that the pH of the sample decreases uniformly. Using a mineral acid such as HCl can result in local gelation as soon as the drop of solution reaches the sample and before mixing can occur. As such, the shear profile used for mixing is highly important, and can be very difficult to reproduce.13,21 This directly translates into a significant difference in the mechanical properties of the gels formed; GdL triggered gels have significantly higher and more reproducible storage and loss moduli (G′ and G′′ respectively). The hydrolysis of GdL has since been used successfully for a number of pH-triggered low molecular weight gels.13–15,22–28
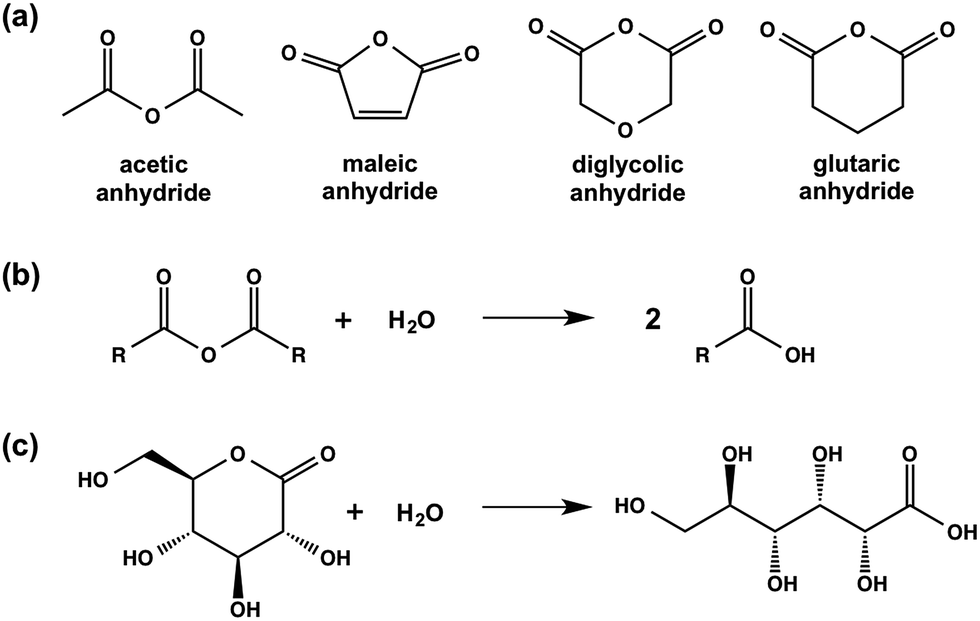 | ||
| Scheme 2 (a) Chemical structures of the anhydrides used in this study; (b) generic hydrolysis of an anhydride to the corresponding carboxylic acid; (c) hydrolysis of GdL to gluconic acid. | ||
In this paper, we focus on the properties of gels formed from LMWG 1 (Scheme 1) via a pH trigger. We discuss the effects of how the pH is adjusted on the self-assembly of 1, the mechanical properties of the resulting gels at the macro- and microscale, and the homogeneity of the gels.
Results and discussion
We have previously reported that LMWG 1 forms gels when the pH of an aqueous solution at a concentration of 1 of 5 mg mL−1 is adjusted from above 10 to below the apparent pKa of 5.0 by adding GdL.14,16 This is typical for this class of LMWG, where assembly begins below the pKa; we have recently shown for a related gelator that the assembled fibres are still charged when a gel is formed.29 For 1, a low viscosity solution is formed at high pH, with a transparent, self-supporting gel being formed at low pH.Here, we investigate the impact of using different methods to adjust the pH on the properties of the resulting gels. First, we utilised a simple addition of HCl. This method results in very quick gelation, with mixing being difficult as described elsewhere.13,21 Second, inspired by the effectiveness of GdL, we expanded on the concept of hydrolysis and screened a series of organic anhydrides able to hydrolyse in water to the carboxylic acids. We describe the use of acetic anhydride, glutaric anhydride, diglycolic anhydride, and maleic anhydride (the chemical structures of these anhydrides are shown in Scheme 2).
Acetic anhydride and glutaric anhydride dissolve well in water. As a result, on addition to a solution of 1, a homogeneous solution is formed, which slowly forms a gel. Maleic anhydride and diglycolic anhydride are significantly less water-soluble. Thus, addition of any of these anhydrides to a solution of 1 results in an inhomogeneous, turbid mixture, which rapidly forms a turbid gel, followed by a degree of clarification.
The rates of pH change can be visualised by the addition of a pH indicator. Here, we used bromophenol blue. Comparative photographs of the solutions are shown in Fig. 1. Immediately after the addition of the trigger (3 molar equivalents of trigger compared to 1), the pH of samples containing HCl, maleic anhydride, and diglycolic anhydride has dropped below 4.6 as indicated by the colour change of the solution to yellow (the pH indicator is blue above a pH of 4.6 and yellow below a pH of 3.0) (Fig. 1b). These solutions also become turbid. The pH has clearly changed for solutions containing acetic anhydride and glutaric anhydride whose colour goes to a lighter blue, although not to the same degree, whilst that of the solution triggered by GdL is almost unchanged.
 | ||
| Fig. 1 Photographs of gels formed from 1 using different triggers in the presence of bromophenol blue (blue above a pH of ∼4.6, and yellow below a pH of ∼3.0). In each case, from left to right the triggers are acetic anhydride, maleic anhydride, glutaric anhydride, diglycolic anhydride, GdL and HCl. (a) shows the solution of 1 with bromophenol blue before addition of trigger; (b) shows the samples immediately after addition of the triggers; (c) after 10 min; (d) after 60 min; (e) after 16 hours. | ||
For solutions containing HCl, maleic anhydride, and diglycolic anhydride, the turbidity decreases and self-supporting gels are formed after 10 minutes (Fig. 1c). Self-supporting gels form between 10 minutes and 1 hour from the samples triggered by acetic anhydride and glutaric anhydride (Fig. 1d); at this time, the sample containing GdL is still above a pH of 7, and no self-supporting gel is formed. After 16 hours, all samples formed self-supporting gels and the pH was below 4.0, as shown by the colour of the samples (Fig. 1e). These observations show that the rates of both pH change and of gel formation are very dependent on the trigger used. Full pH data for analogous samples are shown in Fig. S1, ESI.†
Throughout the discussion below, we routinely use 3 molar equivalents of the anhydride relative to 1. However, we note that it is possible to target different rates of pH change and different final pH values by varying the amount of anhydride added. Data showing the rate of pH change and final pH values for 1 and 5 molar equivalents of each anhydride are shown in Table S1, ESI.† As for GdL, the rate of hydrolysis for each of the anhydrides is temperature dependent,30 meaning that the kinetics of gelation can also be controlled by this parameter (Fig. S3, ESI†). We note that the rheological data collected over time shows that the same assembly steps are occurring when all of the hydrolysable triggers are used,15 although the rates are dependent on trigger and conditions used. The amount of trigger necessary to result in gel formation depends on the amount needed to lower the pH sufficiently. For example, for acetic anhydride, at least 0.2 equivalents are needed, which lowers the pH to around 4.55 (Fig. S4, ESI†).
For a LMWG such as 1, formation of a gel is typically a result of the self-assembly of the gelator into one-dimensional fibrous structures.1,2 We probed the primary assembly of 1 in each case using infrared spectroscopy. In all cases, the IR spectra obtained from the gels were very similar, with peaks at 1630 cm−1, 1640 cm−1 and a minor shoulder 1652 cm−1 in the amide I region (Fig. S5, ESI†). Such peak positions would typically be associated with beta sheets and random coils.31 However, we highlight that interpreting such spectra for short dipeptides with N-protecting groups is difficult using data collected from proteins or longer peptides.32,33 Nonetheless, the similarity in the spectra for gels formed with all triggers implies that the molecular arrangement in the primary self-assembled structures is very similar.
We used electron microscopy and small angle neutron scattering (SANS) to probe the next length scale. Scanning electron microscope (SEM) images showed that, as expected, all gels are the result of an entangled mesh of fibres (Fig. 2). Such images are potentially prone to drying artifacts. Indeed in the cases of the samples triggered by glutaric anhydride and diglycolic anhydride (Fig. 2c and d respectively), non-fibrous structures which we attribute to the formation of aggregates of the corresponding acids can be seen in the images. As such, we utilised SANS to probe the structures formed in situ.
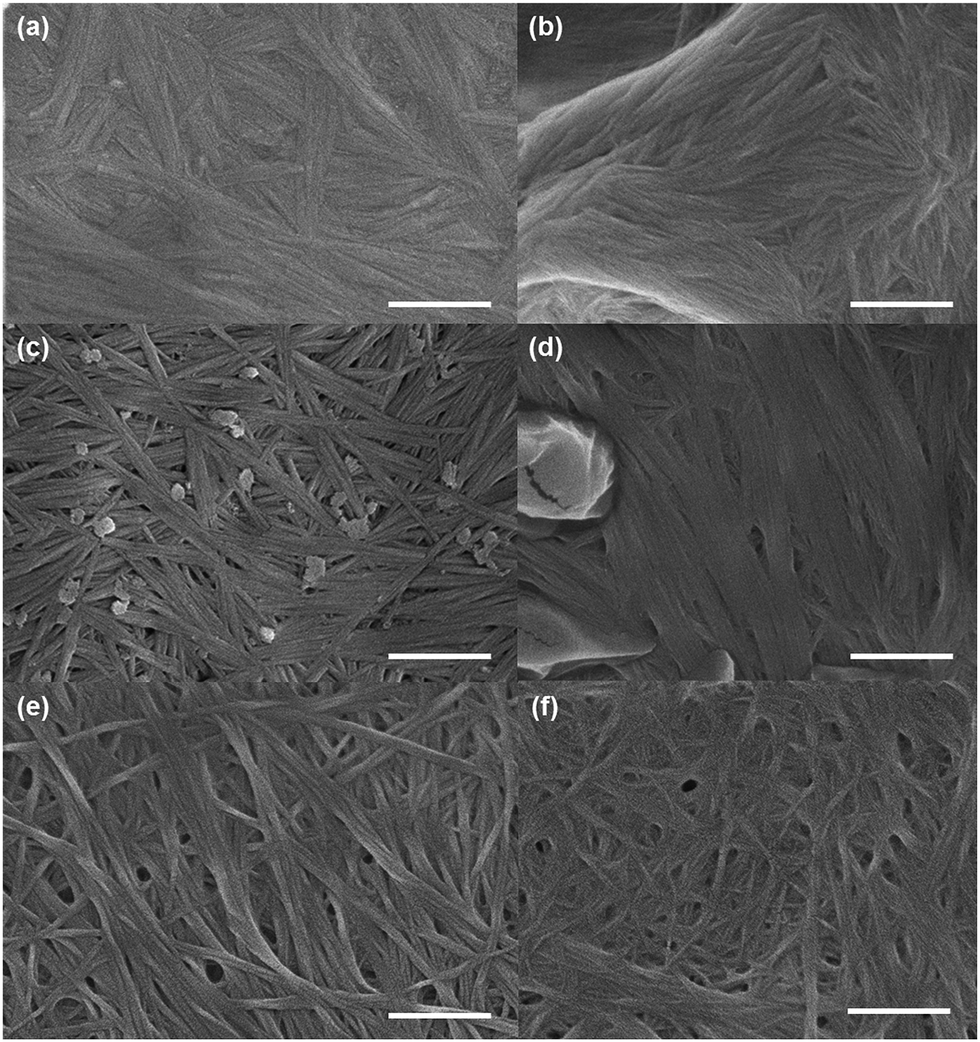 | ||
| Fig. 2 Representative SEM images of gels of 1 triggered using (a) acetic anhydride; (b) maleic anhydride; (c) glutaric anhydride; (d) diglycolic anhydride; (e) GdL; (f) HCl. In all cases, the scale bar represents 500 nm. | ||
SANS can probe structures formed over length scales from just a few to hundreds of nanometres.34–37 The instrument used for these experiments typically measures features between ∼2–80 nm, ideal for the fibre radius. The distance between cross-links in the network are unlikely to fall in this length range, and hence cannot be accurately determined using this instrument. The influence of the network structure on the data through the rest of the Q range can be seen and compared between the samples. The SANS profiles for the gels fall into two distinct groups (Fig. 3 and S8, ESI†). The gels triggered with HCl (in this experiment, the deuterated analogue DCl was used instead), maleic anhydride and diglycolic anhydride, i.e. those which formed quickly (see above), show a small contribution from the Q−N component of the model fit. This represents the mass fractal (2 ≤ N ≤ 3) nature of the gel network, with an exponent N of 3 ± 0.2. We interpret this as areas of more dense clustered regions in the network of fibres. For the second group, containing the GdL, acetic anhydride and glutaric anhydride triggered gels, N is determined to be lower at 2.7 ± 0.1 but with a higher scale factor (proportional to number density) suggesting a more evenly distributed density of cross-links within the network.
 | ||
| Fig. 3 (a) Example scattering patterns from gels formed on triggering with GdL and diglycolic anhydride. The fits to the data are shown as dotted lines. (b) Scattering from GdL-triggered gels in 2 mm and 5 mm cuvettes. (c) Scattering from diglycolic anhydride-triggered gels in 2 mm and 5 mm cuvettes. | ||
Through the Kratky–Porod worm-like chain component of the customized model, we are also able to measure the radius of the flexible cylindrical sub-units from the position of the interference fringes in the data. The two groups have different radii, approximately 6 nm for those which form quickly, and approximately 4 nm for the others, with marginally larger polydispersities in the radii for those in the first group. While the uncertainties on these values could be considered large in the region of 1 nm, this is driven by including polydispersity in the model fits, which is required to improve the fit to the experimental data.
Interestingly, the effect of mixing can also be probed using SANS. When gels were formed in a larger (5 mm) pathlength cuvette the data from the GdL triggered sample could be fitted to a radius of 4.4 ± 0.5 nm without any radius polydispersity. The diglycolic anhydride sample, also formed in both cuvette sizes, could also be fitted with a radius of 4.4 ± 1.0 nm in the 5 mm cuvette; but it still required radius polydispersity in order to obtain a good fit. However, the mass fractal exponent, N, is lower for this sample in the 5 mm cuvette (N = 2.8 ± 0.1) than in the 2 mm one. This suggests a more even distribution of cross-links within the structure in the gel formed in the 5 mm cuvette, suggesting that there is some improvement in the distribution of the anhydride throughout the sample. This again implies that mixing is key, especially in the gels that are formed more quickly. A full description of the model and the fitting process along with the full set of parameters are provided in the ESI.†
The method used to adjust pH not only affects gelation rates (full rheological time courses are shown in Fig. S10, ESI†), but also the turbidity of the resulting gels. After 24 hours, the turbidities of the gels are different (Fig. 4) however, imaging of the gels under a 365 nm UV lamp shows that bulk phase separation does not occur. This indicated that there are variations in the structural homogeneity within the samples over longer length scales, in agreement with the SANS data above. We have previously shown that differences in structural homogeneity for gels formed from an Fmoc-dipeptide using either GdL or HCl translate directly into differences in their mechanical properties.13 We therefore investigated the mechanical properties of the gels formed from 1 using the different anhydrides as triggers.
 | ||
| Fig. 4 Visual assessment of the bulk heterogeneity of the gel after 24 hours. Top: Photographs of gels (left) under natural light and (right) under a 365 nm light of 1 triggered using (a) acetic anhydride; (b) maleic anhydride; (c) glutaric anhydride; (d) diglycolic anhydride; (e) GdL; (f) HCl. In all cases, the scale bar represents 1 cm. | ||
First, the rheological data show that gels formed using the different triggers have different G′ and G′′ values from one another. The pronounced variation in G′ values of the different gels obtained by simply changing the trigger again highlights that the way the gelation is triggered is really important to the final mechanical properties of the gels. The errors from repeat measurements are generally relatively low, except for the data for gels formed using HCl and diglycolic anhydride which are significantly less reproducible than when using the other triggers. Strain sweeps show that these gels are typical LMWG gels.38,39 Both G′ and G′′ are essentially constant at low strain, before there is a rapid decrease in G′. On close examination, there are again two different types of behaviour for these gels. For gels triggered using GdL, glutaric anhydride, and acetic anhydride, there is a critical strain above which G′ starts to decrease (see for example Fig. 5a; other examples are shown in Fig. S11, ESI†). Gels triggered with HCl, maleic anhydride, and diglycolic anhydride show a subtly different behaviour, with G′ decreasing slowly (Fig. 5b shows data for diglycolic anhydride). We have previously correlated these two different types of rheological behaviour with different types of network; gels showing a slow decrease in G′ being indicative of a less homogeneous network.40 Hence, the rheological data indicate that the gels that are formed more rapidly are more heterogeneous, in agreement with the SANS data above.
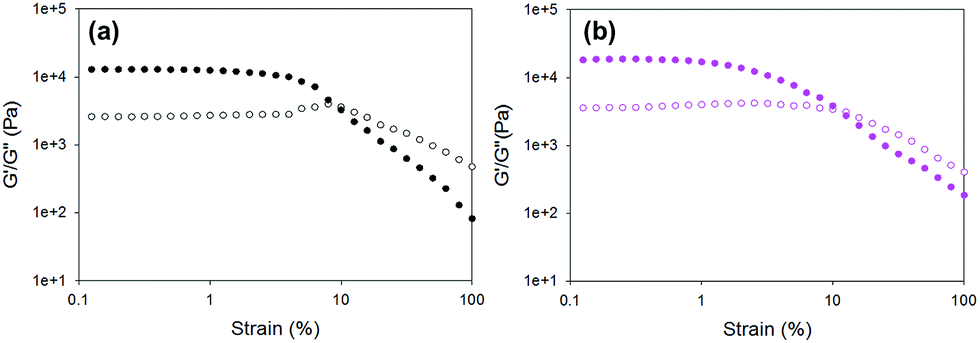 | ||
| Fig. 5 Example strain sweeps for gels formed using 1 triggered by (a) GdL; (b) diglycolic anhydride. | ||
For all gels, the frequency sweep data show that both G′ and G′′ are essentially independent of frequency up to 100 rad s−1 (Fig. 6 and Fig. S12, ESI†), which is the typical range over which such data are collected.14,38,39 However, we note that from approximately 100 rad s−1 to 628 rad s−1 all gels show frequency hardening behaviour (Fig. 6) with G′ and G′′ increasing with frequency. This is most pronounced in the gels formed by HCl. tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ (G′′/G′) for all gels (calculated from data collected at 10 rad s−1) are between 0.17 and 0.24 (Table S4, ESI†), which are quite typical for such low molecular weight gels.
δ (G′′/G′) for all gels (calculated from data collected at 10 rad s−1) are between 0.17 and 0.24 (Table S4, ESI†), which are quite typical for such low molecular weight gels.
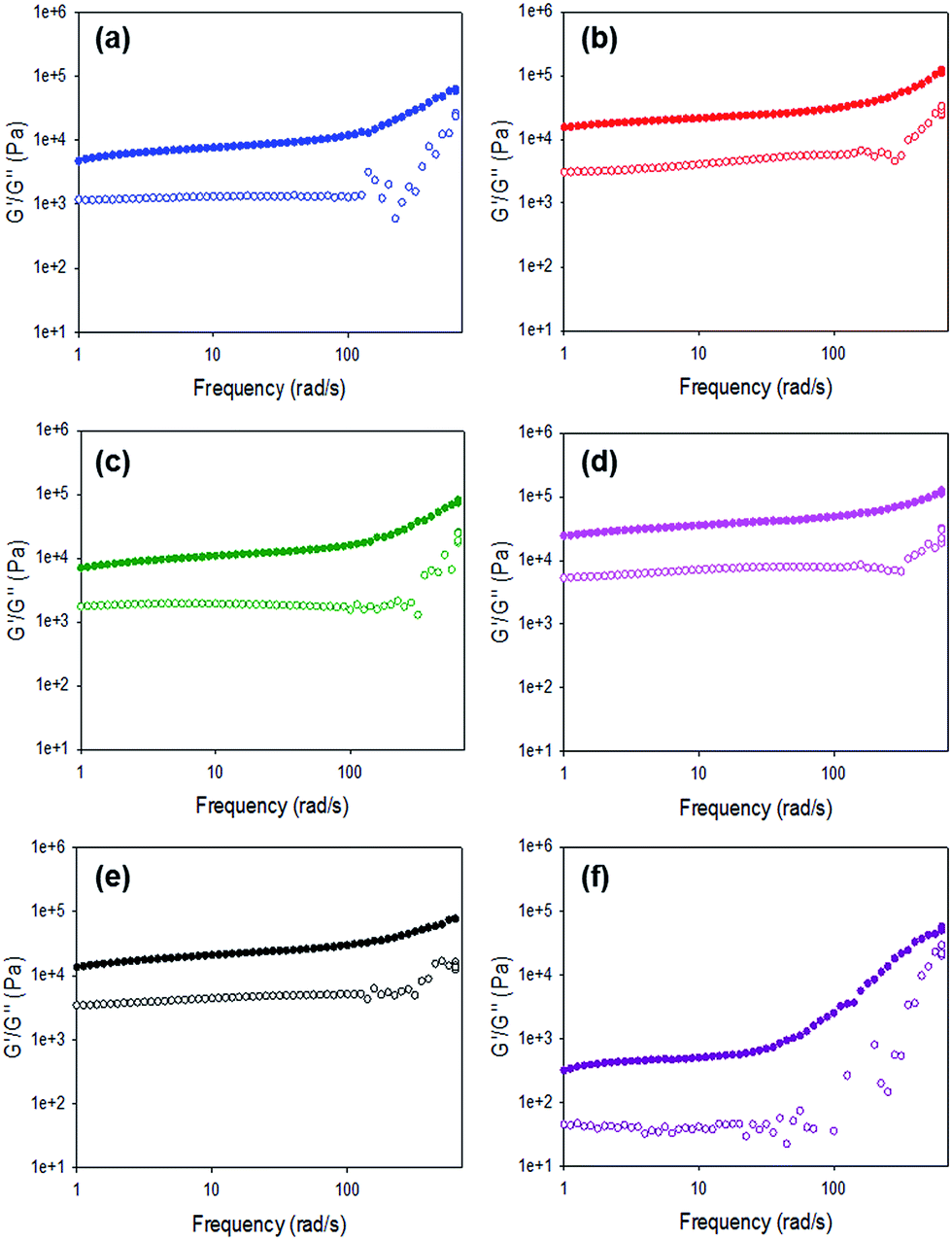 | ||
| Fig. 6 Frequency sweeps for gels formed using 1 triggered by (a) acetic anhydride; (b) maleic anhydride; (c) glutaric anhydride; (d) diglycolic anhydride; (e) GdL; (f) HCl. | ||
These rheological measurements are carried out on the bulk gels. To further probe heterogeneity in the gels, we utilised nanoindentation. Nanoindentation is an extremely powerful technique for characterising the mechanical properties of a wide range of materials because it offers a high spatial resolution (in the order of microns) and can probe small volumes of materials. Although a well-established technique for engineering materials, conventional nanoindentation poses numerous challenges when applied to highly compliant materials such as hydrogels.41 Developments in dynamic nanoindentation methods have shown that the technique can be used to measure the constitutive behaviour of viscoelastic polymers42 with good agreement with more conventional dynamic mechanical analysis (DMA) measurements.43 Dynamic nanoindentation has also been applied to oligo(ethylene glycol)-based hydrogel samples.44 Our methodology builds on this work and extends the methodology to these highly compliant LMWGs with a much higher spatial resolution (100 μm) than previous studies.
With dynamic nanoindentation, the overall micromechanical heterogeneity in G′ was found to be the greatest in the maleic anhydride (standard deviation, SD = 24.2 kPa) and HCl triggered gels (SD = 19 kPa). In contrast, GdL (SD = 10.7 kPa) and acetic anhydride (SD = 10.8 kPa) triggered gels exhibited much less variation in G′ from one indent location to the next. A similar trend was found by comparing inter-specimen variation. HCl, maleic anhydride and diglycolic anhydride triggered gels exhibited the greatest inter-specimen variation with the maximum differences in the mean values being 48.88, 30.06 and 27.3 kPa respectively. In contrast, the GdL and acetic anhydride triggered gels had the lowest inter-specimen variation (maximum differences in the mean values 18.57 and 21.83 kPa respectively). These trends fit those observed in the turbidity measurements (Fig. 4). These effects appear to be driven by differing microstructural features in the gels which is in agreement with SEM (Fig. 2) and SANS data. The SEM data (Fig. 2) and SANS data clearly show that there are significant differences amongst the gels in terms of their microstructure, with the SANS data demonstrating that the HCl, maleic anhydride and diglycolic anhydride gels are the most heterogeneous. Local variations, for example, in porosity, fibre size, and fibre orientation are likely to contribute to the variation in micromechanical properties. As noted above, SEM has been used to examine the dried samples. To better probe hydrated samples, and also to capture a length scale more in line with that of the nanoindentation measurements, we examined the gels using confocal microscopy. Nile blue can be added to the solutions pre-gelation, allowing the fibrous networks to be visualised.45 Representative data for each gel is shown in Fig. 7.
 | ||
| Fig. 7 Representative confocal fluorescence microscopy images of gels of 1 triggered using (a) acetic anhydride; (b) maleic anhydride; (c) glutaric anhydride; (d) diglycolic anhydride; (e) GdL; (f) HCl. In all cases, the scale bar is 100 μm. | ||
As can be seen, the fibrous network formed when gelation is triggered with either acetic anhydride or GdL is less distinct than when using the other triggers. This can be attributed to thinner fibres being formed during the slower gelation (the resolution of confocal microscopy is limited by the diffraction of light). This can be linked to the slower kinetics of gelation. Similar thickness fibres can be seen when the other triggers are used. All structures visualised using this technique will be of significantly higher diameters than the structures probed by SANS, but of the same order of magnitude as suggested by SEM. Hence, again, this implies that the SANS can probe the primary fibre thickness, but not that of the aggregated fibres. To relate the confocal data to the nanoindentation data, the mean intensity of nine regions of interest – 100 μm diameter circles to simulate the area of the gel contacted by the flat punch indenter (example images are shown in Fig. S10, ESI†) – was measured. The standard deviations (σ) between the individual ROIs and an overall mean from all of the regions of interest from each sample were calculated (μ). The coefficient of variation (CV) was then calculated by CV = σ/μ. When looking at the CV for the ‘indents’, it is clear that the gels triggered with diglycolic anhydride and HCl are significantly less homogeneous (CV = 0.36 and 0.32 respectively, as compared to 0.11 for acetic anhydride, 0.12 for maleic anhydride, 0.19 for glutaric anhydride and 0.19 for GdL). Similarly, the CV for the entire gels again show that the gels triggered with diglycolic anhydride and HCl are significantly less homogeneous (CV = 0.42 and 0.51 respectively) as compared to the others (CVs of between 0.24 and 0.29).
Fig. 8 gives a comparison of our macromechanical rheology measurements (628 rad s−1, approx. 100 Hz) with our dynamic nanoindentation measurements (110 Hz). The overall trend in G′ (highest to lowest) is similar with both techniques, with the exception of the acetic acid gel. The mean values for G′ and G′′ obtained with nanoindentation are 2–4 times greater as compared to rheology for each gel with the exception of the acetic anhydride triggered gel (Table 1).
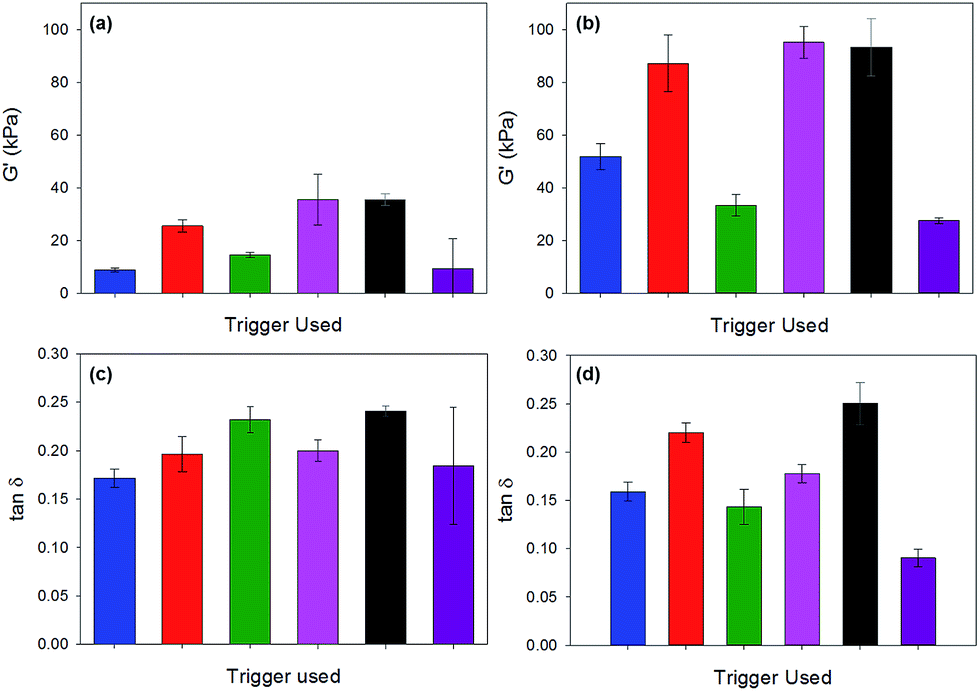 | ||
| Fig. 8 Comparison of G′ and tanδ as measured by rheology (a) and (c), and by nanoindentation (b) and (d). Colour code: blue is acetic acid; red is maleic anhydride; green is glutaric anhydride; pink is diglycolic anhydride; black is GdL; purple is HCl. | ||
| Acetic | Maleic | Glutaric | Diglycolic | GdL | HCl | |
|---|---|---|---|---|---|---|
| G′ | 7.7 | 2.9 | 3.5 | 2.4 | 2.9 | 4 |
| G′′ | 6.6 | 2.6 | 2.1 | 2.2 | 2.4 | 3.2 |
As shown in Table 1, G′ and G′′ for acetic anhydride triggered gels are around seven times higher when measured with nanoindentation as compared to rheology. The tanδ values were approximately the same for all gels with the exception of the glutaric anhydride and maleic anhydride gels. For both of these gels, tanδ values were measured to be approximately 40% lower with nanoindentation as compared to rheology.
Kaufman et al. compared nanoindentation values with macroscale compression tests for pHEMA gels and found a greater discrepancy with the most compliant (i.e. the least cross-linked gels).41 They hypothesised that this could be related to the resolution of the instrument used, or an effect of surface properties (nanoindentation) versus the bulk properties of the hydrogels. Baniasadi and Minary-Jolandan have used a range of techniques including AFM-based nanoindentation, rheology and tensile testing to compare the micro- and macro-mechanical response of alginate-collagen fibril composite hydrogels.46 The overall trends were similar but it was not possible to directly compare the values obtained with the different techniques they utilised. However, dynamic nanoindentation, as used in our current study, has been shown to provide data for a standard thermoplastic material to within 15% of DMA measurements.43 Our data suggest that the micromechanical behaviour of the gels investigated in this study differs significantly from the bulk properties. This is likely to be dominated by the distribution of the fibres and the density of the mesh network, as is suggested by the confocal microscopy (Fig. 7). The tanδ values demonstrate that the ratio of the elastic and viscous portions of deformation remain approximately the same at the micron-level.
Finally, we highlight again that the homogeneity inherent in some of the gels where slow hydrolysis is used is a result of the kinetics of the pH change. For example, when acetic acid is used directly to adjust the pH as opposed to its situ formation on hydrolysis of acetic anhydride, the gels are significantly less homogeneous and the mechanical properties more variable (Fig. S14, ESI†).
Experimental
Materials
Dipeptide 1 was prepared as described previously.14 All other chemicals were purchased from Sigma Aldrich and used as received. Deionised water was used throughout.Hydrogel preparation
A stock solution of 1 at a concentration of 5 mg mL−1 and approximately pH 10–11 was prepared by adding a dilute sodium hydroxide solution (1 molar equivalent of a 0.1 M solution) to a suspension of 1 in water with stirring until full dissolution occurred (for SANS samples, D2O, NaOD and DCl were used). The gelator stock solution was added to a weighed amount of GdL or anhydride (in the case of acetic anhydride or HCl, this was instead added to the solution) and all were gently swirled. The sample was then left to stand to allow gelation to occur over several hours. For nanoindentation measurements, gels were prepared in moulds. The mould was prepared by cutting the top off a 20 mL (2 cm diameter) plastic syringe. 1 mL of the gelator solution and trigger were added into the syringe. These were covered with Parafilm and left to gel overnight. The gels were removed by carefully pushing the plunger. They were sufficiently stiff to be transferred onto the nanoindentation sample holder or a glass slide. On a few occasions, the HCl and the glutaric anhydride formed gels that could not be removed from the moulds. These were not used for measurements.Imaging
For samples to be photographed, gels were prepared in moulds and placed on glass microscope slides. Images were collected in daylight and under 365 nm illumination. For pH change images (Fig. 1), 2 drops of a bromophenol blue indicator solution was added to the gelator solution in 14 mL glass sample tubes before the trigger was added. The trigger was then added to the solutions and photographs were taken periodically for 24 hours.Rheology
Time course rheological measurements were carried out using an Anton Paar Physica MCR101 rheometer using parallel plate geometry. Time sweep measurements were performed at a constant frequency of 10 rad s−1 and a strain of 0.5% at 25 °C. G′ and G′′ were measured over time. Samples were prepared by pipetting 2 mL of gelator solution onto the bottom plate and adding the trigger and mixing before lowering the top plate. A 50 mm sandblasted plate was used to minimise slippage and mineral oil was placed around the plate to minimise drying out. Strain and frequency sweeps were performed using a vane and cup geometry. Gels were prepared in 2 mL Sterilin cups and left to gel overnight. Strain sweeps were carried out at 10 rad s−1 from 0.1–1000% strain. Frequency sweeps were carried out at 0.5% strain and measured between 1–628 rad s−1. For the data used to probe gel reproducibility, frequency sweeps were performed between 1–100 rad s−1. All measurements were performed at 25 °C.pH measurements
A calibrated FC200 pH probe (HANNA instruments) with a 6 mm × 10 mm conical tip was used for the pH measurements. The stated accuracy of the pH measurements is ±0.1. Kinetic pH measurements during gelation were taken by monitoring the pH in situ and by taking pH measurements every 60 s for 18 hours at 25 °C unless otherwise stated. Temperature was controlled by placing the samples in a water bath at a set temperature whilst gelation occurred.Scanning electron microscopy
SEM images were recorded using a Hitachi S-4800 FE-SEM at 3 keV. A portion of the hydrogel was placed on a glass cover slip attached to a sticky carbon tab. The hydrogel was air-dried directly on the coverslip. The samples were gold coated for 3 minutes at 15 mA using a sputter coater (EMITECH K550X) prior to imaging.Confocal microscopy
A Zeiss LSM510 on a Zeiss Observer Z1 (Zeiss, Jena, Germany) were used for imaging. The gel samples were prepared at a concentration of 1 of 5 mg mL−1 containing Nile blue (20 μL of a 0.1 M solution added per mL of gelator solution) in CELLview Culture dishes, (35 mm diameter) and were excited at 633 nm and detected with a Zeiss Meta detector. A spectral filter of 650–710 nm was used to obtain the Nile blue emission. The images obtained are from 2 μm think slices within the first 40 μm of the surface of the gel. Data were captured using Zeiss Zen software (Zeiss, Jena, Germany) and analysed using Zeiss LSM image browser (version 4.2.0.121). Images were further analysed using ImageJ (version 1.48v), the mean intensity at each of the nine regions of interest (ROIs), 100 μm diameter circles (to simulate the area of the gel contacted by the indenter) were measured. The standard deviations (σ) between the individual ROIs and an overall mean from all of the nines ROIs from each sample were calculated (μ). The coefficient of variation (CV) was then calculated by CV = σ/μ.Nanoindentation
The hydrogels were tested with a Nanoindenter G200 system equipped with a DCM-II ultra-low load head (Keysight Technologies, USA). A 100 μm flat-ended cylindrical punch tip (Synton-MDP Ltd, Nidau, Switzerland) was used for all of the experiments.The hydrogels were characterised with a dynamic indentation method, which utilises the Keysight Technologies DCM II actuator along with the Continuous Stiffness Measurement (CSM) to determine their micromechanical complex shear modulus. The test procedure and theory have been detailed elsewhere.47
16 indentations were made on each sample in a 4 × 4 array with 200 μm spacing between each indent. The tip was cleaned after each indent to prevent any material being transferred to the subsequent indent by indenting a piece of double-sided Scotch tape which was mounted on an adjacent sample puck. As described in our preliminary study,47 a pre-compression of 5 μm was applied to the samples. Once the indenter was fully in contact with the sample surface, the indenter vibrated at a frequency of 110 Hz (the resonant frequency of the indenter) and with an oscillation amplitude of 500 nm. The surface detection was determined by a phase shift of the displacement measurement. In order to accurately detect the surface, the phase shift was monitored over a number of data points. This procedure helped to exclude random spikes, which were found to occur in some instances over short time intervals (e.g. 2 data points) and thereby lead to an erroneous surface detection. Once the surface detection requirement had been fulfilled over the predefined number of data points the initial contact was determined from the first point in that sequence. A Poisson's ratio of 0.5 was assumed for each of the gel samples. G′, G′′ and the loss factor (tanδ) i.e. ratio of G′′/G′ were calculated for each indentation.
FTIR
IR spectra were collected on a Bruker Tensor 27 FTIR spectrometer at a resolution of 2 cm−1 with spectral averaging over 64 scans. Measurements were collected using the ATR accessory. Gels were prepared in sample tubes and were measured wet and had the trigger in water background subtracted. The trigger background was prepared by placing the same amount of anhydride, GdL or HCl into 2 mL of water, leaving them for the same time as the gels were left for and then measuring the IR spectra. This background spectrum was then subtracted from the corresponding gel spectrum.Small angle neutron scattering
Small angle neutron scattering (SANS) measurements were performed on the fixed-geometry, time-of-flight LOQ diffractometer (ISIS Spallation Neutron Source, Oxfordshire, UK). A white beam of radiation with neutron wavelengths spanning 2.2 to 10 Å was used to access a Q [Q = 4πsin(θ/2)/λ] range of 0.008 to ≥0.3 Å−1 (at 25 Hz), with a fixed sample to main detector distance of 4.1 m. Samples were prepared as previously described directly into 2 mm or 5 mm path length, UV-spectrophotometer grade, quartz cuvettes (Starna and Hellma respectively) and left to form gels overnight. These were subsequently mounted in aluminium holders on top of an enclosed, computer-controlled, sample chamber with temperature maintained at 25 ± 0.5 °C by use of a thermostatted circulating bath pumping fluid through the base of the sample chamber. Experimental measuring times were approximately 60 minutes.
All scattering data were (a) normalized for the sample transmission, (b) background corrected using a quartz cell, of the same path length, filled with D2O (this also removes the inherent instrumental background arising from vacuum windows etc.) and (c) corrected for the linearity and efficiency of the detector response using the Mantid framework.48,49 The data were put onto an absolute scale by reference to the scattering from a partially deuterated polystyrene blend.50 The instrument-independent data were then fitted to a customised model in the SasView software package51 combining an absolute power law and a (Kratky–Porod) flexible cylinder.52,53 The Q-dependent power law (Q−N) accounts for the mass fractal contribution to the scattering intensity which is superimposed on that from the fibrils. The fibrils themselves are represented as a flexible worm-like chain of cylindrical Kuhn segments. The modelling is discussed further in the ESI.†
Conclusions
We have shown that the hydrolysis of a number of anhydrides can be used to trigger the gelation of a pH sensitive low molecular weight gelator. The primary assembled structures of the gels obtained by this method and those of GdL or HCl triggered gels are the same. However, the homogeneity in the distribution of fibres and the crosslinks, and consequently the homogeneity at different length scales and the mechanical properties of the gels, depend on the trigger used. We thus demonstrate how the properties of gels obtained from the same stock solution of a single gelator can be tuned by the choice of pH change trigger.A comparison of rheological and nanoindentation data shows that the micromechanical behaviour of the gels studied differs significantly from their bulk properties. This is likely to be dominated by the distribution of the fibres and the density of the mesh network, which again is controlled by the choice of trigger. This also highlights that simple rheological data may not be sufficient to probe and capture the subtleties of the mechanical properties of low molecular weight gels.39
Acknowledgements
ED thanks the EPSRC for a DTA studentship. LM and AMC thank the EPSRC for funding. DA thanks the EPSRC for a Fellowship (EP/L021978/1). TM thanks the EPRSC (EP/M01973X/1) and the Royal Society (RG140213) for funding. RA is grateful to the Royal Society for funding (Research Grant RG103629). Experiments at the ISIS Pulsed Neutron and Muon Source were supported by a beam-time allocation from the Science and Technology Facilities Council (Experiment number RB1510065). We acknowledge the Liverpool Centre for Cell Imaging (CCI) for continuous support in this project. This work benefitted from the SasView software, originally developed by the DANSE project under NSF award DMR-0520547.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- N. Zweep and J. H. van Esch, in Functional Molecular Gels, The Royal Society of Chemistry, 2014, pp. 1–29 Search PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- B. Ding, Y. Li, M. Qin, Y. Ding, Y. Cao and W. Wang, Soft Matter, 2013, 9, 4672–4680 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024–2027 RSC.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed.
- Z. Yang, G. Liang, M. Ma, Y. Gao and B. Xu, J. Mater. Chem., 2007, 17, 850–854 RSC.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- K. A. Houton, K. L. Morris, L. Chen, M. Schmidtmann, J. T. A. Jones, L. C. Serpell, G. O. Lloyd and D. J. Adams, Langmuir, 2012, 28, 9797–9806 CrossRef CAS PubMed.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- C. Tang, R. Ulijn and A. Saiani, Eur. Phys. J. E, 2013, 36, 1–11 CrossRef PubMed.
- Y. Pocker and E. Green, J. Am. Chem. Soc., 1973, 95, 113–119 CrossRef CAS PubMed.
- W. Helen, P. de Leonardis, R. V. Ulijn, J. Gough and N. Tirelli, Soft Matter, 2011, 7, 1732–1740 RSC.
- H. Huang, S. Lu, X. Zhang and Z. Shao, Soft Matter, 2012, 8, 4609–4615 RSC.
- R. C. T. Howe, A. P. Smalley, A. P. M. Guttenplan, M. W. R. Doggett, M. D. Eddleston, J. C. Tan and G. O. Lloyd, Chem. Commun., 2013, 49, 4268–4270 RSC.
- E. R. Draper, J. J. Walsh, T. O. McDonald, M. A. Zwijnenburg, P. J. Cameron, A. J. Cowan and D. J. Adams, J. Mater. Chem. C, 2014, 2, 5570–5575 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Angew. Chem., Int. Ed., 2014, 53, 12461–12465 CAS.
- B. C. Baker, A. L. Acton, G. C. Stevens and W. Hayes, Tetrahedron, 2014, 70, 8303–8311 CrossRef CAS.
- R. Li, C. C. Horgan, B. Long, A. L. Rodriguez, L. Mather, C. J. Barrow, D. R. Nisbet and R. J. Williams, RSC Adv., 2015, 5, 301–307 RSC.
- A. D. Martin, A. B. Robinson, A. F. Mason, J. P. Wojciechowski and P. Thordarson, Chem. Commun., 2014, 50, 15541–15544 RSC.
- M. Wallace, J. A. Iggo and D. J. Adams, Soft Matter, 2015, 11, 7739–7747 RSC.
- C. A. Bunton, N. A. Fuller, S. G. Perry and V. J. Shiner, J. Chem. Soc., 1963, 2918–2926 RSC.
- J. T. Pelton and L. R. McLean, Anal. Biochem., 2000, 277, 167–176 CrossRef CAS PubMed.
- X. Mu, K. M. Eckes, M. M. Nguyen, L. J. Suggs and P. Ren, Biomacromolecules, 2012, 13, 3562–3571 CrossRef CAS PubMed.
- K. L. Morris, L. Chen, A. Rodger, D. J. Adams and L. C. Serpell, Soft Matter, 2015, 11, 1174–1181 RSC.
- J.-B. Guilbaud and A. Saiani, Chem. Soc. Rev., 2011, 40, 1200–1210 RSC.
- D. J. Pochan, L. Pakstis, B. Ozbas, A. P. Nowak and T. J. Deming, Macromolecules, 2002, 35, 5358–5360 CrossRef CAS.
- R. A. Hule, R. P. Nagarkar, A. Altunbas, H. R. Ramay, M. C. Branco, J. P. Schneider and D. J. Pochan, Faraday Discuss., 2008, 139, 251–264 RSC.
- R. A. Hule, R. P. Nagarkar, B. Hammouda, J. P. Schneider and D. J. Pochan, Macromolecules, 2009, 42, 7137–7145 CrossRef CAS PubMed.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- S. Sathaye, A. Mbi, C. Sonmez, Y. Chen, D. L. Blair, J. P. Schneider and D. J. Pochan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 34–68 CrossRef CAS PubMed.
- E. R. Draper, K. L. Morris, M. A. Little, J. Raeburn, C. Colquhoun, E. R. Cross, T. O. McDonald, L. C. Serpell and D. J. Adams, CrystEngComm, 2015, 17, 8047–8057 RSC.
- J. D. Kaufman, G. J. Miller, E. F. Morgan and C. M. Klapperich, J. Mater. Res., 2008, 23, 1472–1481 CrossRef CAS PubMed.
- J. L. Loubet, W. C. Oliver and B. N. Lucas, J. Mater. Res., 2000, 15, 1195–1198 CrossRef CAS.
- E. G. Herbert, W. C. Oliver and G. M. Pharr, J. Phys. D: Appl. Phys., 2008, 41, 074021 CrossRef.
- P. O. Guglielmi, E. G. Herbert, L. Tartivel, M. Behl, A. Lendlein, N. Huber and E. T. Lilleodden, J. Mech. Behav. Biomed. Mater., 2015, 46, 1–10 CrossRef CAS PubMed.
- J. Raeburn, L. Chen, S. Awhida, R. C. Deller, M. Vatish, M. I. Gibson and D. J. Adams, Soft Matter, 2015, 11, 3706–3713 RSC.
- M. Baniasadi and M. Minary-Jolandan, Materials, 2015, 8, 799 CrossRef.
- R. Akhtar, E. R. Draper, D. J. Adams and H. Pfaff, Complex Shear Modulus of Hydrogels Using a Dynamic Nanoindentation Method, Mechanics of Biological Systems and Materials, Proceedings of the 2015 Annual Conference on Experimental and Applied Mechanics, ed. S. A. Tekalur, P. Zavattieri and C. S. Korach, vol. 6, pp. 141–145, http://www.springer.com/uk/book/9783319214542, DOI:10.1007/978-3-319-21455-9.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 2014, 764, 156–166 CrossRef CAS.
- https://www.mantidproject.org.
- G. D. Wignall and F. S. Bates, J. Appl. Crystallogr., 1987, 20, 28–40 CrossRef CAS.
- https://www.sasview.org.
- J. S. Pedersen and P. Schurtenberger, Macromolecules, 1996, 29, 7602–7612 CrossRef CAS.
- W.-R. Chen, P. D. Butler and L. J. Magid, Langmuir, 2006, 22, 6539–6548 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further rheological data and detailed description of fitting of the SANS data. See DOI: 10.1039/c5ra22253b |
| This journal is © The Royal Society of Chemistry 2015 |