
Effective parameters on selective catalytic hydrodeoxygenation of phenolic compounds of pyrolysis bio-oil to high-value hydrocarbons
Abstract
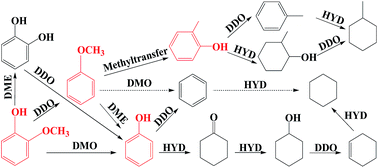
Pyrolysis bio-oil is recognized as a renewable and carbon-neutral fuel which could be a potential alternative for depleting fossil fuels. However, bio-oil is highly oxygenated and needs to be upgraded prior to be used as fuel additive. Catalytic hydrodeoxygenation (HDO) is an efficient technique for bio-oil upgrading. The reaction pathway for HDO of bio-oil is unknown since it is a mixture of hundreds of different compounds. The study on mechanism of transformation of these compounds could be helpful to propose an overall pathway for HDO of bio-oil. Phenols which are derived from pyrolysis of lignin fraction of biomass are considered as attractive model compounds for study of bio-oil HDO since they are highly stable in HDO reaction. Reaction pathway and product selectivity in HDO of phenols are highly affected by type of catalyst promoters and supports, catalyst preparation procedure, solvent type, chemicals used as co-feed and operating conditions (i.e., temperature and pressure). The effects of these factors on selective production of high-value hydrocarbons of aromatics and alicyclics from HDO of phenol, cresol, guaiacol and anisole are discussed in this review.
 Please wait while we load your content...
Please wait while we load your content...

