DOI:
10.1039/C5RA20066K
(Paper)
RSC Adv., 2015,
5, 103989-103998
Synthesis of mesoporous MCM-41 supported reduced graphene oxide-Fe catalyst for heterogeneous Fenton degradation of phenol
Received
28th September 2015
, Accepted 25th November 2015
First published on 26th November 2015
Abstract
A new heterogeneous Fenton catalyst, mesoporous MCM-41 supported reduced graphene oxide-Fe (rGO-Fe/MCM-41), was synthesized via a hybrid hydrothermal-calcination treatment. The physicochemical characteristics of the catalyst were evaluated by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), infrared spectroscopy (FT-IR) and surface area (BET) analysis. The results indicated that the rGO-Fe/MCM-41 possessed a mesoporous structure. The effective reduction of GO to rGO and a high degree of α-Fe2O3 loading were observed. After the incorporation of rGO, the activity and stability of the catalyst in phenol degradation significantly increased. The kinetics of phenol degradation fit the first order kinetic model well. The effects of Fe and GO dosage, as well as calcination temperature, were investigated. The XRD and the Raman scattering demonstrated that the reduction of GO was more effective, and the α-Fe2O3 crystal structure was formed when calcination temperature is 550 °C, which beneficially increased the catalytic activity.
1. Introduction
In recent years, advanced oxidation processes (AOPs) have been accepted as an efficient way to remove hazardous contaminants.1 Specifically, the Fenton reaction is highly attractive due to its high efficiency, low cost and wide application range (non-selectivity) by the production of highly reactive hydroxyl radicals.2 Recent reports have noted that the application of the homogeneous Fenton reaction consisting of a homogeneous solution of Fe2+/Fe3+ and hydrogen peroxide is limited because the Fe3+ non-recyclable soluble iron salts will produce abundant iron sludge that requires further treatment. To overcome this problem, the heterogeneous Fenton reaction was given more attention. In the heterogeneous Fenton reaction, H2O2 was decomposed into hydroxyl radicals by metal oxides on the surface of the catalyst.3–6 Hence, developing a high activity Fenton catalyst is the key for the increase of Fenton reaction efficiency.
As reported, various heterogeneous Fenton catalysts such as iron/copper/transition metal oxides immobilized on zeolites, clays and carbon materials have been developed for the degradation of organic pollutants.3,7 Iron-containing supported solid materials with reactive components, such as Fe, Fe2O3, Fe3O4 and FeOOH, were widely studied because of their high efficiency and low cost.8–11 In the reaction, the redox cycle of Fe2+/Fe3+ activates H2O2 to produce hydroxyl radicals. Methods of increasing the electron transport speed are notably important to accelerate this reaction.
Graphene, a novel one-atom-thick two-dimensional graphitic carbon system, has drawn tremendous attention due to its excellent electronic properties and possible applications in various fields.12 Graphite oxide (GO), as the derivative of graphene, contains various hydrophilic oxygen-containing functional groups. Therefore, GO is water-soluble and more flexible than grapheme in the fabrication of different functional materials.13 As reported, GO can be easily reduced to graphene oxide (rGO) by hydrothermal treatment or high-temperature calcination under inert gases, or in the presence of metal. Compared with GO, rGO possess higher electrical conductivity and thermal stability, which may promote the electron transport speed in the Fenton process.14 Additionally, rGO also can prevent the aggregation of Fe and further enhance the catalytic activity. Recently, Huang et al. successfully synthesized FeOOH/rGO as a heterogeneous Fenton catalyst for the removal of organic dye.11 The results showed that the catalyst possesses excellent catalytic activity for the Fenton reaction. However, no support was used in their study, which leads to the coverage of certain active sites. Thus, the cost of the catalyst increases.
In recent years, mesoporous supports were widely used in the fields of catalysis because it is easier for products to diffuse and thus benefit the reaction. Among them, MCM-41 has drawn considerable attention because of its wide mesopore sizes distribution (2–10 nm) and a large specific surface area (>1000 m2 g−1).15,16 To further increase the activity of the catalyst, Fe2O3/rGO supported MCM-41 was developed in our work. Its application to degrade organic pollutants has never been reported before to the best of our knowledge. The catalysts were then characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction spectroscopy (XRD), Fourier transform infrared spectroscopy (FTIR), Brunauer–Emmett–Teller (BET) and X-ray photoelectron spectroscopy (XPS). The effects of iron dosage, graphene dosage and calcination temperature were optimized.
2. Experimental
2.1. Materials
The methanol (CH4O) used in this experiment was chromatography grade, and all other reagents were analytical reagent grade. The water for analytical laboratory use is ultra-pure water.
2.2. Catalyst synthesis
2.2.1. Synthesis of Fe/MCM-41. MCM-41 was prepared by the following two methods. One is as previously described by Amaresh C. Pradhan and K. M. Parida.8 Firstly, a certain amount of cetyltrimethyl ammonium bromide (CTAB) was dissolved in deionized water at room temperature. Then, the appropriate amount of 25% aqueous NH3 was added to the above solution. In this process, the pH is 8 to 9. Next, tetraethyl orthosilicate (TEOS) was added to the solution dropwise under vigorous stirring (240–300 rpm) for 2 to 3 h. After that, the solution was aged at room temperature for 24 to 48 h. Next, the compound was filtered, dried and calcined (550 °C in a muffle furnace for 6 h). The material produced by this method is the carrier, MCM-41@.The second method is as followed. 6–10 weight portions of sodium silicate were dissolved in 20–30 weight portions of deionized water at 40 °C, adjusting the pH to 8 to 10 by 5 mol L−1 sulfuric acid (H2SO4). Next, it was stirred at 300–360 rpm for 0.5 h until the solution became viscous and gelatin-like. 4 to 6 weight portions cetyltrimethyl ammonium bromide (CTAB) was added to the above suspension under vigorous stirring until the suspension became a viscous gelatin. Next, the viscous gelatin was put into a reaction kettle at 100 to 130 °C to crystallize for 16 to 36 h. Finally, the mixture was filtered, air-dried and calcined at 550 °C in a N2-tube furnace for 4 to 6 h. The sample is MCM-41.
Two kinds of MCM-41 prepared by above two methods were used as carrier to obtain Fe modified MCM-41. The method is as followed. 1 g of MCM-41 (or MCM-41@) was impregnated with 5% ferric nitrate (Fe(NO3)3·9H2O), stirred well at 35 °C and subjected to ultrasonication. Next, it was shaken at room temperature for 12 h. Finally, it was filtered, dried and calcined (550 °C in a muffle furnace for 6 h). The resulting material of this method is Fe/MCM-41 and Fe/MCM-41@.
2.2.2. Synthesis of GO. According to the modified Hummers method,17 40 weight portions of 98% sulfuric acid (H2SO4) were dissolved in 2 weight portions of graphite and 1 weight portion of sodium nitrate (NaNO3) in an ice-bath under stirring for 15 min. Next, 6 weight portions of potassium permanganate (KMnO4) were added slowly to the suspension, stirring for 15 min. The ice-bath was then removed and the temperature of the suspension rose to 35 °C, where it was maintained for 4 to 6 h; it was subsequently warmed to approximately 97 °C. 75 weight portions of deionized water were slowly stirred into the paste, keeping the temperature at 90 °C for 15 min. The suspension was then diluted with 100 weight portions of deionized water and treated with 20 weight portions of 30% hydrogen peroxide (H2O2) until the suspension turned bright yellow. 900 weight portions of 5% hydrochloric acid (HCl) were used to purge the suspension. It was then filtered, and the yellowish-brown filter cake was washed with deionized water to remove the loosely attached particles. Finally, the washed samples were filtered, and GO was obtained by ultrasonication with 450 weight portions of deionized water added. The final concentration of GO is 7.75 mg mL−1.
2.2.3. Synthesis of rGO-Fe/MCM-41 and Fe/rGO. Synthesis route for rGO-Fe/MCM-41 is shown in Scheme 1. First, 6–10 weight portions of sodium silicate were dissolved in 20–30 weight portions of deionized water at 40 °C, adjusting the pH to 8 to 10 by 5 mol L−1 sulfuric acid (H2SO4). Next, it was stirred at 300–360 rpm for 0.5 h until the solution became viscous and gelatin-like. 4 to 6 weight portions cetyltrimethyl ammonium bromide (CTAB) and 10–40 weight portions graphene oxide were added to the above suspension under vigorous stirring until the suspension became a dark gray viscous gelatin. Next, the viscous gelatin was put into a reaction kettle at 100 to 130 °C to crystallize for 16 to 36 h. Finally, the mixture was filtered, air-dried and calcined at 550 °C in a N2-tube furnace for 4 to 6 h. The sample is rGO modified MCM-41. Next, 1 g of rGO modified MCM-41 was impregnated with 5% (2% or 10%) ferric nitrate (Fe(NO3)3·9H2O), shaking the mixture at room temperature for 12 h, and it was filtered, dried and calcined at 550 °C in a N2-tube furnace for 4 to 6 h. The sample is recorded as rGO-Fe/MCM-41.
 |
| | Scheme 1 Synthesis route of rGO-Fe/MCM-41. | |
30 weight portions graphene oxide and 5% ferric nitrate (Fe(NO3)3·9H2O) was ultrasonic mixed for 30 min. Then the mixture was shaked 12 h at room temperature, then filtered, dried and calcined at 550 °C in a N2-tube furnace for 4 to 6 h. The sample is recorded as Fe/rGO.
2.3. Catalyst characterization techniques
The physiochemical properties of the catalysts were characterized. Surface morphology was observed via a scanning electron microscope and dispersive X-ray spectrum (SEM-EDX, HITACHI S-4700N). Transmission electron microscopy (TEM) experiments were conducted on a Hitachi H-800 microscope (Japan) operated at 200 kV. Powder X-ray diffraction (XRD) analyses were obtained using Fe Kα radiation. The electronic states were investigated by X-ray photoelectron spectroscopy (XPS, VG Scientific Ltd ESCALAB MK II). FTIR spectra of the samples were recorded on a FTIR spectrophotometer (Nicolet Nexus 670) in the range of 400 to 4000 cm−1. Raman spectra were recorded on a laser confocal micro-Raman scattering spectrometer (LabRAM Aramis, HJY Company).
2.4. Fenton degradation procedure
A 500 mL glass beaker filled with 200 mL of 100 mol L−1 phenol was used as the Fenton reactor. The initial pH was adjusted using HCl, as measured by a PB-10 (SARTORIUS) pH meter. After stabilizing the pH, 0.1 g of catalyst was added to the solution, and the reaction was initiated by adding a certain amount of H2O2 (30 wt% solution). All samples were taken at given time intervals during the reaction and mixed immediately with NaOH, which was used as a radical scavenger. Next, the samples were filtered through a 0.45 μm membrane. All of the experiments were performed at 25 °C.
2.5. Analytical methods
The concentration of phenol was determined by HPLC (UltiMate3000, DIONEX) analysis with an UV detector adjusted to 270 nm. The mobile phase was a mixture of 50% methanol and 50% ultrapure water (0.1% phosphoric acid) with a flow rate of 1 mL min−1. The extent of mineralization was determined by a Hach COD analyzer (DRB200, United States). The samples were first modified with 50 g L−1 Na2CO3 to remove residual H2O2 in the filtrate to prevent interference in the analysis of COD, the samples were heated in a water bath at 90 °C for 90 min.18 Next, COD was measured by the rapid digestion spectrophotometry method. Spectrophotometry was used for the determination of H2O2 in the Fenton reaction with titanium oxalate. In this method, H2O2 was reacted with titanium in acidic media to form a stable and orange complex for the determination of H2O2 content by spectrophotometry. The O-phenanthroline spectrophotometry method19 was used to determine the content of iron. The data of actual loadings of Fe for all samples were measured by ICP (SPECTRO ARCOS EOP).
The acute toxicity change of the solution during the process of the Fenton catalytic reaction was measured using the luminescent bacteria vibrio fischeri. The solution was withdrawn at different reaction times (0, 30, 60 and 90 min) and later mixed with the luminescent bacteria. After 5 min and 15 min, the luminosity of the bacteria was measured using the photobacteria toxicity system (BHP 9514).
3. Results and discussion
3.1. Characterization of catalyst
3.1.1. SEM-EDX and TEM analysis. The Scanning Electron Microscope (SEM) analysis of rGO-Fe/MCM-41 and Fe/MCM-41 catalysts is shown in Fig. 1. The result demonstrates that the particles are spherical and homogeneously dispersed on the support. The size of Fe/MCM-41 particles (Fig. 1(a)) was approximately 60 to 80 nm. After the incorporation of rGO, the particle size of rGO-Fe/MCM-41 (Fig. 1(b)) decreased to 20 to 40 nm, which is less than half the size of the Fe/MCM-41 catalyst. The BET analysis showed that BET surface areas increased from 348.3 m2 g−1 for the Fe/MCM-41 catalyst to 521.2 m2 g−1 for the rGO-Fe/MCM-41 catalyst. That is, the incorporation of graphene oxide can decrease the particle size. The reason may be due to the result that the oxygen-containing groups of GO remarkably increase favorite nucleation sites for SiO2 nanoparticle growth and the nanoparticle is stabilized by the nanoparticle–GO interaction.20 And the BET surface area of the catalyst enhances because GO possessed a high specific area,21 which may increase the catalytic capacity. The quantitative EDX analysis demonstrated that the atomic ratio of Si/O/C/Fe is 54/89/41/1 for the rGO-Fe/MCM-41 catalyst, and Si/O/Fe is 45/139/1 for the Fe/MCM-41 catalyst. This indicated that the amount of oxygen atoms decreases significantly after the incorporation of rGO, which is due to the high temperature calcination under N2 atmosphere. ICP analysis showed Fe loading dosage is 3.5% for Fe/rGO, 3.4% for Fe/MCM-41 and 3.8% for rGO-Fe/MCM-41.
 |
| | Fig. 1 SEM images of (a) Fe/MCM-41 and (b) rGO-Fe/MCM-41. | |
The results of the BJH analysis showed that average pore sizes were 2.739 nm and 2.745 nm for Fe/MCM-41 and rGO-Fe/MCM-41 catalysts, respectively, which indicated that both of them are mesoporous materials.
The morphology of MCM related materials was also studied by Transmission Electron Microscope (TEM). The TEM image of MCM-41 (Fig. 2(a)) indicates that the size of SiO2 nanoparticles was about 40–70 nm with mesoporous structure. TEM image of Fe/MCM-41 (Fig. 2(b)) revealed that Fe2O3 with a small particle size was embedded on the mesoporous SiO2. Fig. 2(c) and (d) showed that rGO sheets were coated by the mesoporous silica nanoparticles and rGO can keep layered structure in these two materials.
 |
| | Fig. 2 TEM images of (a) MCM-41, (b) Fe/MCM-41, (c) rGO/MCM-41 and (d) rGO-Fe/MCM-41. | |
3.1.2. X-ray diffraction (XRD) analysis. The X-ray diffraction (XRD) analysis was used to identify the structural composition of the catalyst materials. As shown in Fig. 3(a), the characteristic diffraction peak (d100) indicated the symmetry of the materials and that the Fe/MCM-41 and rGO-Fe/MCM-41 catalysts exposes a typical MCM-41 mesoporous structure.22 Compared with MCM-41, the peak intensity at d100 obviously decreased with the addition of rGO, suggesting that there is less ordered structure formed in rGO/MCM-41 catalyst.23 And the addition of Fe can also decrease the ordered structure, which can be inferred by the comparison of d100 peak intensity between MCM-41 and Fe/MCM-41.24 Additionally, the diffraction peak (d100) of was shifted toward higher 2θ values, indicating that the incorporation of rGO into the extra framwork.8 Fig. 3(b) showed the large angle XRD of catalysts, the broad peak at approximately 23° (for line (1)–(4)) indicated the existence of amorphous silica. A well-defined characteristic peak at a 2θ value of 11.6° (d001) can be observed for line (5), which corresponds to GO.25 However, this peak disappeared for the rGO/MCM-41 and rGO-Fe/MCM-41 catalysts, which indicates that GO has been reduced by thermal reduction in the process of preparation.26 For the Fe/MCM-41 catalyst, a small peak at a 2θ value of 35.6° (d110) corresponding to α-Fe2O3 was observed. For the rGO-Fe/MCM-41 catalyst, the α-Fe2O3 crystal structure was determined according to 2θ at 31.9° (d113) and 34° (d116) (JCPDS card no. 40-1139). According to the previous report,27 α-Fe2O3 has been proven to possess excellent catalytic capacity in the heterogeneous Fenton process.
 |
| | Fig. 3 XRD patterns of (a) small angle XRD patterns of synthesized samples; (b) large angle XRD patterns of synthesized samples. | |
According to the Scherrer equation (eqn (1)), the grain sizes of the rGO-Fe/MCM-41 and Fe/MCM-41 were 0.574 nm and 0.543 nm, respectively.
| |
D = Kγ/Bcos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ θ
| (1) |
K is the Scherrer constant of 0.89,
B was the full width at half maximum of the diffraction peak of the sample,
θ was diffraction angle,
γ was X-ray wavelength of 0.154056 nm.
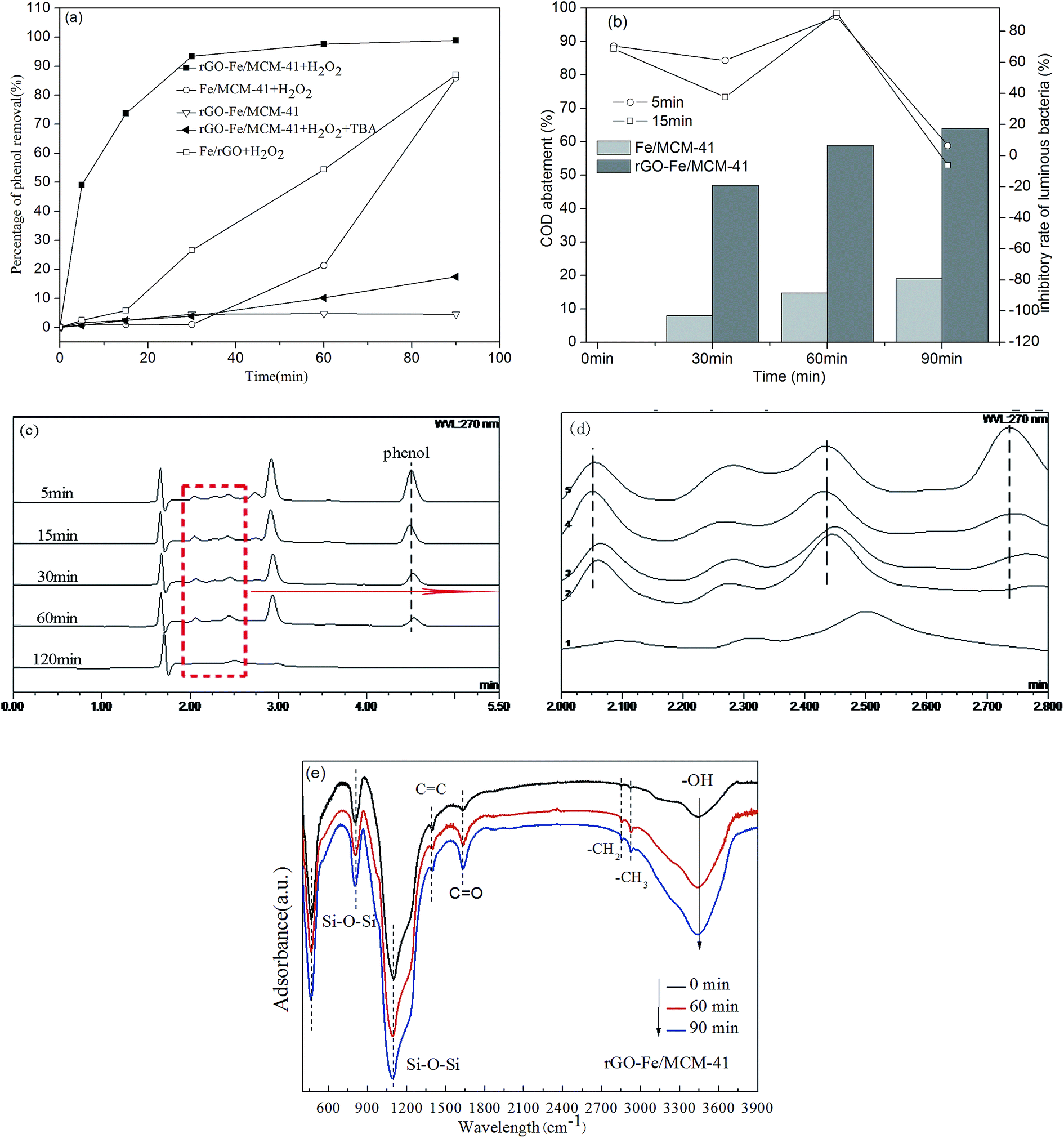
3.1.3. XPS analysis. The binding energy of elements C, Si and Fe on the surface of the rGO-Fe/MCM-41 catalyst and element C of the GO were studied with the XPS spectrum presented in Fig. 4. As shown in Fig. 4(a), there is a small increase in the binding energy of Si 2p (104.1 eV) for the rGO-Fe/MCM-41 catalyst compared with the energy for pure SiO2 (103.5 eV). This upward shifting might be due to the strong interaction between graphene and SiO2.8 In Fig. 4(b), the peaks at 711.09 eV and 724.88 eV for Fe 2p3/2 and Fe 2p1/2 showed Fe in the α-Fe2O3 form. In Fig. 4(c and d), three types of functional groups ofGO,26 namely, sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.4 eV), epoxy C–O (286.7 eV) and carboxyl CO (288.4 eV), were observed. Compared with Fig. 4(d), GO was proven to be partially reduced on the rGO-Fe/MCM-41 catalyst (Fig. 4(c)), which was confirmed by the decreasing area of sp2 CC bonds (from 39237 to 12180 at 284.6 eV)28 and the oxygen-containing carbon (from 50527 to 11559 for the epoxy C–O at 286.5 eV and from 9246 to 6105 for the carboxyl CO at 288.0 eV).
C (284.4 eV), epoxy C–O (286.7 eV) and carboxyl CO (288.4 eV), were observed. Compared with Fig. 4(d), GO was proven to be partially reduced on the rGO-Fe/MCM-41 catalyst (Fig. 4(c)), which was confirmed by the decreasing area of sp2 CC bonds (from 39237 to 12180 at 284.6 eV)28 and the oxygen-containing carbon (from 50527 to 11559 for the epoxy C–O at 286.5 eV and from 9246 to 6105 for the carboxyl CO at 288.0 eV).
 |
| | Fig. 4 XPS of (a) Si 2p spectrum; (b) Fe 2p core-level spectrum; (c) C 1s spectrum of rGO-Fe/MCM-41 catalyst; (d) C 1s spectrum of GO. | |
3.1.4. FTIR spectroscopy analysis. The FTIR spectra of rGO-Fe/MCM-41, rGO/MCM-41, MCM-41, Fe/MCM-41 and GO are shown in Fig. 5. For the MCM-41 and modified MCM-41 materials (lines (a–d)), the band in the range of 3600–3000 cm−1 is related to the Si–OH characteristic peaks. The band at 1640 cm−1 is attributed to the stretching vibration of –OH groups. The band in the range of 1240 to 1080 cm−1 corresponds to the asymmetric stretching vibration mode of Si–O–Si, the band at 803 cm−1 is linked with symmetric Si–O–Si stretching and the band at 465 cm−1 is assigned to a Si–O bending mode.23 No difference was observed in the FTIR spectra of Fe/MCM-41 and MCM-41, which indicates that the introduction of Fe does not disrupt the skeleton of MCM-41. Compared with the FTIR spectra of GO, the carbonyl bands (CO) at 1720 cm−1, –OH at 3425 cm−1 and epoxy C–O at 1401 cm−1 were significantly decreased for rGO/MCM-41 and rGO-Fe/MCM-41, demonstrating that the effective reduction of GO sheets25,26 occured during high temperature calcination under N2 atmosphere.
 |
| | Fig. 5 FTIR spectra of different catalysts. | |
3.2. Performance of the system
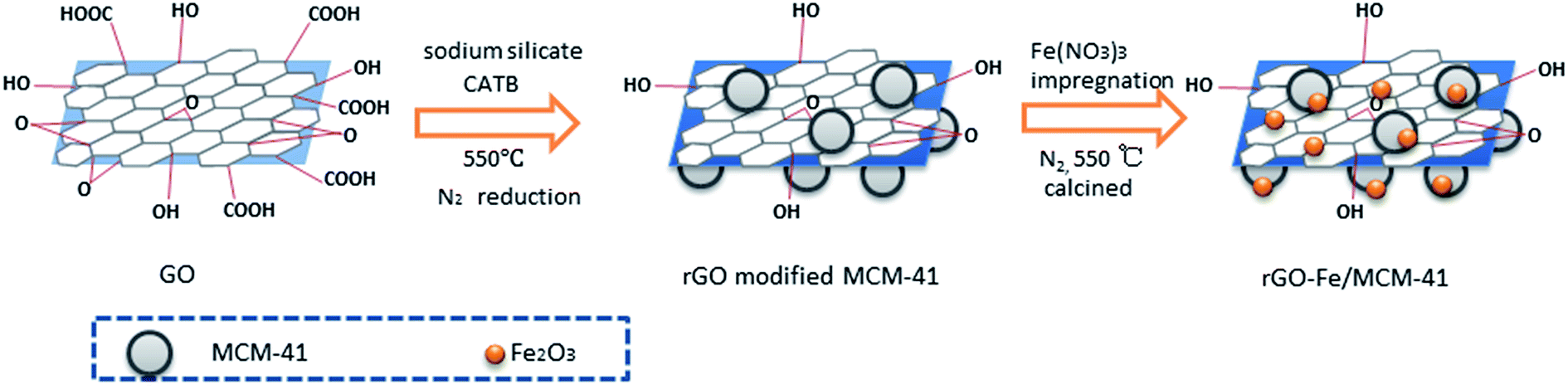
The catalytic ability of Fe/MCM-41 and Fe/MCM-41@ for phenol degradation was compared. The result illustrated that phenol and COD removals were 85.9% and 18.5%, respectively, using Fe/MCM-41 catalyst, which were higher than that using Fe/MCM-41@ catalyst (73% and 15.1% respectively). So the MCM-41 carrier for other catalysts was prepared by the method which is for Fe/MCM-41 catalyst. The phenol and COD removals in different systems using rGO-Fe/MCM-41 or Fe/MCM-41 as the Fenton catalyst are shown in Fig. 6(a and b). If no catalyst was added to the system, only 2.64% of the phenol was removed after a 90 min reaction, which may be due to the volatilization of phenol. When rGO-Fe/MCM-41 was used as the Fenton catalyst, the phenol and COD removals were over 95% and 44%, respectively, after only 30 min of reaction. After 90 min, COD removal reached 62.9%. This result was obviously higher than that in the adsorption experiment. Without H2O2, the adsorptive removal of phenol by rGO-Fe/MCM-41 was only 4.57% after 90 min. So, it can be conferred that the most phenol was catalytically degraded by the rGO-Fe/MCM-41 catalyst. Comparably, when Fe/MCM-41 was used as the Fenton catalyst, there was only 1.0% of phenol removal after 30 min reaction and 85.9% after 90 min reaction. COD removal efficiency was only 18.5% after 90 min reaction. Without carrier, Fe/rGO catalyst possesses lower catalytic activity than rGO-Fe/MCM-41. That is, using Fe/rGO catalyst, 52% of phenol and 17% of COD is removed after 60 min, and 86.1% of phenol and 21% of COD is removed after 90 min reaction. After the dope of rGO, higher activity was observed for rGO-Fe/MCM-41 and Fe/rGO catalysts than Fe/MCM-41 catalyst. So the rGO-Fe/MCM-41 catalyst showed the highest catalytic capacity. The incorporation of rGO may increase the activity of the catalyst, which attributed to higher electron mobility capacity of rGO. Additionally, according to the result of BET analysis, the incorporation of rGO can significantly increase the specific surface area of the catalyst, which leads to active components loading much more uniformly29,30 and promotes a higher degree of adsorption of pollutants on the surface of the catalyst. This would facilitate the acceleration of the catalytic reaction. Simultaneously, rGO also provides higher electron mobility to effectively separate the charges, which thus increases the catalytic capacity.31 Additionally, the dissolved iron concentrations in the solution were 0.825 mg L−1 for the rGO-Fe/MCM-41 catalyst and 1.843 mg L−1 for the Fe/MCM-41 catalyst, respectively, which indicated that the doping of rGO can also increase the stability of the catalyst.
 |
| | Fig. 6 (a) Comparison of phenol removal in different system; (b) comparison of COD removal using rGo-Fe/MCM-41 or Fe/MCM-41 as Fenton catalyst and phenol degradation on luminous bacteria using rGo-Fe/MCM-41; (c and d) phenol degradation liquid phase diagram; (e) FTIR spectra of rGo-Fe/MCM-41. Reaction conditions: pH = 3.0, catalyst dosage = 0.1 g L−1, temperature = 25 °C, initial phenol concentration = 100 mg L−1 and H2O2 dosage = 10 mmol L−1, TBA (C = 10 mmol L−1). | |
As reported, ˙OH was the main oxidative free radical in the Fenton reaction. The possible reactions are shown below in eqn (2)–(10)9,32 To further demonstrate the effect of ˙OH during the degradation of phenol, an additional experiment was carried out. tert-Butyl alcohol (TBA, 10 mmol L−1), a type of radical scavenger, was put into the Fenton reaction system at the beginning with rGO-Fe/MCM-41 as the catalyst. The phenol removal efficiency decreased from 98.8% to 17.4% (Fig. 6(a)) after a 90 min reaction, which demonstrated that the hydroxyl radical played a key role in the degradation of phenol in this system.
| |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) FeII + ˙OH → FeIII + OH− FeII + ˙OH → FeIII + OH−
| (2) |
| | |
FeIII + ˙OOH → FeII + OOH−
| (3) |
| | |
Fe3+ + H2O2 → Fe2+ + ˙OOH
| (4) |
| | |
Fe2+ + H2O2 → Fe3+ + ˙OH− + ˙OH
| (5) |
| | |
Fe2+ + ˙OH → Fe3+ + ˙OH−
| (6) |
| | |
Fe3+ + ˙OOH → Fe2+ + O2 + ˙H+
| (7) |
| | |
Fe2+ + ˙OOH → Fe3+ + OOH−
| (8) |
| | |
H2O2 + ˙OH → H2O + ˙OOH
| (9) |
| | |
˙OOH + ˙OH → H2O + O2
| (10) |
The toxicity change of the intermediate during the Fenton catalytic oxidation of phenol using rGO-Fe/MCM-41 as catalyst was evaluated on luminescent bacteria vibrio fischeri (Fig. 6(b)). The results showed the toxicity change tendency was similar after mixing with luminescent bacteria for 5 min and 15 min. The inhibitory rate initially decreased during the first 30 min reaction and increased significantly to 89% after 60 min of reaction. Finally, after 90 min of reaction, the inhibitory rate was down to approximately zero. At the beginning, the decrease of toxicity may be ascribed to the decomposition of phenol. Next, intermediates were formed,33,34 which was observed in the HPLC spectrum (Fig. 6(c and d)). The retention time of 2.35–2.55 min was associated with benzoquinone, which reached a maximum when the reaction time was 60 min; thus, the toxicity reached its highest value. After 90 min, these peaks decreased significantly. Simultaneously, the peaks in retention time of 2.0 to 2.1 min for hydroquinone, 2.7 to 2.8 min for resorcinol and 2.9 to 3.0 min for catechol disappeared. These results showed that the toxic intermediates can be further degraded, which lead to the decrease in toxicity.
The FTIR spectra of the catalyst at different reaction times were shown in Fig. 6(e). At each reaction time, the FTIR spectra of the catalyst were basically the same, indicating that the structure of the rGO-Fe/MCM-41 catalyst did not change over time. The absorption bands at 3300 to 3600 cm−1 associated with –OH group increased significantly, which showed that more alkaline conditions occurred on the surface of the catalyst. The increase of the bands at 2700 to 3000 cm−1, associated with –CH2, –CH3 groups, and at 1500 to 1700 cm−1 for the CO group35 with the reaction time indicated that many intermediates were formed.
3.3. The optimization of the rGO-Fe/MCM-41 catalyst
3.3.1. Iron loading and GO dosage. The effect of iron loading dosage on the Fenton catalytic oxidation of phenol using the rGO-Fe/MCM-41 catalyst was investigated by varying the iron loading dosage (the actual Fe loading dosages are 1.2%, 3.8% and 8.9% using ICP analysis). The results are shown in Fig. 7(a). The amount of iron has a considerable effect on the catalyst activity. When iron loading dosages were 1.2%, 3.8% and 8.9%, the degradation efficiencies of phenol were 64%, 74% and 91%, respectively, after 15 min of reaction. This may be because increasing the iron loading is equivalent to increasing the number of active sites, which in react with hydrogen peroxide to generate more hydroxyl free radicals.36 After 30 min, the degradation efficiency of phenol was over 91%. However, with the increase in iron loading, more ions dissolved. When the iron loading dosages were 1.2% and 3.8%, the concentration of iron ions in the solution was lower than 1 mg L−1. After the iron loading dosage achieved 8.9%, the concentration of iron ions in the solution was as high as 4.87 mg L−1. Therefore, in this study, an iron loading dosage of 3.8% was chosen as the optimal dosage.
 |
| | Fig. 7 The effect of parameters on phenol degradation: (a) the effect of iron loading; (b) the effect of graphene dosage; (c) the effect of calcination temperature; (d) the removal of COD by different calcination temperature; (e) H2O2 decomposition for different calcination temperature. Reaction conditions: pH = 3.0, catalyst dosage = 0.1 g L−1, temperature = 25 °C, initial phenol concentration = 100 mg L−1 and H2O2 dosage = 10 mmol L−1. | |
Fig. 7(b) showed the effect of GO dosage on phenol oxidation. When GO dosage increased from 10 mL to 30 mL, a positive impact on the phenol degradation was observed. This may be ascribed to the increase of GO promoting electron transfer.14 When GO dosage further increased to 40 mL, there was a negative impact on the phenol degradation, which may be observed because too much GO covers active sites and thus decreases the reaction activity. A GO dosage of 30 mL was found to be optimal.
The kinetics of phenol degradation by the heterogeneous Fenton reaction using different rGO-Fe/MCM-41 catalysts were in good agreement with the first order kinetics. The results also indicated that k values were the highest when iron loading dosages was 8.9%, and the GO dosage was 30 mL.
3.3.2. Calcination temperature. The calcination temperature is an important factor affecting the catalyst activity. In our study, two steps of calcination (the first step is after doping the GO, the second is after rGO modified MCM-41 was impregnate ions) were denoted as calcination I and calcination II. Fig. 7 indicated that the phenol degradation efficiency, COD and decomposition of H2O2 changed significantly with the calcination temperature. Calcination I showed greater influence on the activity than calcination II. When calcination I was performed at 550 °C, the degradation efficiency of phenol and COD reached 87.8% and 42.3% after 30 min of reaction, which was higher than the efficiencies at 400 °C and 750 °C. Simultaneously, the decomposition of H2O2 was the most obvious at 550 °C, which would lead to the formation of more hydroxyl radicals. So calcination I at 550 °C was chosen at the optimum. For calcination II, higher temperatures (550 and 750 °C) were more beneficial for the degradation and decomposition of H2O2. Although there was a slight increase in the degradation and decomposition of H2O2 at 750 °C compared to that at 550 °C, more iron ions were dissolved at 750 °C (Table 1). So a calcination temperature of 550–550 °C was chosen as optimal for the rGO-Fe/MCM-41 catalyst.
Table 1 Iron solution value of different calcination temperature
| T/°C |
400–400 |
400–550 |
400–750 |
550–400 |
550–550 |
550–750 |
750–400 |
750–550 |
750–750 |
| C (Fe)/mg L−1 |
0.698 |
0.712 |
0.767 |
0.634 |
0.825 |
0.974 |
0.612 |
0.411 |
0.149 |
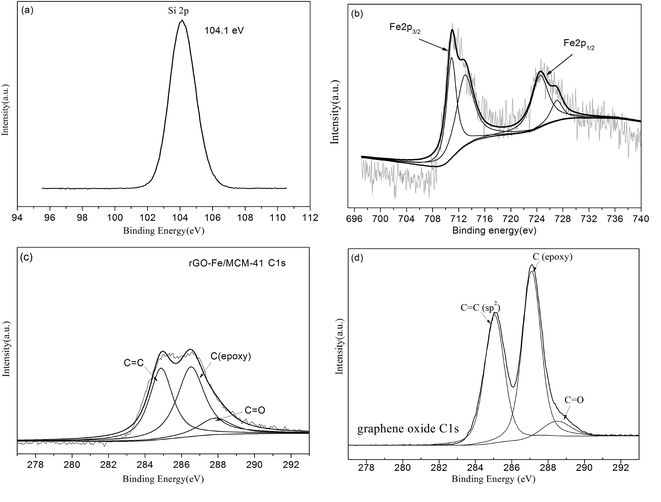
To fully understand the effect of calcination I on the formation of rGO, Raman scattering (Fig. 8(a)) is chosen to characterize the electronic structure of the carbon materials. When calcination II was performed at 550 °C, calcination I temperatures of 400, 550 and 750 °C were chosen. For GO and all the catalysts calcined at different calcination I temperatures, two main bands at 1600 cm−1 (G band) and 1350 cm−1 (D band) were observed.28 Moreover, the intensity ratios (ID/G = 1.053) of the well-documented D band and G band for rGO-Fe/MCM-41 calcined at 550–550 °C is higher than that of GO (ID/G = 1.020) and the other catalysts. According to the literature, this intensity ratio increase suggests the improvement of the disordered graphene sheets26 and the effective reduction of GO during the hydrothermal process.37 In Section 3.1, the occurrence of rGO was also demonstrated by the results of XRD, XPS and FTIR analysis. The reduction of GO into rGO can accelerate electron transfer and thus lead to an increase in phenol degradation.
 |
| | Fig. 8 The effect of calcination temperature for materials: (a) Raman spectra of the first calcination temperature of materials and GO (400–550, 550–550, 750–550); (b) XRD of the second calcination temperature of materials (550–400, 550–550, 550–750). | |
The influence of calcination II was studied using XRD analysis. When calcination I was performed at 550 °C, calcination II temperatures of 400, 550 and 750 °C were chosen. It can be observed that all of the above materials exhibited a strong peak in the 2θ range of 20 to 25°, indicating that the MCM-41 mesoporous structure was not damaged. These peaks were broadened and shifted slightly to higher angle with the increase of calcination II temperature. No other peaks could be found from the XRD of the catalyst calcined at 550 to 750 °C, which may be due to low iron loading and the high degree of dispersion.24 According to the results in Section 3.1.2, α-Fe2O3 crystal structure was determined for the catalyst calcined at 550 to 550 °C, while a low peak at 35.5° (d101) corresponding to α-FeOOH was observed for the catalyst calcined at 550–400 °C. The different calcination temperatures affect the iron crystal morphology, which thus affect the phenol degradation efficiency.
4. Conclusions
The rGO-Fe/MCM-41 Fenton catalyst was successfully synthesized via a hybrid hydrothermal-calcination method. The catalyst had a typical mesoporous structure. XRD, TEM, XPS and FTIR spectroscopy analysis demonstrated the GO was reduced effectively to rGO and the α-Fe2O3 was observed to be well distributed. The incorporation of rGO can decrease the particle size and enhance the BET surface areas, which was beneficial to the catalytic reaction. Compared with the Fe/MCM-41 catalyst, the rGO-Fe/MCM-41 catalyst exhibited a higher activity and stability in phenol degradation. The hydroxyl radical plays a key role in this reaction. The effects of iron dosage, rGO dosage and calcination temperature were investigated. It was found that the increase of iron and rGO dosage had a positive effect on phenol degradation. However, too high of an rGO dosage may cover active sites and thus decrease the reaction activity. The influence of calcination temperature after GO doping was significantly higher than that after Fe doping. More effective reduction of GO and the formation of the α-Fe2O3 crystal structure were both observed at 550 °C.
Acknowledgements
This work was supported by International S&T Cooperation Program of China (2013DFR90290) and the Natural Science Foundation of China (No. 21177013&51578070).
Notes and references
- A. Babuponnusami and K. Muthukumar, Sep. Purif. Technol., 2012, 98, 130–135 CrossRef CAS
 .
. - P. Bautista, A. F. Mohedano, J. A. Casas, J. A. Zazo and J. J. Rodriguez, J. Chem. Technol. Biotechnol., 2008, 83, 1323–1338 CrossRef CAS .
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS .
- I. Fechete, Y. Wang and J. C. Védrine, Catal. Today, 2012, 189, 2–27 CrossRef CAS .
- F. Tomul, Chem. Eng. J., 2012, 185–186, 380–390 CrossRef CAS .
- C. Zhou, L. Sun, A. Zhang, X. Wu, C. Ma, S. Su, S. Hu and J. Xiang, Chemosphere, 2015, 125, 16–24 CrossRef CAS PubMed .
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ChemSusChem, 2011, 4, 1712–1730 CrossRef CAS PubMed .
- A. C. Pradhan and K. M. Parida, J. Mater. Chem., 2012, 22, 7567 RSC .
- X. Hu, B. Liu, Y. Deng, H. Chen, S. Luo, C. Sun, P. Yang and S. Yang, Appl. Catal., B, 2011, 107, 274–283 CrossRef CAS .
- S. Guo, G. Zhang, Y. Guo and J. C. Yu, Carbon, 2013, 60, 437–444 CrossRef CAS .
- G. Huang, C. Zhang, Y. Long, J. Wynn, Y. Liu, W. Wang and J. Gao, Nanotechnology, 2013, 24, 395601 CrossRef PubMed .
- Y. P. L. Hae-Kyung Jeong, R. J. W. E. Lahaye and M.-H. Park, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef .
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef .
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848 CAS .
- F. L. Y. L. Xijun Hu, L. M. Cheung, K. F. Chan, X. S. Zhao and G. Q. Lu, Catal. Today, 2001, 68, 129–133 CrossRef .
- Y. Ling, M. Long, P. Hu, Y. Chen and J. Huang, J. Hazard. Mater., 2014, 264, 195–202 CrossRef CAS .
- R. E. Offeman and W. S. Hummers Jr, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef .
- T. Wu and J. D. Englehardt, Environ. Sci. Technol., 2012, 46, 2291–2298 CrossRef CAS PubMed .
- W. C. Cuili Fu, A. Guo, J. Li and X. Luo, Rock and Mineral Analysis, 2012, vol. 31, pp. 621–626 Search PubMed .
- M. R. Majidi, S. Ghaderi, K. A. Zeynali and H. Dastangoo, Food Anal. Method., 2015, 8, 2011–2019 CrossRef .
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 CAS .
- R. Silva-Rodrigo, H. Castillo Jimenez, A. Guevara-Lara, J. A. Melo-Banda, A. Olivas Sarabia, A. I. Reyes de la Torre, F. Morteo Flores and A. Castillo Mares, Catal. Today, 2015, 250, 2–11 CrossRef CAS .
- S. Samanta, S. Giri, P. U. Sastry, N. K. Mal, A. Manna and A. Bhaumik, Ind. Eng. Chem. Res., 2003, 42, 3012–3018 CrossRef CAS .
- M. Xia, M. Long, Y. Yang, C. Chen, W. Cai and B. Zhou, Appl. Catal., B, 2011, 110, 118–125 CrossRef CAS .
- P. Shi, R. Su, S. Zhu, M. Zhu, D. Li and S. Xu, J. Hazard. Mater., 2012, 229–230, 331–339 CrossRef CAS .
- X.-C. R. Huai-Ping Cong, P. Wang and S.-H. Yu, ACS Nano, 2012, 6, 2693–2703 CrossRef PubMed .
- F. Chen, Y. Li, W. Cai and J. Zhang, J. Hazard. Mater., 2010, 177, 743–749 CrossRef CAS .
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice Jr and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS .
- S. H. Hsieh, W. J. Chen and C. T. Wu, Appl. Surf. Sci., 2015, 340, 9–17 CrossRef CAS .
- Y.-H. Zhao, Z.-K. Wu and S.-L. Bai, Composites, Part A, 2015, 72, 200–206 CrossRef CAS .
- S. G. Babu, R. Vinoth, D. P. Kumar, M. V. Shankar, H. L. Chou, K. Vinodgopal and B. Neppolian, Nanoscale, 2015, 7, 7849–7857 RSC .
- M. J. M. Rafael Gonzalez-Olmos, A. Georgi, F.-D. Kopinke and I. Oller, Appl. Catal., B, 2012, 125, 51–58 CrossRef .
- A. Gentili, S. Marchese and D. Perret, TrAC, Trends Anal. Chem., 2008, 27, 888–903 CrossRef CAS .
- A. B. Ahmed, B. Jibril, S. Danwittayakul and J. Dutta, Appl. Catal., B, 2014, 156–157, 456–465 CrossRef CAS .
- Y.-C. Lee, S.-J. Chang, M.-H. Choi, T.-J. Jeon, T. Ryu and Y. S. Huh, Appl. Catal., B, 2013, 142–143, 494–503 CrossRef CAS .
- H. Lan, A. Wang, R. Liu, H. Liu and J. Qu, J. Hazard. Mater., 2015, 285, 167–172 CrossRef CAS PubMed .
- J. Cao, G. Q. Qi, K. Ke, Y. Luo and W. Yang, J. Mater. Sci., 2012, 47, 5097–5105 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.