
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of click thiol-ene/thiol-epoxy thermosets by controlled photo/thermal dual curing sequence
Dailyn
Guzmán
a,
Xavier
Ramis
 b,
Xavier
Fernández-Francos
b and
Angels
Serra
*a
b,
Xavier
Fernández-Francos
b and
Angels
Serra
*a
aDepartment of Analytical and Organic Chemistry, University Rovira i Virgili, C/Marcel·lí Domingo s/n, 43007, Tarragona, Spain. E-mail: angels.serra@urv.cat; Tel: +34-977559558
bThermodynamics Laboratory, ETSEIB University Politècnica de Catalunya, Av. Diagonal 647, 08028, Barcelona, Spain
First published on 20th November 2015
Abstract
A new sequential two steps photo and thermal process for the preparation of click thiol-ene/thiol-epoxy thermosets is described. Commercially available diglycidyl ether of bisphenol A (DGEBA), triallylisocyanurate (TAIC) and pentaerythritol tetrakis (3-mercaptopropionate) (PETMP) were combined to produce tailored materials with a 75, 50 and 25% of thiol-ene/thiol-epoxy networks. A photoinitiator was used to trigger the radical thiol-ene polymerization and a latent amine precursor was used to start the base-catalyzed thiol-epoxy click reaction. Neat thiol-ene and thiol-epoxy materials were prepared and taken as the references. The use of a latent amine precursor in adequate proportion and under suitable reaction conditions allowed us to reach a dual system with two well-defined steps, stable intermediate materials and well-controlled structure after the first curing stage and at the end of the curing process. This process overcomes some limitations observed in analogous curing systems reported previously such as the absence of latency for the second curing stage leading to unstable materials in the intermediate stage. Both chemical reactions were studied by FTIR and calorimetry. The latency of the different formulations was studied by DSC and rheometry. The materials prepared were characterized by thermal mechanical analysis and thermogravimetry.
1. Introduction
Dual curing combines two different and compatible polymerization reactions, which take place separately in a well-controlled way. This process is used to obtain stable materials after the first stage that maintain the ability, upon application of a second stimulus, to further react and achieve the final properties desired.1,2 Dual processes usually combine two different curing methodologies, being usually photocuring followed by thermal curing.3,4 The interest in such complex curing procedures is because of the processing advantages, such as: (a) application one day and bonding of the parts at later date; (b) application at one location and shipping them to a 2nd location for assembly; (c) avoiding manufacturing bottlenecks; (d) flip chip processes-flip chip and wafer passivation enabling 3D stacking, (e) gasket and perimeter sealing of glasses in LCDs displays and (f) automated in line processes assembly.Click reactions have been extensively employed in material science because they are considered easy and fast methods of obtaining new substances, generally without the need of solvents.5–7 The orthogonality of click reactions, their simplicity, efficiency and environmentally-friendly conditions make them highly advisable for combination in two curing step procedures. This orthogonality allows these reactions to be combined in a convenient way so as to reach technological applicability.8 Thiol-click reactions are suitable candidates since thiols are able to react under mild conditions by forming radical or anionic species selecting the appropriate conditions and catalysts,9 making it possible to prepare thermosets in a controlled manner.
Thiol-ene and thiol-epoxy reactions are one of the most used in the last years to prepare new polymeric structures, linear, branched or crosslinked.10–12 Radical thiol-ene polymerization is a step-wise process consisting in the alternation between thiol radical propagation through the -ene functional group and the chain transfer reaction, which involves the abstraction of hydrogen radical from the thiol by the carbon radical. This mechanism is depicted in Scheme 1. Studies of these reactions reveal that the thiol-ene can proceed without photoinitiator,13 but their use assures a complete reaction, especially in thick specimens. One of the most commonly used photoinitiators is 2,2-dimethoxy-2-phenylacetophenone (DMPA).14 One advantage of these reactions is that they are not inhibited by oxygen. During propagation carbon radical is capable of generating peroxides, but this reaction is slow and does not contribute much to the global process.15 Although the thiol-ene reaction is a valuable strategy for the production of crosslinked polymers, it usually leads to low Tg materials.
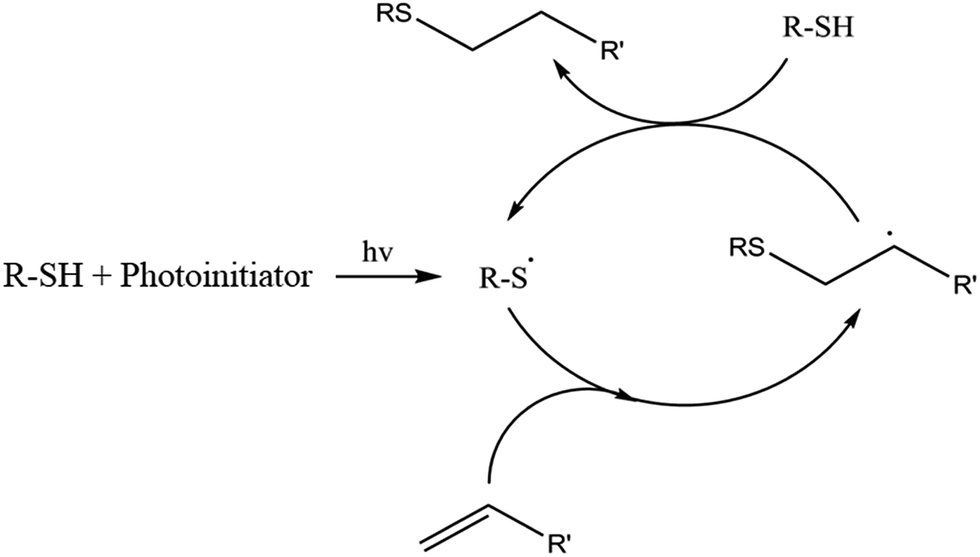 | ||
| Scheme 1 Proposed mechanism of thiol-ene reaction. | ||
In the present work the thiol-ene is combined with a thiol-epoxy click reaction16 that can be carried out under moderate temperature conditions to overcome vitrification during curing.17 This reaction usually requires tertiary amines for activation. The reaction mechanism is step-wise and consists, essentially, in a nucleophilic attack to the oxirane group by the thiol. However, the thiol group can be transformed into a more nucleophilic thiolate anion, by the presence of amines acting as a base. The true mechanism of formation of the propagating species is complex and may involve either proton exchange between the thiol and the amine or nucleophilic attack of the amine to the epoxy, depending on the basicity and nucleophilicity of the tertiary amine, or the presence of proton donors acting as co-catalysts.18 The formed nucleophilic thiolate anion attacks the oxirane ring, forming an alkoxide ion, according to the mechanism presented below. Proton transfer takes place between the protonated amine or a thiol leading eventually to the formation of another thiolate anion.19–21 The mechanism is depicted in Scheme 2.
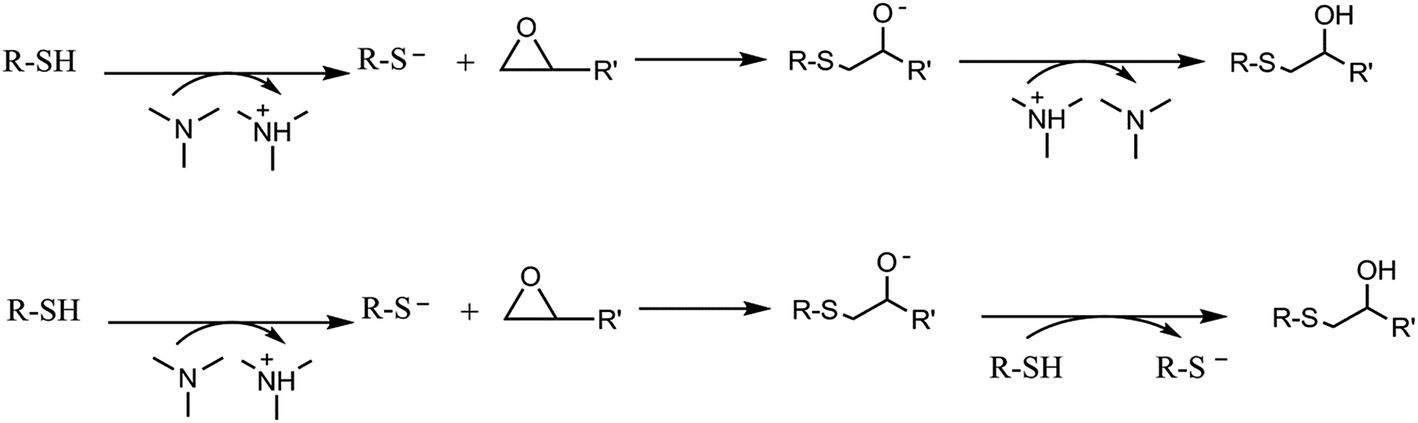 | ||
| Scheme 2 Proposed mechanism of thiol-epoxy reaction. | ||
One of the problems that arise when using this methodology is that the use of tertiary amines leads to a too short pot life.12 In a previous work we proposed the use of latent catalysts that are activated only by thermal stimuli,22 with the purpose of preparing stable one-pot curing mixtures and facilitating their application in a more comfortable way. Compounds able to release amines such as encapsulated imidazoles or ureas can be of interest not only to increase pot-life of thiol-epoxy formulations but also to permit controlled activation of the thiol-epoxy reaction in a dual-curing process.
The strategy of combining these two thiol-click reactions has been studied previously by different research groups. An advantage coming from this combination is that the thiol component can participate in both processes and therefore the networks arising form thiol-ene and thiol-epoxy reactions are covalently interconnected. Another advantage comes from the fact that both thiol-ene and thiol-epoxy reactions are step-wise so that relevant network build-up parameters during both curing stages such as gel point conversion, gel fraction or crosslinking density can be easily calculated using well-established methods,23,24 thus making it possible to tailor the curing process and the material properties in the intermediate stage and at the end of it in order to fit different processes and material requisites in a flexible way.
Cariosca et al.25 studied thiol-ene/thiol-epoxy hybrid systems for dental applications. They used a tertiary amine as the catalyst and they reported homopolymerization of epoxide side-reaction to occur, because of the low activity of thiols after the first curing stage. Carlborg et al.26 prepared thermosets from non-stoichiometric thiol-ene/thiol-epoxy mixtures using a two-stage UV curing with a photoinitiator and a photobase that were activated at different wavelengths. This methodology allowed achieving a dual curing system with an intermediate stage stable for only 24 h and they also reported that epoxy homopolymerization took place. However, it has been shown that the epoxy-thiol reaction takes place preferently while there is still remaining thiol, so that homopolymerization is not observed in epoxy-thiol systems.18 Saharil et al.27 used non-stoichiometric two-stage thiol-ene/thiol-epoxy systems for the manufacture of microfluidic chips. In the first step the photocuring was carried out, whereas the second stage was done at 70 °C. This paper reported that the intermediate material was stable for 1–2 days, but the thiol-epoxy reaction occurred slowly at room temperature.
In the present work, a new dual curing system combining sequential thiol-ene/thiol-epoxy reactions, overcoming the above stability limitations and without the occurrence of undesired side-reactions, is described. The first stage is a radical thiol-ene reaction initiated by 2,2-dimethoxy-2-phenylacetophenone (DMPA) and the second one is a base-catalyzed thiol-epoxy reaction initiated by a latent catalyst (an encapsulated imidazole, LC-80 or a urea, PDU-250) that release the amine at a given temperature leading to the formation of the thiolate active species. What is sought by this methodology is to define a dual-curing system with a controlled curing sequence, with no overlapping between both curing reactions, and with sufficient stability in the intermediate stage, so that the second curing process is only activated at the time deemed appropriate. There is an important advantage coming from the fact that the first process is activated by UV-light and the second one is thermal and latent. This would make it possible, for instance to apply the material to a substrate and irradiate to promote the first reaction, then join two parts that adhere by contact with the partially cured material so that the final reaction and permanent bonding is triggered by temperature only when it is considered safe, something not possible if the second reaction is activated by UV light as well, or else is not stable enough. The proportions of -ene and epoxy compound in the formulation have been varied so as to control the extent of each process and consequently the characteristics of the intermediate and final materials. Both curing stages have been studied by DSC and FTIR. The latency and stability at the intermediate stage, as well as that of the unreacted formulation, have been evaluated. The thermal and mechanical properties of the intermediate and final materials have also been characterized.
2. Experimental part
2.1. Materials
Diglycidylether of bisphenol A (DGEBA, GY240, Huntsman, epoxy equivalent 182 g eq.−1) was dried at 80 °C under vacuum for 6 h. The latent amine precursors were PDU-250 (N,N-dimethyl phenyl urea) and an encapsulated imidazole (Technicure® LC-80) both from AC Catalysts. Pentaerythritol tetrakis(3-mercaptopropionate) (PETMP), 1,3,5-triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TAIC) and 2,2-dimethoxy-2-phenylacetophenone (DMPA) from Sigma-Aldrich were used as received. The structures of the compounds used are shown in Scheme 3.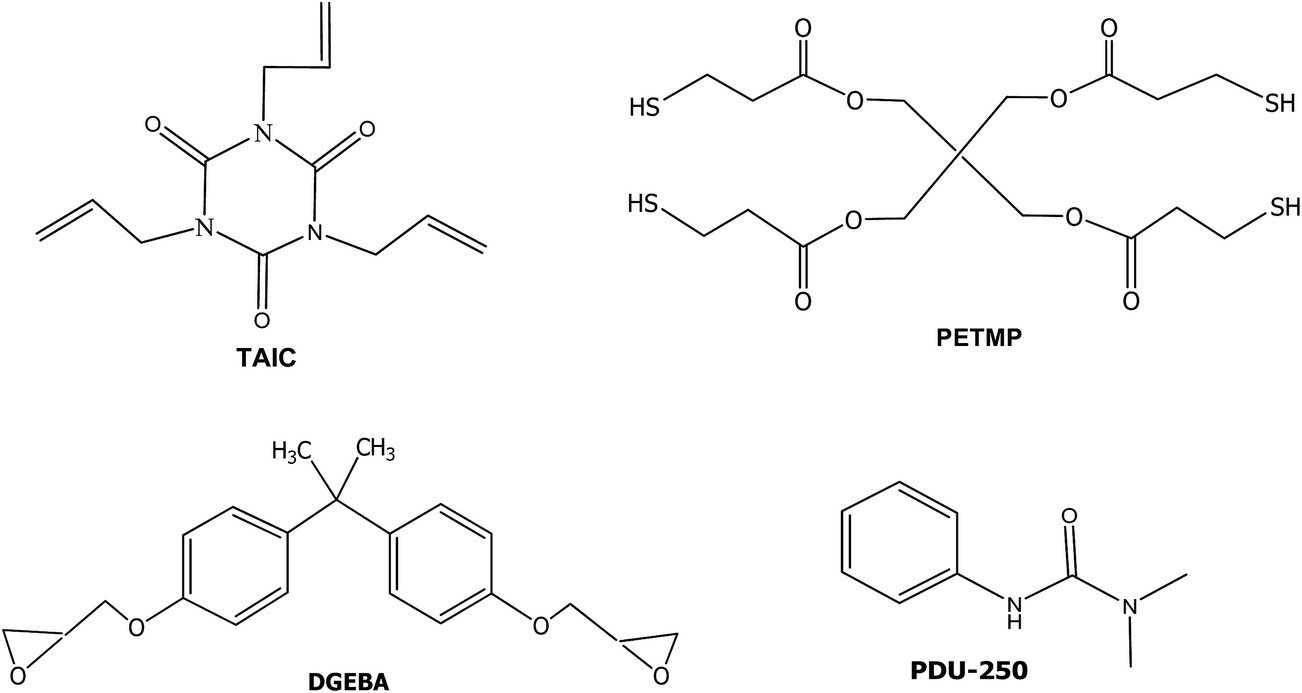 | ||
| Scheme 3 Chemical structures of the starting compounds. | ||
2.2. Preparation of the curing mixtures
For the preparation of the curing mixtures it was initially necessary to prepare two different stoichiometric mixtures: TAIC-PETMP (thiol-ene) and DGEBA-PETMP (thiol-epoxy) both without any catalyst. Different proportions of these mixtures were mixed just before needed with a spatula to prepare homogeneous formulations (named as 75A%–25E%, 50A%–50E% and 25A%–75E%), being A the thiol-ene mixture and E the thiol-epoxy. To these formulations 0.1 phr of DMPA (parts per hundred of total mixture) as photoinitiator was added and different proportions of PDU-250 or 1 phr of LC80 as latent amine catalyst. For comparison purposes, neat thiol-ene and thiol-epoxy formulations were prepared with the corresponding catalysts.The different formulations have been chosen so that they have significant differences between them at the intermediate stage, in terms of network structure and properties. The critical gelation ratio rc for a step-wise reaction is given by the following expression:
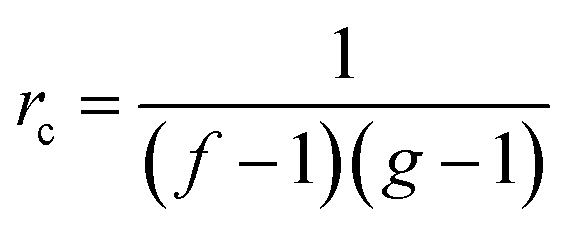 | (1) |
2.3. Characterization techniques
Samples of the different compositions were photocured at 35 °C, in a Mettler DSC-821e calorimeter appropriately modified with a Hamamatsu Lightning cure LC5 (Hg–Xe lamp) with two beams, one for the sample side and the other for the reference side. 5 mg samples were cured in open aluminium pans in nitrogen atmosphere. Two scans were performed on each sample, the second one needed to subtract the thermal effect of the radiation. The method consisted of 1 min without irradiation for temperature stabilization, followed by 1 min irradiation and finally 0.50 min without irradiation. The light intensity used was 21 mW cm−2 (total light intensity measured with a pan filled with carbon black).Larger rectangular specimens (40 mm × 10 mm × 1.5 mm) were prepared using a suitable mould. The samples were irradiated in an ultraviolet oven Dymax ECE 2000 UV-curing flood system (UV-intensity of 105 mW cm−2, 365 nm) for a period of 30 s on each face, waiting for 5 min in between to control the temperature of the sample.
The curing kinetics of the following thermal thiol-epoxy reaction was analyzed by differential scanning calorimetry (DSC) in a Mettler DSC-821e apparatus calibrated using an indium standard (heat flow calibration) and an indium–lead–zinc standard (temperature calibration). Samples of ca. 10 mg weight were analyzed under N2 atmosphere (100 mL min−1 flow rate). Dynamic curing was studied at a heating rate of 10°C min−1 from 30 to 300 °C, and isothermal experiments were carried out at 120 °C.
The glass transition temperatures (Tgs) of the samples after irradiation were determined by a dynamic DSC scan at 20 °C min−1 from −50 °C to 50 °C. The Tgs of the final thermosets were determined after two consecutive heating dynamic scans at 20°C min−1 starting at −50 °C in a Mettler DSC-822e device. The Tg value was taken as the middle point in the heat capacity step of the glass transition.
The unreacted mixtures were stored in an oven at controlled temperature of 35 °C for latency tests. The viscosity of the mixtures was measured at different storage times in a TA Instruments ARG2 rheometer equipped with disposable parallel plates. A sample gap of 0.4 mm was exactly set with an accuracy of 0.001 mm. A shear rate of 100 s−1 was applied at 35 °C after 5 min of temperature stabilization. A maximum of 2 min was needed to measure a steady value of viscosity. The values were corrected for non-uniform stresses in the parallel plates. The remaining enthalpy after storage was evaluated by DSC and compared with the enthalpy of a fresh prepared sample to determine the degree of curing occurred during storage.
A Bruker Vertex 70 FTIR spectrometer equipped with an attenuated total reflection accessory (ATR) (Golden Gate, Specac Ltd. Teknokroma) which is temperature controlled (heated single-reflection diamond ATR crystal) equipped with a liquid nitrogen-cooled mercury–cadmium–telluride (MCT) detector was used to register the FTIR spectra of uncured samples, after 1 min of UV irradiation and on fully cured samples. The spectra were registered in the wave number range between 4000 and 600 cm−1 with a resolution of 4 cm−1 and averaged over 100 scans. UV-curing was performed using a Hamamatsu Lightning cure LC5 (Hg–Xe lamp) with one beam conveniently adapted to the ATR accessory. A wire-wound rod was used to set a sample thickness of 50 μm. OPUS software was used for the analysis of the spectra. The spectra were corrected for the dependence of the penetration depth on the wavelength and normalized with respect to the absorbance of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO peaks between 1900 and 1560 cm−1 (neglecting the contribution of the overlapping tiny signal associated with the allyl group). The normalized thiol band at 2570 cm−1 was integrated and the thiol conversions after the photocuring and after thermal curing were determined as:
C and CO peaks between 1900 and 1560 cm−1 (neglecting the contribution of the overlapping tiny signal associated with the allyl group). The normalized thiol band at 2570 cm−1 was integrated and the thiol conversions after the photocuring and after thermal curing were determined as:
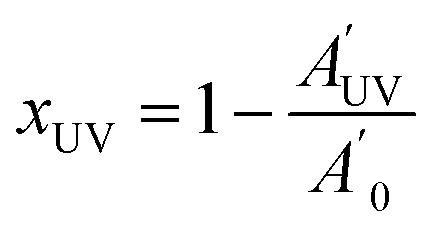 | (2) |
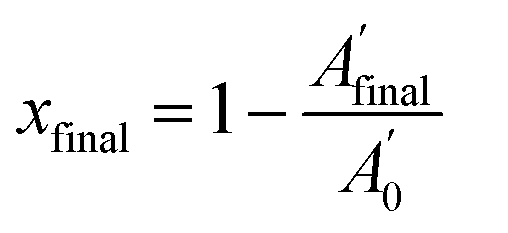 | (3) |
The thermal stability of cured samples was determined by thermogravimetric analysis (TGA), using a Mettler–Toledo TGA/DSC 1 stare system. All experiments were performed under inert atmosphere (N2 at 50 mL min−1). Pieces of the cured samples with an approximate mass of 10 mg were degraded between 30 and 600 °C at a heating rate of 10 K min−1.
Dynamic mechanical thermal analyses (DMTA) were carried out with a TA Instruments DMA Q800 analyzer. The samples were photocured as described before and then isothermally cured in a mould at 120 °C for 1 h with a postcuring at 150 °C for 1 h. Three point bending clamp was used on the prismatic rectangular samples (10 × 7.4 × 1.6 mm3) that were analyzed first of all at 5 °C min−1 to delete the thermal history and then at 2 °C min−1 from 30 to 120 °C at a frequency of 1 Hz with and oscillation amplitude of 10 μm. Young modulus was determined by the stress/strain test, under the same clamp and geometry testing conditions, at 30 °C using a force ramp of 1 N min−1 and upper force limit of 18 N.
3. Results and discussion
3.1. Calorimetric study of the curing process
An important issue concerning dual-curing processes with a photo-activated first stage and a thermally activated second stage is that the photocuring conditions and the combination of catalysts needs to be optimized in order to prevent the occurrence of any thermal reaction (in this case thiol-epoxy or epoxy homopolymerization) during the first photocuring step. While this may not be of importance when small samples are studied under well-controlled temperature conditions in a DSC, photocuring of thicker samples such as those prepared for further thermal and mechanical analysis needs to be carried out carefully, that is, using the minimum possible amount of photoinitiator and adjusting the exposure time and intensity so as to avoid an excessive temperature increase (due to the exothermicity of the thiol-ene reaction and the thermal effect of the UV light) leading to premature activation of the latent catalysts used for the second stage. Therefore, the suitable proportion of DMPA as photoinitiator was determined. The photocalorimetric study of formulations containing 0.1 or 1 phr of DMPA in different thiol-ene/thiol-epoxy formulations produced no relevant difference, possibly due to the opposing effects of increased photoinitiator in reaction rate and penetration depth. Thus, and in order to prevent undesired excessive exothermicity effects, the amount of DMPA was fixed at 0.1 phr for all the studied formulations.After that, the optimization of the catalyst in the thermal step was carried out using two formulations containing 50% of allyl (TAIC) and 50% of epoxy (DGEBA) compounds (from now 50%A–50%E) with stoichiometric amounts of tetrathiol (PETMP) and 1 phr of different base precursor (LC-80 or PDU-250).
To test whether the thermal thiol-epoxy reaction started during photocuring we determined the remaining heat of the formulation 50%A–50%E after irradiation in the UV chamber, and compared it with that of non-irradiated formulation. The dynamic DSC thermograms of the irradiated and non-irradiated samples are represented in Fig. 1.
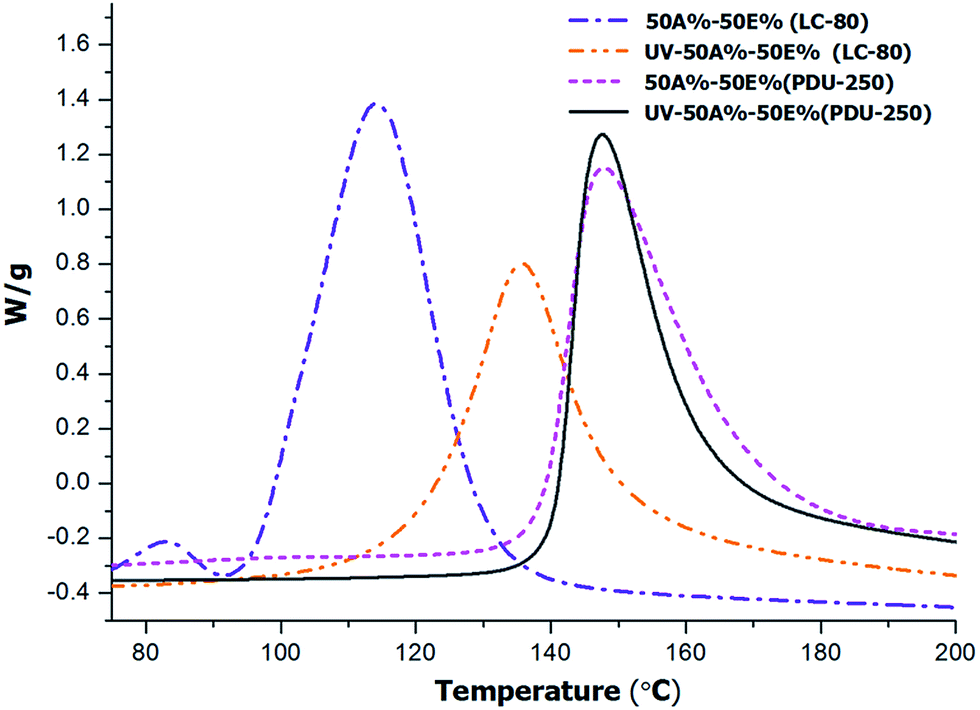 | ||
| Fig. 1 DSC thermograms corresponding to the 50%A–50%E before and after UV-irradiation for formulations containing 1 phr of PDU-250 or LC-80. | ||
In agreement with a previous study,17 some relevant differences were observed depending on the type of latent catalyst used. From the analysis of the non-irradiated samples, it is seen that LC-80 is clearly more active because of the lower curing temperature in comparison with PDU-250. However, this is also a disadvantage since the exotherm of the irradiated sample with LC-80 is entirely different from that of the initial mixture. This puts in evidence that the latent catalyst is unintentionally activated during the photoirradiation stage, leading to an overlapping of both processes. Possibly there is a too high temperature increase during the photocuring due to the exothermicity of the reaction or the thermal effect of the UV light leading to partial melting of the encapsulating polymer and early release of the imidazole catalyst. A temperature of 78 °C could be measured by spirometry in thiol-ene processes initiated by DMPA28 which indicates that the thiol-ene process is quite exothermic. In the case of the formulation with PDU-250, reaction enthalpy remains almost unchanged on comparing the exotherms before and after being exposed to photoirradiation, which indicates that the catalyst is not released due to its higher activation temperature, so that the epoxy-thiol reaction is not initiated during the first photocuring stage. Thus, PDU-250 has been selected as latent curing agent to further studies. It was not observed a significant influence of the catalyst on the Tg of the cured material, in both cases producing a value around 60 °C.
Another point to be optimized is the curing rate of the second thermal process. Although initially 1 phr of PDU-250 catalyst in reference to the global mixture was used, it was put in evidence that the quantity of PDU-250 required was different depending on the formulation. In a previous paper, we found an optimum value of 2 phr (in reference to the amount of DGEBA) for a DGEBA–thiol mixture in order to reduce curing time and temperature,17 although it was observed that a smaller amount such as 0.5 phr would also lead to complete curing but in a longer curing process. In the dual-curable formulations, the presence of a primary network originated after the photocuring stage and the dilution of the remaining epoxy and thiol groups depending on the formulation composition requires to find out the adequate proportion of amine precursor to be added to the formulation to promote complete and quick curing but, at the same time, ensuring enough stability in the intermediate stage. Different tests were done changing the proportion of PDU-250 in reference to the global mixture in non-irradiated samples. The most suitable proportions are the ones detailed in Table 1. The ratio PDU-250/DGEBA in the 75A%/25E% sample is quite high but lower proportions of catalyst led to a lower reaction heat than expected and the curing began at such high temperature that epoxides might homopolymerize. The sample 50A%/50E% was the one that cured completely in the adequate range of temperatures with less catalyst.
| Sample | PDU-250a (phr) | Initial mixture | 1st stage | 2nd stage | |||||
|---|---|---|---|---|---|---|---|---|---|
| ΔHb (J g−1) | ΔHc (kJ eq.−1) | ΔHd (J g−1) | ΔHe (kJ eq.−1) | T g f (°C) | ΔHg (J g−1) | ΔHh (kJ eq.−1) | T g i (°C) | ||
| a Parts of PDU-250 per hundred parts of the global mixture. b Enthalpy evolved by gram of curing of the initial mixture (without photocuring). c Enthalpy by equivalent epoxy of curing of the initial mixture (without photocuring). d Enthalpy evolved by gram of sample in an isothermal photocuring scan at 35 °C. e Enthalpy by equivalent of allyl group in an isothermal photocuring scan at 35 °C. f Glass transition temperature of the material after completion of the thiol-ene reaction. g Enthalpy evolved by gram of sample in dynamic thermal curing process after photoirradiation h Enthalpy evolved by epoxy equivalent in dynamic thermal curing process after photoirradiation i Glass transition temperature of the material after photo and thermal curing. | |||||||||
| Neat 100A% | — | — | — | 243 | 50 | 65 | — | — | 65 |
| 75A%/25E% | 2.5 | 86.2 | 105.0 | 229 | 64 | 24 | 94.0 | 114.5 | 63 |
| 50A%/50E% | 1 | 182.2 | 112.1 | 165 | 68 | 0 | 185.6 | 114.2 | 61 |
| 25A%/75E% | 2 | 302.9 | 126.0 | 83 | 70 | −19 | 301.0 | 125.2 | 60 |
| Neat 100E% | 2 | 409.7 | 124.8 | — | — | — | 406.5 | 123.8 | 57 |
In order to further understand the evolution of both processes and the possible overlapping among them, we first determined the reaction enthalpy of the mixtures without photoirradiation by DSC in order to evaluate the heat evolved in the thermal process without any influence of the photopolymerization stage. Fig. 2 shows the thermograms of curing of the initial non-irradiated mixtures with the compositions detailed in Table 1.
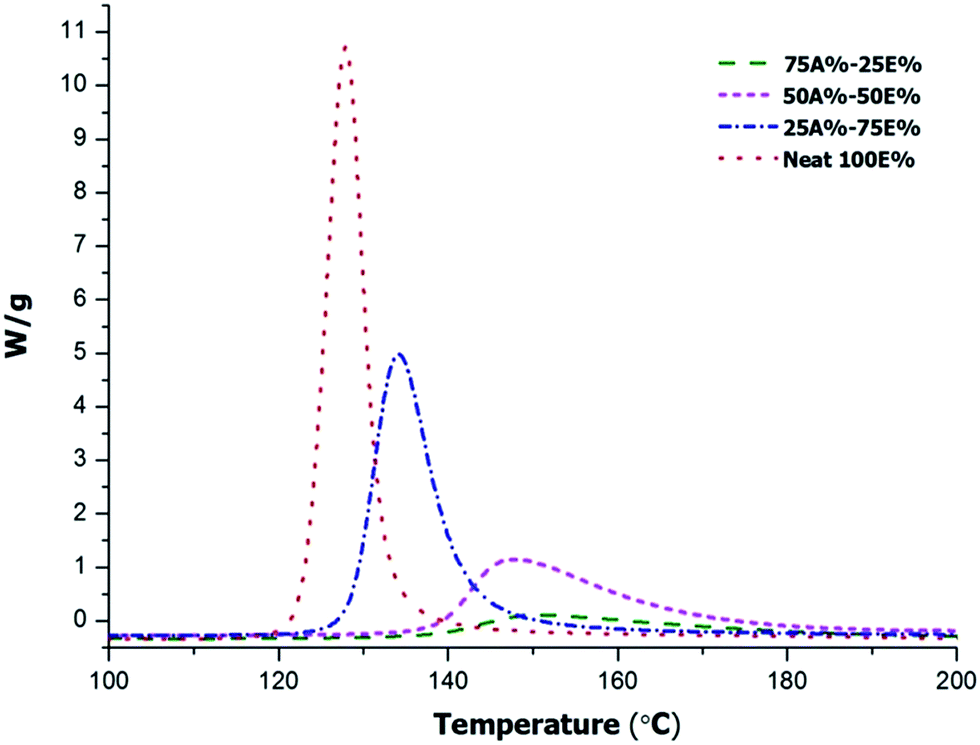 | ||
| Fig. 2 Dynamic DSC thermograms at 10°C min−1 of non-irradiated thiol-ene/thiol-epoxy mixtures with different proportions of epoxy and allyl compounds. | ||
In the curves we can see how the curing reaction is accelerated on increasing the thiol-epoxy proportion within the system. The composition of the formulation also influences the onset temperature of the reaction. It is important to note that although the formulation 75%A–25E% contains a higher proportion of PDU-250, the curing starts at higher temperature, above 140 °C because of the significant dilution of epoxy groups in the mixture, resulting in lower initiation and propagation rates. In Table 1 the enthalpies determined are collected in the first column. Although the enthalpy per gram changes in the different formulations, the value by epoxy equivalent ranges between 105–126 kJ eq.−1, since the only process is the thiol-epoxy reaction. In a previous study we determined a reaction enthalpy between 120–128 kJ eq.−1 in pure thiol-epoxy formulations.17 From the values of the table it seems that the increase of the proportion of TAIC in the formulation apparently reduces the conversion of epoxide, although there might also be some error associated with the measurement of the reaction heat in a smaller exotherm (i.e. baseline determination).
The first stage of the dual curing, corresponding to the thiol-ene reaction, was studied by photocalorimetry. In the exotherms in Fig. 3 we can appreciate that on increasing the content of allyl compound in the formulation the photocuring occurs faster. However, the reaction rate of the mixture with 100% thiol-ene (Neat 100A%) and the area below the exotherm (i.e. the reaction heat) is comparable to that of the 75A%–25E% formulation. In terms of reaction heat per amount of reacted allyl group, the reaction heat might be even lower taking into account the higher concentration of reactive allyl groups. The reason for this unexpected effect is unclear but the lower reaction heat may be caused by vitrification of the material at room temperature, as will be discussed below.
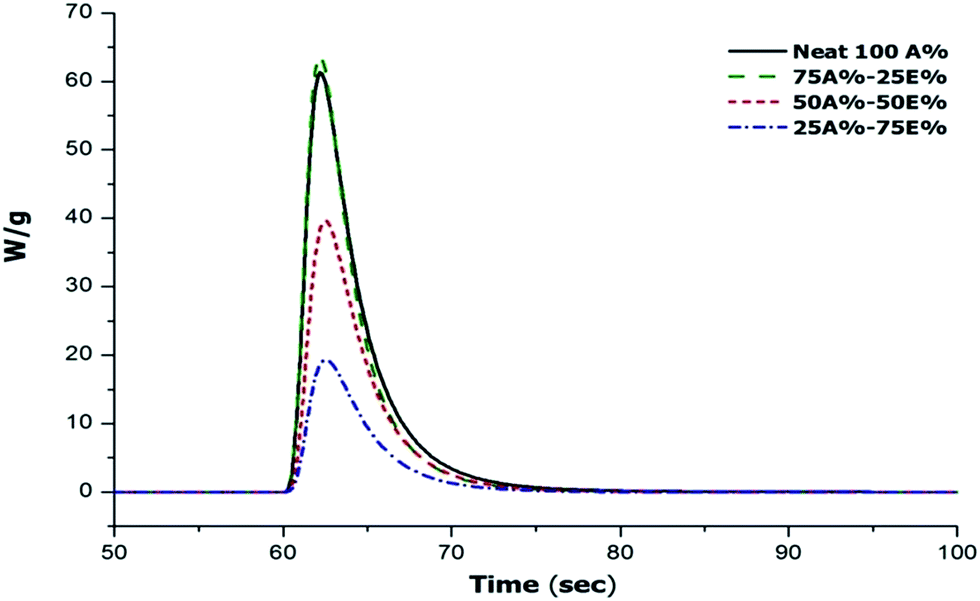 | ||
| Fig. 3 DSC thermograms corresponding to the photocuring stage at 35 °C of the mixtures thiol-ene/thiol-epoxy with different proportions. | ||
Table 1 shows the values of the heat released during the photocuring reaction measured by photocalorimetry at a controlled temperature of 35 °C. Upon decreasing the proportion of the allyl component in the formulation, the heat released during photocuring decreases but, in terms of the heat released by allyl equivalent they are similar, regardless of the proportion of the thiol-ene reaction, between 64–70 kJ eq.−1. The only exception is the lower value obtained for the neat 100A%, because of the before mentioned vitrification of the sample. The Tg values of the material at the intermediate stage were measured by DSC for the materials previously photocured in a curing chamber. In the case of the Neat 100A% formulation, it was determined after a second DSC run because of the release of some remaining heat during the first DSC scan (devitrification process). Indeed, Table 1 shows that the Tg of the completely cured neat allyl formulation is 65 °C while, in contrast, all the other formulations have a Tg in the intermediate stage below photocuring temperature, decreasing with increasing proportion of the epoxy component, because of the lower crosslinking density and the presence of significant soluble fraction containing unreacted DGEBA and thiol monomers with a plasticizing effect.
After studying both processes separately, the sequential dual-curing process was studied in order to confirm that no overlapping between both processes occurred. In Table 1 the values of the heat evolved in the thermal process in previously irradiated samples are detailed in the 2nd stage column. No relevant differences could be observed between the measured heat and that corresponding to non-irradiated samples, indicating that epoxy-thiol reaction did not take place and therefore there is no overlapping between both stages. This confirms the dual character of the curing obtained by using a latent amine precursor.
3.2. Study of the reactive processes by FTIR
Although the results of DSC analysis indicated that the dual curing process could take place in a controlled and sequential manner, with no overlapping between processes, it did not provide evidence that other side reactions such as the epoxy homopolymerization did not occur. Cariosca et al.25 determined that homopolymerization could take place to a measurable extent by monitoring the thiol band by FTIR. In the present study, we have followed a similar approach and we have irradiated the samples directly on the ATR/FTIR device with a UV lamp and then we performed the analysis of fully cured samples. The study was conducted qualitatively and quantitatively. Qualitative results are shown in the spectra of Fig. 4 and 5 for two different formulations, 75A%–25E% and Neat 100A% respectively.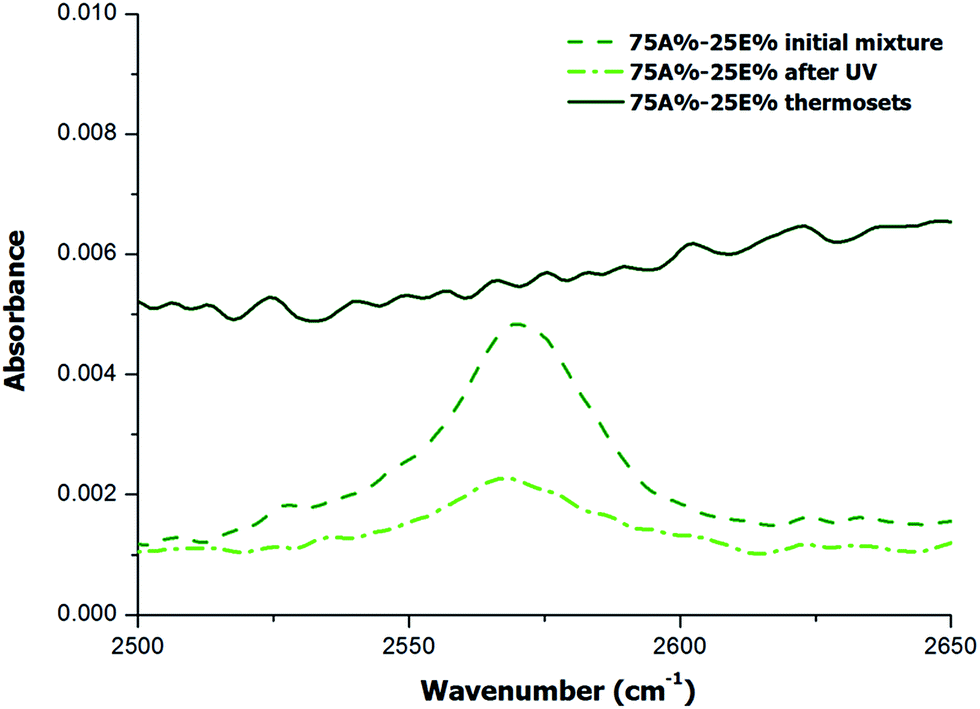 | ||
| Fig. 4 FTIR region corresponding to S–H stretching absorption band for the formulation with 75% of thiol-ene of the initial mixture, after irradiation and after complete curing. | ||
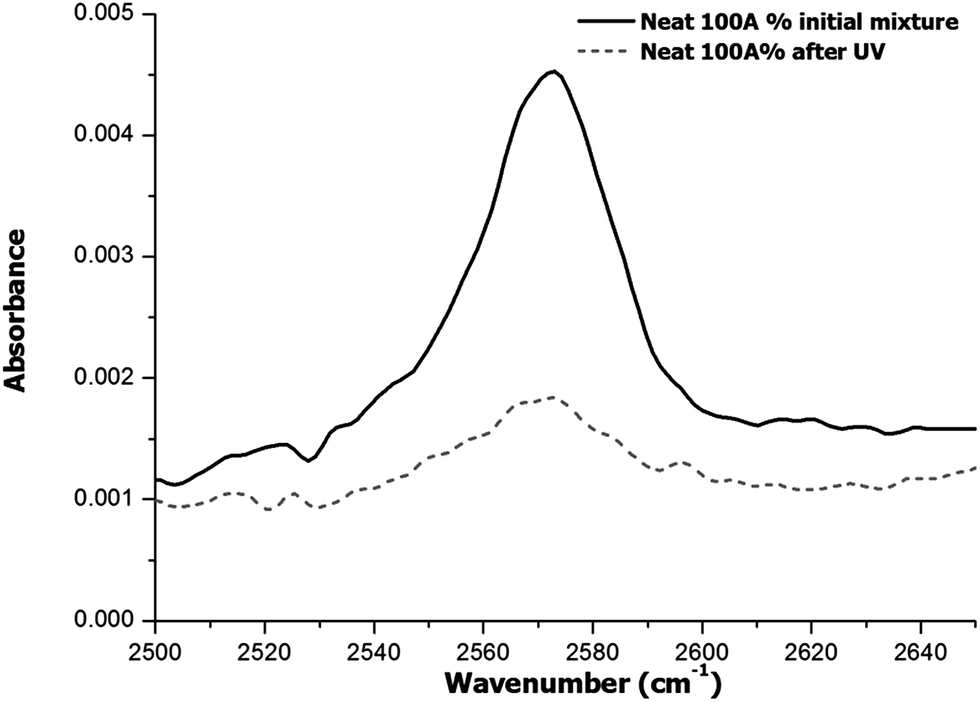 | ||
| Fig. 5 FTIR region corresponding to S–H stretching absorption band for the formulation with 100% of thiol-ene of the initial mixture and after irradiation. | ||
In the case of the formulation 75A%–25E%, Fig. 4 shows that the band about 2570 cm−1 assigned to SH stretching absorption band decreases after UV irradiation and finally disappears when the material is thermally cured. This result is in line with the expected reaction mechanism for both stages, first the thiol-ene reaction and second the thiol-epoxy reaction. Given that no unreacted thiol group was detected, and the fact that the amount of thiol is stoichiometric with respect to allyl and epoxy groups, the occurrence of epoxy homopolymerization can be ruled out. In contrast, in the spectrum of the photocured neat thiol-ene formulation (Fig. 5) the absorption of S–H stretching absorption band does not disappear completely, which indicates that the process has not been fully reacted due to the vitrification, as discussed above and in agreement with the lower heat released per allyl group (see Table 1).
Table 2 shows the results of the quantitative analysis. All calculations were done using eqn (2) and (3) detailed in the experimental part.
| Sample | A′0 | A′UV | A′final | x UV | x final |
|---|---|---|---|---|---|
| Neat 100A% | 0.101 | 0.029 | 0.007 | 0.71 | 0.93 |
| 75A%/25E% | 0.137 | 0.040 | 0 | 0.71 | 1 |
| 50A%/50E% | 0.159 | 0.081 | 0 | 0.49 | 1 |
| 25A%/75E% | 0.244 | 0.181 | 0 | 0.26 | 1 |
In the case of the neat 100% thiol-ene sample, the thiol conversion after the photocuring process is about 0.71, because of vitrification. This is in excellent agreement with the measured heat of 50 kJ mol−1, in comparison with the 64–70 kJ mol−1 for complete reaction of allyl groups in the other formulations. After heating, this material devitrifies so that the thiol-ene reaction can resume and proceed up to almost complete conversion, reaching a final value of 0.93. Some topological restrictions may be operative, leading to incomplete cure, but it must be acknowledged there may be some experimental error in its determination because the thiol peak is very small. For the formulations with different contributions of thiol-ene and thiol-epoxy reaction, the thiol conversion after the photocuring process is in good agreement with the expected reaction of thiol groups in the first curing stage by thiol-ene reaction, taking into account the composition and experimental error in the integration of such small peaks. At the end of the curing process, after heating the samples to promote epoxy-thiol reaction, no thiol was detected. Therefore it can be assumed that the system reacted quantitatively to full conversion of thiol, allyl and epoxy groups so that no homopolymerization could be detected. This is in contrast with previous works reporting a certain amount of epoxy homopolymerization that the authors attributed to the reduced mobility of the thiol groups in the intermediate material.25 It was attempted to obtain further ascertainment of the above hypothesis on the curing mechanism by analysing the epoxy signal at 915 cm−1, but the overlapping of other signals appearing during curing at the region around 900 cm−1 made it difficult to compare the epoxy and thiol conversions in the second curing stage in quantitative terms, to verify there is a one to one reaction. However, it has been recently shown that epoxy homopolymerization should not take place while there is remaining thiol, because the presence of remaining thiol groups promotes fast formation of thiolate anion to the detriment of epoxy homopolymerization.18 Therefore it is quite safe to assume that epoxy homopolymerization does not take place in stoichiometric formulations such as the ones studied in this work.
3.3. Storage stability of the formulations without irradiation
Curing formulations that show no activity under normal conditions but show activity by external stimulation can be considered latent.29 Latency is an important concept in terms of storage stability and the possibility of preparing one-pot formulations. The latency of pure thiol-epoxy formulations with different proportions of amine precursors was previously studied in our research team and the period of save storage determined, but the addition of thiol-ene and DMPA could affect the results.17,22 The latency studies were done by calorimetry (determining the residual heat) and by rheometry (measuring the viscosity changes). The data obtained are collected in Table 3.| Sample | η a (Pa s) | η b (Pa s) | ΔHc (J g−1) | ΔHd (J eq.−1) |
|---|---|---|---|---|
| a Viscosity of the initial mixture. b Viscosity of the initial mixture after one day of storage. c Remaining heat of the initial mixture determined by DSC in a dynamic scan. d Remaining heat after one day of storage determined by DSC in a dynamic scan. e The viscosity could not be measured because of gelation. | ||||
| Neat 100A% | 1.7 | 7.6 | — | — |
| 75A%/25E% | 0.5 | e | 82 | 83 |
| 50A%/50E% | 0.5 | e | 193 | 194 |
| 25A%/75E% | 0.5 | 13 | 320 | 323 |
| Neat 100E% | 0.5 | 0.6 | 408 | 410 |
In the table it is shown that the viscosity increases with only one day of storage, making it impossible its measurement in formulations with high thiol-allyl proportion. However, it remains practically unchanged in the case of neat epoxy-thiol formulation. After two days of storage the viscosity of any sample with allyl compound could not be measured. If we look to the difference in heat evolved after one day of storage, corresponding to the epoxy-thiol reaction, there is no difference for all the formulations prepared, giving very similar values to those shown in Table 1. This clearly indicates that whereas the second stage may keep its latent characteristics during this period the first photochemical stage has a low latency and that the reaction starts even without the application of any light source. This study indicates that it is not possible to prepare one pot-formulations with this curing system, but they could be stored for several days at room temperature without adding the catalysts, since uncatalyzed formulations keep good processability. It should be commented that PDU-250 is advantageous in comparison with other amine catalysts, such as those used by other authors in similar curing systems.25
3.4. Stability of the materials after the photoirradiation stage
To test the stability of the intermediate materials with the storage time, it was necessary to track the remaining heat by DSC after irradiation, since rheological measurements were not possible due to thiol-ene crosslinking after the first stage. Therefore, we proceeded to irradiate all the mixtures in the UV chamber. The resulting samples were stored at room temperature and then the remaining enthalpy was periodically measured. In this way, the advancement of the epoxy-thiol process could be evaluated. It should be pointed out, for comparison purposes, that mixtures of DGEBA/thiol containing 2 phr of PDU-250 (in reference to DGEBA) showed a latency of only 2 days at 35 °C.22 The results obtained for the present formulations are represented in Fig. 6.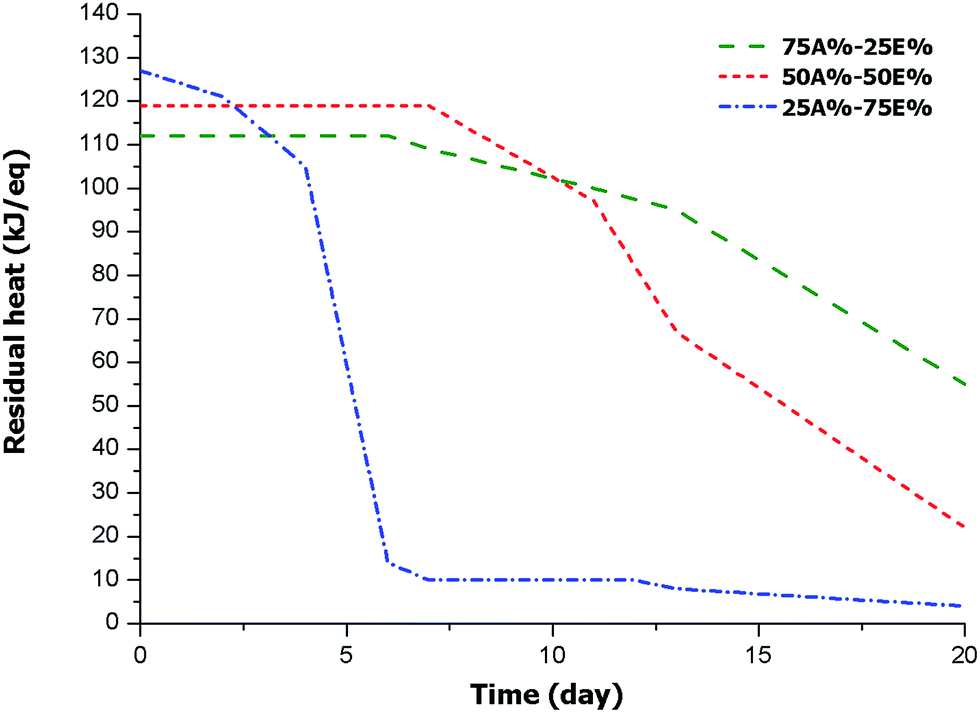 | ||
| Fig. 6 Plot of remaining enthalpy of curing against storage time for the materials obtained from different formulations. | ||
The plot shows the influence of the proportion of thiol-ene system in the stability of the intermediate material. For the 25A%–75E% formulation, with lower amount of thiol-ene, there is a significant decrease in remaining heat after the second day of storage because of the activation of the thiol-epoxy reaction. After six days of storage the process stops (reaching a conversion of 89%), possibly due to the vitrification. The other two formulations behave in a different manner, showing a longer stability. The sample 50A%-50%E is completely stable up one week and then the reaction begins and reaches a conversion of 18% after 11 days. After three weeks, the epoxy conversion is 81%. The formulation 75A%–25%E with less epoxy-thiol proportion shows a higher stability. After one week of storage, a conversion of 11% could be measured. Epoxy-thiol reaction proceeds further but at a slow rate, so that after three weeks the epoxy conversion is only 51%. In general, the lower the proportion of epoxide in the formulation, the longer the storage time of the intermediate material. This reflects the effect of the primary thiol-ene network formed in the photoirradiation stage in the immobilization of the thiol-epoxy reactive groups. According to these results, the dual system proposed could be of interest for assembling devices, for flip chip processes, for perimeter sealing and especially for automated in line assemblies, but it shall not apply to uses that require a long transport or long time between both curing stages. However, the curing system proposed keeps the characteristics of the intermediate material much longer than those previously described.25,26
3.5. Characterization of the materials
The materials obtained after dual curing were transparent but coloured because of the thermal treatment (Fig. 7). | ||
| Fig. 7 Photographs of the materials obtained. | ||
There are few differences in the transparency due to the good compatibility between thiol-ene and thiol-epoxy structures by the covalent bonding between them in the network.
The thermosets obtained from the different formulations were studied by TGA to evaluate their thermal stability. The materials were photocured and then thermally cured at 120 °C for 1 h with a post-curing at 150 °C for 1 h. Fig. 8 shows the derivative curves registered in inert atmosphere. Other relevant degradation parameters, the onset temperature of degradation (T5%, temperature at which a mass loss of 5% is observed) and degradation peak temperature, are presented in Table 4.
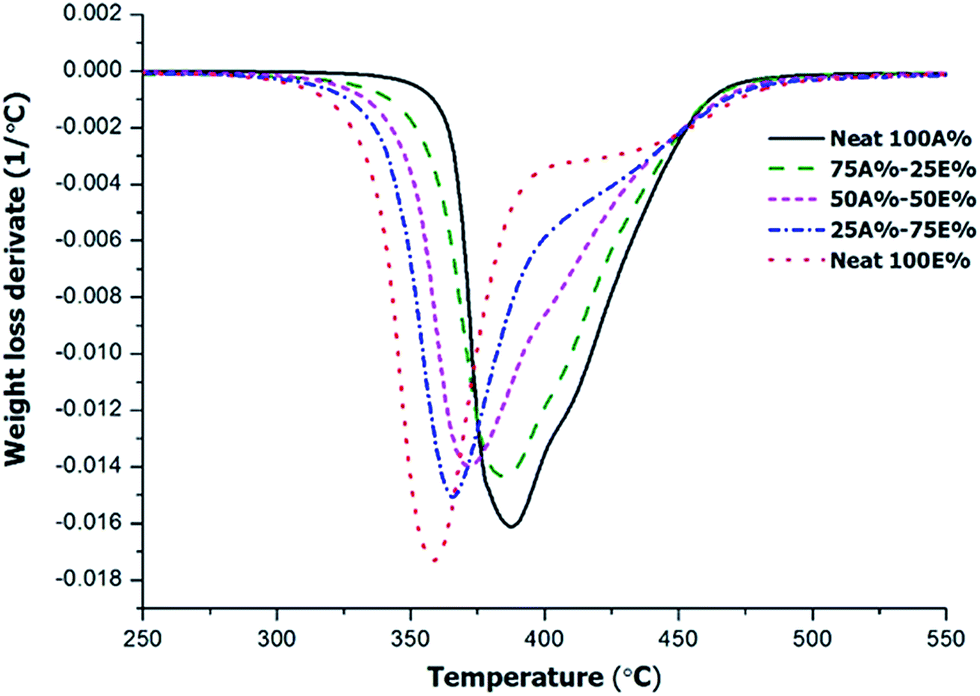 | ||
| Fig. 8 DTG curves under N2 at 10 K min−1 of the materials obtained. | ||
| Notation | TGA | DMTA | |||
|---|---|---|---|---|---|
| T 5% a (°C) | T max b (°C) |
T
tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ
c (°C) δ
c (°C) |
E r d (Pa) | E e (MPa) | |
|
a Temperature of 5% of weight loss in N2 atmosphere.
b Temperature of the maximum rate of degradation in N2 atmosphere.
c Glass transition temperature determined by DMTA.
d Storage modulus in the rubbery state determined at tanδ + 50 °C.
e Young modulus at 30 °C.
|
|||||
| Neat 100A% | 372 | 388 | 74 | 30.0 | 1177.0 |
| 75A%/25E% | 350 | 386 | 73 | 24.8 | 1122.0 |
| 50A%/50E% | 348 | 373 | 72 | 22.3 | 1029.0 |
| 25A%/75E% | 338 | 367 | 68 | 15.1 | 933.6 |
| Neat 100E% | 330 | 359 | 62 | 11.4 | 920.9 |
The curves show unimodal/bimodal shapes, with an overlapping between two processes, indicating a complex degradation profile. Unlike other epoxy systems combining two curing processes, such as epoxy-amine/epoxy homopolymerization30 or dual-curing combining aza-Michael and radical homopolymerization of acrylates,31 it is not possible to assign these processes to the individual contribution of each one of the networks being formed in the dual-curing process. This may be explained by the fact that the thiol is present in both networks so that the thermal stability of the materials are intermediate between both. In fact, as a general trend, it can be observed that the whole degradation peak is shifted at lower temperatures with increasing proportion of epoxide in the formulation. The shoulder at higher temperatures remains in the same position and decreases in intensity with increasing epoxy content, but is present already in the neat thiol-epoxy formulation. The global stability trend is also observed in Table 4. The thermal stability of all materials is remarkable in any case (T5% above 330 °C) but both the T5% and the peak degradation temperature decrease with increasing epoxy in the formulation. It should be considered the different structure of the monomers that leads to a higher crosslinking density for thiol-ene network due the compact structure of TAIC.
The thermomechanical characteristics of the materials were tested by DMTA. Fig. 9 shows the tanδ curves for the thermosets and the relevant parameters are collected in Table 4.
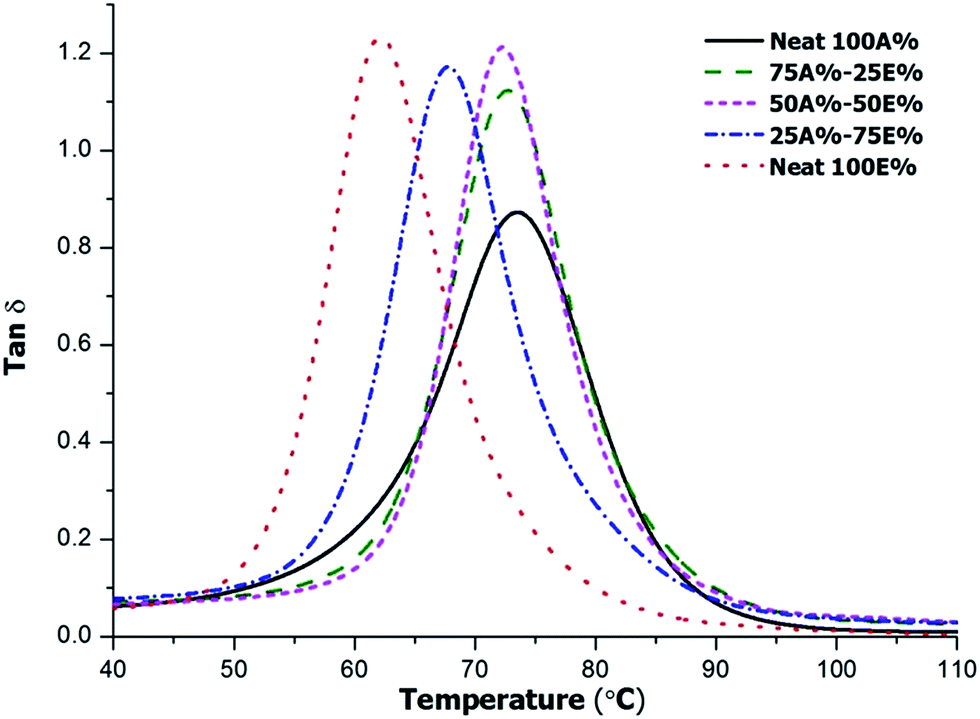 | ||
| Fig. 9 Plot of tanδ against temperature of the different materials prepared. | ||
As we can see, the curves are unimodal and quite narrow indicating the homogeneity of the samples. The maximum of tanδ is shifted to higher temperatures on increasing the proportion of allyl compound in the formulation as the result of the higher functionality and stiffness of its structure in comparison with DGEBA. The values obtained follow the same trend as those obtained by DSC, which are collected in Table 1. Similarly, in a previous work, we were able to increase the Tgs of thiol-epoxy thermosets by substituting bisphenol A epoxide by triglycidyl isocyanurate due the stiffness and higher functionality of this compound.17
The Young modulus of the material at 30 °C shows an increase in stiffness on increasing the proportion of allyl compound in the formulation. This could be expected taking into account the increase in Tg with the increase in the more rigid allyl component, so that the measurement of the Young modulus is less affected by the slow relaxation of the network, given the proximity of the measurement temperature and the Tg of all the materials. There is also a noticeable increase in the moduli in the rubbery state on increasing the proportion of allyl that could be expected taking into account the increase in crosslinking density given the higher functionality obtained when the allyl component is used instead of DGEBA.
As it has been demonstrated in this study the structure and proportion of comonomers can be varied to tailor the characteristics of the dual-cured materials in both the intermediate stage and at the end of the curing process, although the flexible structure of the commercially available thiol compounds is the main limitation of these processes to reach materials with a higher Tg and mechanical characteristics.
4. Conclusions
A new dual curing procedure based in the combination of UV-induced thiol-ene and thermal thiol-epoxy click reactions has been established by the use of a latent amine precursor that can catalyze the thermal process. The application of this procedure makes it possible to obtain tailored intermediate and final materials by just changing the formulation composition, and to store them for a certain period of time in the intermediate state before the final application and completion of the curing process.From the DSC analysis of different TAIC/DGEBA formulations with the stoichiometric amount of tetrathiol using LC-80 or PDU-250 as latent catalysts it was possible to determine that PDU-250 was a more suitable catalyst to suppress the overlapping between photo and thermal processes as it was more resistant to the possible temperature overshot occurring during the first photocuring stage. The amount of PDU-250 was adjusted to reach complete curing in all formulations. FTIR confirmed that homopolymerization of epoxides did not occur.
The materials in the intermediate stage, after photoirradiation, showed higher storage stability on increasing the proportion of allyl compound in the formulation. The new dual curing system could be applied to assemble devices, in flip chip processes, in perimeter sealing and especially in automated in line assemblies.
The increasing presence of the allyl compound in the formulation increased the Tg in the intermediate stage due to the higher extent of cure and also that of the final material due to the increase in crosslinking density and the lower mobility of the resulting network.
Thermosetting materials exhibited good transparency and thermal stability, with an increase in the initial degradation temperature with the amount of TAIC in the formulation. The moduli of these materials were also affected by the ratio of this compound in agreement with its more rigid structure and the increase in Tg.
Acknowledgements
The authors would like to thank MINECO (MAT2014-53706-C03-01, MAT2014-53706-C03-02) and Generalitat de Catalunya (2014-SGR-67) for the financial support.References
- D. P. Nair, N. B. Cramer, J. C. Gaipa, M. K. McBride, E. M. Matherly, R. R. McLeod, R. Shandas and C. N. Bowman, Adv. Funct. Mater., 2012, 22, 1502–1510 CrossRef CAS
.
- C.-H. Park, S.-W. Lee, J.-W. Park and H.-J. Kim, React. Funct. Polym., 2013, 73, 641–646 CrossRef CAS
- R. Acosta Ortiz, A. E. Garcia Valdez, L. Berlanga Duarte, R. Guerrero Santos, L. R. Ovando Flores and M. D. Soucek, Macromol. Chem. Phys., 2008, 209, 2157–2168 CrossRef
- D. Foix, X. Ramis, A. Serra and M. Sangermano, Polymer, 2011, 52, 3269–3276 CrossRef CAS
- C. E. Hoyle and C. N. Bowman, Angew. Chem., 2010, 49, 1540–1573 CrossRef CAS PubMed
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC
- U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3147–3165 CrossRef CAS
- Y. Jian, Y. He, Y. Sun, H. Yang, W. Yang and J. Nie, J. Mater. Chem. C, 2013, 1, 4481–4489 RSC
- V. S. Khire, T. Y. Lee and C. N. Bowman, Macromolecules, 2008, 41, 7440–7447 CrossRef CAS
- B.-S. Chiou, R. J. English and S. A. Khan, Macromolecules, 1996, 29, 5368–5374 CrossRef CAS
- Y.-H. Li, D. Wang and J. M. Buriak, Langmuir, 2010, 26, 1232–1238 CrossRef CAS PubMed
- N. B. Cramer, J. P. Scott and C. N. Bowman, Macromolecules, 2002, 35, 5361–5365 CrossRef CAS
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS
- M. Kharasch, W. Nudemberg and G. Mantell, J. Org. Chem., 1951, 16, 524–532 CrossRef CAS
- A. Brandle and A. Khan, Polym. Chem., 2012, 3, 3224–3227 RSC
- D. Guzmán, X. Ramis, X. Fernández-Francos and A. Serra, Polymers, 2015, 7, 680–694 CrossRef
- R. Meizoso Loureiro, T. Carballeira Amarelo, S. Paz Abuin, E. R. Soulé and R. J. J. Williams, Thermochim. Acta, 2015, 616, 79–86 CrossRef
-
Y. Tanaka and R. S. Bauer, in Curing Reactions, ed. C. A. May, Marcel Dekker, New York, 2nd edn, 1988, ch. 3, pp. 310–312 Search PubMed
- T. Ware, D. Simon, K. Hearon, T. H. Kang, D. J. Maitland and W. Voit, Macromol. Biosci., 2013, 13, 1640–1647 CrossRef CAS PubMed
-
E. M. Petrie, Epoxy adhesive formulations, McGraw-Hill Companies Inc, USA, 2006 Search PubMed
- D. Guzmán, X. Ramis, X. Fernández-Francos and A. Serra, Eur. Polym. J., 2014, 59, 377–386 CrossRef
-
J. P. Pascault, H. Sautereau, J. Verdu and J. J. Williams, Thermosetting Polymers, Marcel Dekker, New York, USA, 2002 Search PubMed
- D. R. Miller, E. M. Valles and C. W. Macosko, Polym. Eng. Sci., 1979, 19, 272–283 CAS
- J. A. Carioscia, J. W. Stansbury and C.
N. Bowman, Polymer, 2007, 48, 1526–1532 CrossRef CAS PubMed
- C. F. Carlborg, A. Vastesson, L. Yitong, W. van der Wijngaart, M. Johansson and T. Haraldsson, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2604–2615 CrossRef CAS
- F. Saharil, F. Forsberg, Y. Liu, P. Bettotti, N. Kumar, F. Niklaus, T. Haraldsson, W. van derWijngaart and K. B. Gylfason, J. Micromech. Microeng., 2013, 23, 025021, DOI:10.1088/0960-1317
- M. Sangermano, M. Cerrone, G. Colucci, I. Roppolo and R. Acosta Ortiz, Polym. Int., 2010, 59, 1046–1051 CAS
- T. Endo and F. Sanda, Macromol. Symp., 1996, 107, 237–242 CrossRef CAS
- X. Fernandez-Francos, D. Santiago, F. Ferrando, X. Ramis, J. M. Salla, À. Serra and M. Sangermano, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1489–1503 CrossRef CAS
- G. Gonzalez, X. Fernandez-Francos, A. Serra, M. Sangermano and X. Ramis, Polym. Chem., 2015, 6, 6987–6997 RSC
| This journal is © The Royal Society of Chemistry 2015 |