
The pore network and the adsorption characteristics of mesoporous silica aerogel: adsorption kinetics on a timescale of seconds†
 *a
*a
Abstract
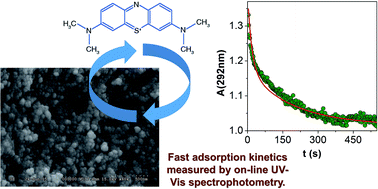
Mesoporous silica aerogel particles of ca. 5 μm in diameter can be conveniently produced by grinding in an aqueous phosphate buffer at pH 7. The pores in the suspended aerogel particles are spherical and their diameter is 18–20 nm, as measured by NMR cryoporometry. NMR diffusiometry revealed that diffusion of water is hindered inside the pores of the aerogel. In spite of steric hindrance, bulk water and pore water exchange rapidly on the millisecond timescale in the suspension, indicating a highly interconnected pore network. The adsorption of methylene blue (MB), as a model compound, was studied on the silica aerogel particles. The process was followed by on-line UV-Vis spectrophotometry after injecting the dye into the aerogel suspension. Biphasic kinetics were observed with the first process complete in ca. 80 s and the second in ca. 600 s. A detailed kinetic model was developed for the interpretation of the results. It postulates a relatively fast adsorption process with Langmuir-type kinetics, and the aggregation of aerogel particles covered by the dye on a longer timescale. The aggregates are involved in a reversible sedimentation process which actually removes MB from the suspension.
 Please wait while we load your content...
Please wait while we load your content...

